

Abstract
The genotype data of 7054 single nucleotide polymorphism (SNP) loci in 40 tomato lines, including inbred lines, F1 hybrids, and wild relatives, were collected using Illumina's Infinium and GoldenGate assay platforms, the latter of which was utilized in our previous study. The dendrogram based on the genotype data corresponded well to the breeding types of tomato and wild relatives. The SNPs were classified into six categories according to their positions in the genes predicted on the tomato genome sequence. The genes with SNPs were annotated by homology searches against the nucleotide and protein databases, as well as by domain searches, and they were classified into the functional categories defined by the NCBI's eukaryotic orthologous groups (KOG). To infer the SNPs' effects on the gene functions, the three-dimensional structures of the 843 proteins that were encoded by the genes with SNPs causing missense mutations were constructed by homology modelling, and 200 of these proteins were considered to carry non-synonymous amino acid substitutions in the predicted functional sites. The SNP information obtained in this study is available at the Kazusa Tomato Genomics Database (http://plant1.kazusa.or.jp/tomato/).
Keywords: single nucleotide polymorphism (SNP), Solanum lycopersicum, infinium assay, goldengate assay, homology modelling
1. Introduction
The emergence of massive parallel sequencers known as next generation sequencers (NGSs) has drastically changed DNA sequencing strategy and has accelerated the collection of genome sequences not only of unicellular and model plants, but also of plants that have large and complicated genomes. Moreover, NGSs can be used to detect huge numbers of sequence variations or polymorphisms, e.g. single nucleotide polymorphisms (SNPs), insertions and deletions (indels), and copy-number variations (CNV), among different species, germplasms and varieties.1 The usual strategy for detecting such variations using NGSs is to re-sequence portions of the genome by mapping the sequence reads from the NGSs onto the accurate reference genome sequences.2 Along with the advances associated with NGSs, high-throughput SNP genotyping methods based on microbeads or microarrays have also been developed, e.g. GoldenGate and Infinium assays (Illumina Inc., San Diego, CA, USA) and GeneChip Mapping/SNP arrays (Affymetrix, Santa Clara, CA, USA). Using various combinations of these technologies for large-scale DNA sequencing and genotyping, genome-wide genetic studies have been extended from models to various plant species.3
SNPs are the most abundant DNA polymorphisms in genome sequences and are thought to play major roles in the induction of phenotypic variations. In addition, the positions of SNPs on genome sequences affect gene expression and functions. In particular, nonsense mutations in exon regions are considered to be the most critical to phenotypic variations, and missense mutations accompanied by non-synonymous amino acid substitutions might influence protein functions. Moreover, the substitutions at splice junction sites produce truncated proteins. SNPs in upstream and downstream regions might cause phenotypic variations because they often activate or suppress gene expression by causing substitutions in regulatory sequences, e.g. promoters and terminators. Therefore, investigations of the SNPs related to functional changes of genes are considered essential for clarifying the functional genomics of organisms, including plants. In human genetics, the categorized SNPs associated with human diseases have been released from the F-SNP database (http://compbio.cs.queensu.ca/F-SNP/).4 Based on the support vector machine models, the sequence profiles and the structural stability of proteins whose functions are changed by non-synonymous substitutions have also been determined and are reported on the SNP3D website (http://www.snps3d.org).5 In plants, the functional SNPs on ‘granule bound starch synthase I’ (GBSSI) have been screened by computational analyses including sequence alignments and the modelling of three-dimensional structures.6
Cultivated tomato (Solanum lycopersicum) is one of the world's most important vegetable crops. It is characterized by a self-crossing reproduction system and a diploid genome (2n = 2X = 24) consisting of approximately 950 Mb. The whole-genome sequence of tomato was published recently by the International Tomato Genome Consortium,7 and a total of 34 727 protein-coding genes were predicted by the International Tomato Annotation Group (ITAG) (http://www.uk-sol.org). Using the whole-genome sequence as a reference, a large number of SNPs in tomato have been identified by the re-sequencing strategy.8 In a subsequent study, 7720 genome-wide SNPs were genotyped for 426 tomato lines, consisting of 410 inbred and 16 hybrid lines, using a high-throughput genotyping system, Illumina's Infinium assay. The SNPs on the assay were called ‘scorable SNPs’ because they could be used to score the tomato germplasm in genotyping.9 In our previous study, we discovered 1337 SNPs by a comparative analysis of transcriptome sequences derived from Micro-Tom and 19 tomato lines [SOL Genomics Network (SGN),10 http://solgenomics.net; MiBASE,11 http://www.pgb.kazusa.or.jp/mibase/] and used them for polymorphism analysis of 27 tomato lines by the GoldenGate assay.12 Although a large number of SNPs in tomato have been discovered in previous studies, their functions have not been surveyed.
In this study, we used the Infinium assay developed by Sim et al.9 to perform SNP genotyping for 40 tomato lines, including the 27 lines used in our previous study.12 The SNP genotype data obtained in the present and previous studies12 were merged. To infer their effects on gene functions, SNPs can be classified into six categories: cSNPs (SNPs in a coding region that cause amino acid substitution), sSNPs (SNPs in a coding region that do not cause amino acid substitution), iSNPs (SNPs in an intron region), uSNPs (SNPs within the 1 Kb region upstream from the start codon), dSNPs (SNPs within the 1 Kb region downstream from the stop codon), and gSNPs (SNPs in intergenic regions). Furthermore, the three-dimensional structures of proteins encoded by genes having cSNPs were constructed by homology modelling. Based on the constructed protein structures, candidates for the functional sites were predicted to identify the SNPs that directly affect protein functions. We expected that the SNPs located on the candidate functional sites of proteins would be applicable to gene functional analysis and molecular marker-assisted selection as functional markers.
2. Materials and methods
2.1. Plant materials
Forty tomato lines, consisting of 30 lines of cultivated tomato (S. lycopersicum), 2 lines of S. lycopersicum var. cerasiforme, and 8 lines of wild relatives (2 each of Solanum pimpinellifolium, Solanum pennellii, Solanum peruvianum, and Solanum chilense), were used for the genotyping assays. The 30 S. lycopersicum lines included 5 ‘Micro-Tom’ lines that were obtained from different resource centres. Micro-Tom is the experimental line most frequently used in the study of cultivated tomato. Natural variations were found in our previous study within two lines, one derived from NIVTS, Japan and the other from INRA, France.12 Two lines of ‘M82’ derived from different sources (NIVTS and INRA) were also included in the 30 S. lycopersicum lines to investigate the existence of SNPs within M82 because M82 has been widely used in tomato genetics. The names of the lines, accession numbers, sources, and breeding types are listed in Table 1. All lines were grown in a greenhouse, and the DNA of each line was isolated from leaves using a DNeasy Plant Mini Kit (Qiagen, Hilden, Germany).
Table 1.
Descriptions of the plant materials
| Species | Line names | Accession Number | Sourcesa | Breeding type | Array type | Descriptions | Referencesb |
|---|---|---|---|---|---|---|---|
| S. lycopersicum | |||||||
| Micro-Tom MM | INRA | Inbred | I & G | A parental line of MMF2 | 1 | ||
| Micro-Tom KDRI | KDRI | Inbred | I | Genome sequenced line | 2 | ||
| Micro-Tom NBRP (TOMJPF00001) | TOMJPF00001 | NBRP | Inbred | I | |||
| Micro-Tom AM | NIVTS | Inbred | I & G | A parental line of AMF2 | 1 | ||
| Micro-Tom TGRC (LA3911) | LA3911 | TGRC | Inbred | I | |||
| Ailsa Craig | NIVTS | Inbred | I & G | A parental line of AMF2 | 1 | ||
| Aichi First | NIVTS | Inbred | I & G | 1 | |||
| Best Of All | LS3908 | NIVTS | Inbred | I & G | 1 | ||
| Earliana | LA3238 | TGRC | Inbred | I & G | 1 | ||
| Fruit | LS1100 | NIVTS | Inbred | I & G | 1 | ||
| Furikoma | NIVTS | Inbred | I & G | 1 | |||
| Heinz 1706-BG | LA4345 | TGRC | Inbred | I & G | Genome sequenced line | 1,3 | |
| LA925 | LA925 | CU | Inbred | I & G | A parental line of Tomato-EXPEN2000 | 1,4,5 | |
| M82_MM | INRA | Inbred | I & G | A parental line of MMF2 | 1 | ||
| M82 | NIVTS | Inbred | I | ||||
| Marglobe | LA0502 | TGRC | Inbred | I & G | 1 | ||
| Money Maker | LA2706 | TGRC | Inbred | I & G | 1 | ||
| Tomato Chuukanbohon Nou 11 | NIVTS | Inbred | I & G | 1 | |||
| Tomato Chuukanbohon Nou 9 | NIVTS | Inbred | I & G | 1 | |||
| Ponderosa | LS1728 | NIVTS | Inbred | I & G | 1 | ||
| Rio Grande | LA3343 | TGRC | Inbred | I & G | 1 | ||
| Rutgers | LA1090 | TGRC | Inbred | I & G | 1 | ||
| San Marzano | LS4956 | NIVTS | Inbred | I & G | 1 | ||
| Geronimo | De Ruiter Seeds Co. | F1 hybrid | I & G | 1 | |||
| Labell | De Ruiter Seeds Co. | F1 hybrid | I & G | 1 | |||
| Matrix | De Ruiter Seeds Co. | F1 hybrid | I & G | 1 | |||
| Momotaro 8 | Takii Seeds Co. | F1 hybrid | I & G | 1 | |||
| Reika | Sakata Seeds Co. | F1 hybrid | I & G | 1 | |||
| Sweet 100 | Vilmorin Seeds Co. | F1 hybrid, cherry type | I & G | 1 | |||
| Regina | Sakata Seeds Co. | Inbred, cherry type | I & G | 1 | |||
| S. lycopersicum var. cerasiforme | |||||||
| var. cerasiforme (TOMJPF00009) | TOMJPF00009 | NBRP | I | ||||
| var. cerasiforme (WIR25/30) | WIR25/30 | NIVTS | I | Canadian line | |||
| S. chilense | |||||||
| S. chilense (LA1958) | LA1958 80L | NIVTS | I | ||||
| S. pennellii | |||||||
| S. pennellii (LA716) | LA716 | CU | I & G | A parental line of Tomato-EXPEN2000 | 1,4,5 | ||
| S. pennellii (TOMJPF00008) | TOMJPF00008 | NBRP | I | ||||
| S. peruvianum | |||||||
| S. peruvianum (TOMJPF00010) | TOMJPF00010 | NBRP | I | ||||
| S. peruvianum (WIR2020) | WIR2020 | NIVTS | I | American line | |||
| S. pimpinellifolium | |||||||
| S. pimpinellifolium (TOMJPF00012) | TOMJPF00012 | NBRP | I | ||||
| S. pimpinellifolium (PI344102) | PI344102 | NIVTS | I | American line | |||
| S. pimpinellifolium (LS1513) | LS1513 | NIVTS | I | Peru line |
aCU, Cornell University; INRA, Institut National de la Recherche Agronomique; KDRI, Kazusa DNA Research Institute; NBRP, National BioResource Project, University of Tsukuba; NIVTS, National Institute of Vegetable and Tea Sciences, National Agriculture and Food Research Organization; TGRC, Tomato Genetic Resource Center, University of California, Davis.
b1: Shirasawa et al. (2010) DNA Res., 17, 381-391.; 2: Aoki et al. (2010) BMC Genomics, 11, 210.; 3: Tomato Genome Consortium (2012) Nature, 485, 635-641.; 4: Shirasawa et al. (2010) Theor. Appl. Genet., 121,731-739.; 5: Fulton et al. (2002) Plant Cell, 14,1457-1467.
2.2. SNP genotyping
Genome-wide SNP genotyping was performed using the Infinium assay (Illumina Inc.) that was developed by the Solanaceae Coordinated Agricultural Project (SolCAP),8 according to the manufacturer's standard protocol. The probe sequences and SNP information are available from SolCAP (http://solcap.msu.edu) and in Supplementary Table S1. The data obtained from the assays were analysed using the Genotyping Module of the GenomeStudio software (Illumina Inc.).
2.3. Genetic diversity analysis and detection of linkage disequilibrium (LD)
The Hamming distances13 between each pair of the 40 tomato lines tested were calculated to generate the distance matrix based on the genotyping data obtained in this study. A neighbour-joining tree was generated using MEGA 5 software.14 The heterozygosity (HZ) values were calculated by the equation
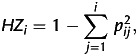 |
where pij is the frequency of the jth of the i allele.
Linkage disequilibrium (LD) in the tomato lines was detected by the r2 and D′ values calculated using the Tassel program.15
2.4. Classification of SNPs according to their locations on the genome sequence
The locations of SNPs on the tomato genome genotyped using the two assay platforms, Infinium and GoldenGate, were identified by the following method. The probe sequences in both assays were searched against the tomato genome sequence SL2.40 using Blastn16 with an E-value cutoff of 1E-20. According to the relative positions between the SNPs and the genes predicted on the tomato genome sequence, the SNPs were classified into six groups, as described above: cSNPs, sSNPs, iSNPs, uSNPs, dSNPs, and gSNPs.
2.5. Inference of SNP effects on protein and gene functions
The amino acid sequences of the genes were searched against the Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do) by Blastp16 with an E-value cutoff of 1E-10. The amino acid sequences were further aligned on those of the homologous proteins by Fasta36 program,17 and homology modelling was performed by Modeller 9.10 software18 based on the alignment with default parameters. The functional sites on the constructed protein structures were predicted using FPocket software19 with default parameters. The functions of proteins were predicted by combining the results obtained by the Blastp searches with an E-value cutoff of 1E-10 against the databases NR (non-redundant amino acid sequence database released by NCBI) (http://www.ncbi.nlm.nih.gov), TAIR10,20 KOG,21 and KEGG.22 The functional domains of proteins were searched against Pfam database release 26.023 using HMMscan program.24
3. Results and discussion
3.1. SNP genotyping by high-throughput assay technologies
Of the 7720 scorable SNPs defined by the Infinium assay, 7617 (98.7%) were successfully genotyped, and 6374 (82.6%) showed polymorphism among the 40 tested lines. In our previous study using the GoldenGate assay, 916 of 1338 SNP loci (68.5%) showed polymorphism among the 27 tomato lines.12 Among the 916 SNP loci on the GoldenGate assay, 236 were duplicated on the Infinium assay; therefore, the genotype data of the specific 680 SNP loci on the GoldenGate assay were analysed with the 6374 SNPs of the Infinium assay. In total, polymorphic data were successfully obtained at 7054 SNP loci. The numbers of SNPs identified between each pair of the 40 lines are shown in Supplementary Table S2. The average number of polymorphic loci between each pair of the 40 lines was 4270, whereas that between each pair of the 30 cultivated tomatoes, the 2 lines of S. lycopersicum var. cerasiforme, and the 5 lines of Micro-Tom, was 930.
The heterozygous alleles were found at 2292, 967, 921, 901, 772, and 1200 SNP loci within the F1 hybrid varieties of ‘Sweet 100’, ‘Geronimo’, ‘Matrix’, ‘Labell’, ‘Momotaro 8’, and ‘Reika’, respectively. In two of the five lines of Micro-Tom, i.e. ‘Micro-Tom NBRP’ and ‘Micro-Tom KDRI,’ all genotypes were completely identical. In the other pairs of Micro-Tom, the numbers of identified SNPs ranged from 275 (between ‘Micro-Tom TGRC’ and Micro-Tom KDRI/Micro-Tom NBRP) to 1008 (between ‘Micro-Tom MM’ and Micro-Tom KDRI/Micro-Tom NBRP).
3.2. Classification of tomato lines based on genotyping data
The genetic distances between all the combinations of any two lines were calculated based on the genotyping data of the 7054 SNP loci, and a dendrogram was then constructed (Fig. 1). Of the 40 lines examined, 36 were classified into 4 large clusters, which consisted of 8 wild relatives (Cluster 1), 5 lines of Micro-Tom (Cluster 2), 5 F1 hybrid cultivars, and an inbred line, ‘Tomato Chuukanbohon Nou 11′ (Cluster 3), and the 15 inbred lines and 2 lines of S. lycopersicum var. cerasiforme (Cluster 4). Because Momotaro 8 was used as a backcross parent in the breeding program of Tomato Chuukannbohon Nou 11,25 75% of the genome of the Tomato Chuukanbohon Nou 11 might be identical to that of Momotaro 8, one of the tested F1 cultivars. Two cherry types, ‘Sweet 100’ and ‘Regina,’ and the two lines of M82 were separated from the four clusters. Sweet 100 showed the largest genetic distances in comparison with the other cultivated tomato lines. The graphical genotypes of the 40 tomato lines with the 7054 SNPs are shown in Supplementary Fig. S1. Most of the SNP loci were located on the ends of chromosomes corresponding to the euchromatic regions, where the gene density is higher than that on the heterochromatic regions, except for the three regions of the heterochromatins of chromosomes 3, 11, and 12. The biased locations of the SNPs on the genome have been found. These results are valid because most of the SNPs on the Infinium and GoldenGate assays were selected from the transcriptome sequences.9,12
Figure 1.
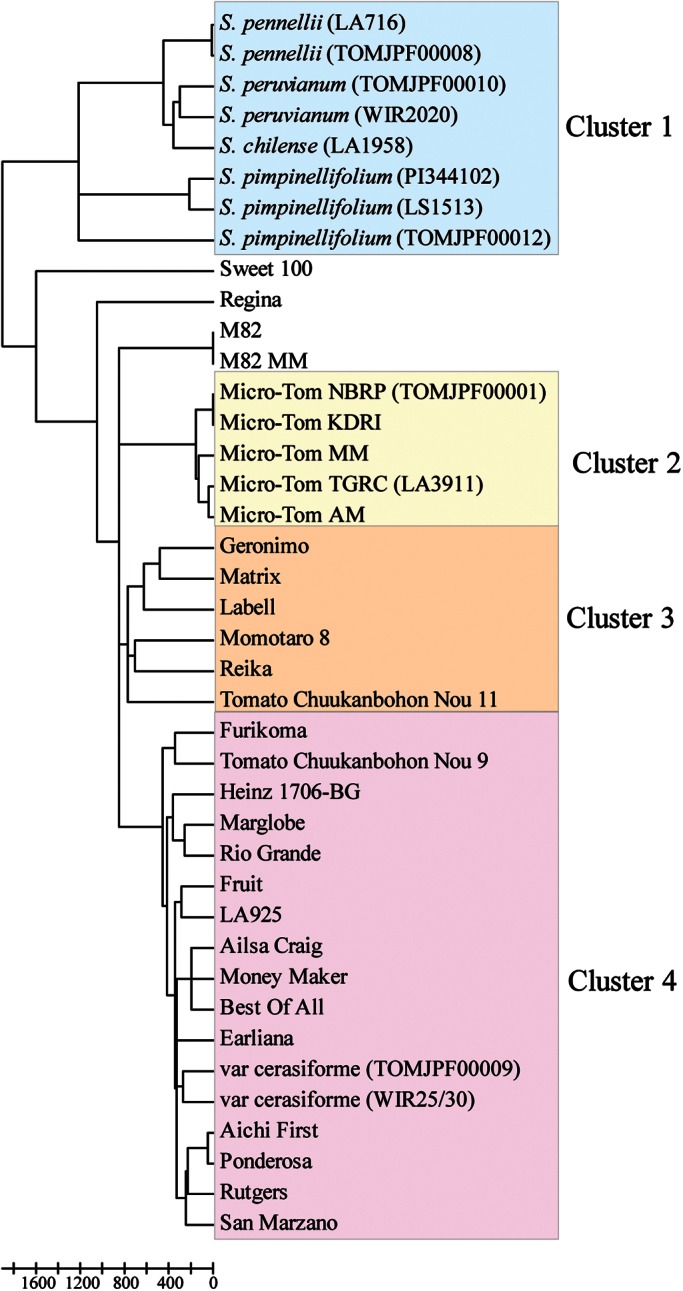
A dendrogram for the 40 lines of tomatoes based on the 7054 SNP genotypes.
The HZ value of the 7054 SNP loci was 0.30 on average, ranging from 0.25 for chromosome 10 to 0.37 for chromosome 11 (Table 2). The average HZ value of Cluster 1, which consisted of the eight wild species, was the highest among the four clusters. In Cluster 2, which included the five lines of Micro-Tom, the HZ values were greater than zero, except for chromosome 11. In other words, SNPs were observed on 11 of 12 chromosomes within the Micro-Tom lines. Micro-Tom was developed by Scott and Harbaugh in 198926 and has been used as a model line in tomato genetics since 1997.27 The heterozygous regions in the five Micro-Tom lines might be caused by the existence of heterozygous regions in the first generation of Micro-Tom or mutations that arose during generation changes. In Cluster 3, which included the F1 hybrid cultivars, high HZ values were found in the large regions of chromosomes 6 (8.7–30.0 Mb), 9 (4.8–58.9 Mb), and 11 (7.7–49.3 Mb) and in small segmental regions on all chromosomes. On the other hand, in Cluster 4, which included the 16 inbred lines, high HZ values were found in chromosomes 3 (60.8–61.3 Mb), 7 (60.6–61.0 Mb), and 11 (3.5–4.6 Mb). The high HZ regions in chromosomes 3 (60.8–61.3 Mb) and 7 (60.6–61.0 Mb) overlapped between Clusters 3 and 4, whereas those in chromosome 11 were specific to Cluster 4. The tested F1 hybrids might be an introgressed disease-resistance gene, I-2,28 from wild relatives in the process of breeding. The gene is located on the specific high-HZ regions of Cluster 3 on chromosome 11. Therefore, we inferred that the genomic regions derived from wild relatives remained in the genomes of F1 hybrids and caused high heterozygosity in the regions (Supplementary Fig. S1).
Table 2.
Average HZ values in each chromosome investigated on the 40 tomato lines and the 4 clusters
| Chromosome | Total | Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 |
|---|---|---|---|---|---|
| 1 | 0.28 | 0.15 | >0.00 | 0.11 | 0.13 |
| 2 | 0.30 | 0.20 | 0.01 | 0.09 | 0.09 |
| 3 | 0.28 | 0.16 | 0.03 | 0.10 | 0.15 |
| 4 | 0.31 | 0.18 | 0.08 | 0.17 | 0.08 |
| 5 | 0.32 | 0.20 | 0.02 | 0.09 | 0.05 |
| 6 | 0.28 | 0.23 | 0.06 | 0.22 | 0.07 |
| 7 | 0.26 | 0.18 | 0.06 | 0.09 | 0.07 |
| 8 | 0.27 | 0.18 | 0.01 | 0.05 | 0.13 |
| 9 | 0.29 | 0.17 | >0.00 | 0.18 | 0.11 |
| 10 | 0.25 | 0.21 | >0.00 | 0.08 | 0.09 |
| 11 | 0.37 | 0.20 | 0.00 | 0.18 | 0.11 |
| 12 | 0.29 | 0.16 | 0.19 | 0.16 | 0.13 |
| 0 | 0.19 | 0.00 | 0.00 | 0.05 | 0.00 |
| Total | 0.30 | 0.19 | 0.04 | 0.13 | 0.10 |
The LD on the 40 lines and on each cluster across the chromosomes is shown in Supplementary Fig. S2. Although significant LD was not clearly observed along with the diagonal lines on any of the LD maps of the 40 tomato lines, high LD values between adjacent SNPs were segmentally observed across the genomes. In Cluster 1, high LD values were observed on all of the chromosomes, suggesting that the wild relatives were independently evolved with few inter-crossings. In Cluster 2, small LD blocks were found on chromosomes 3, 4, 6, 7, and 12 that were caused by heterozygous regions within the five Micro-Tom lines. In Clusters 3 and 4, no significant LD was observed between adjacent SNPs in most of the genomic regions. This suggested that the genomes of cultivated tomato have been well admixed in germplasm collections. Large LD blocks were found on chromosomes 4, 6, 9, and 11 of Cluster 3, whereas a clear large LD block was observed on chromosome 11 in Cluster 4. Within each block, higher LD values were observed along with the oblong bars, and not diagonally. This implied that selection biases had occurred in partial regions of the genomes during the breeding processes. For example, the genes controlling disease resistance are located on chromosomes 6 (Cf-2 and Mi), 9 (Tm22), and 11 (I-2), where LD blocks were observed (Supplementary Fig. S2). The selection of specific alleles of these genes might induce the LD blocks.
3.3. Classification of SNPs based on the positions on the tomato genome
The SNPs were classified into six categories according to their positions on the tomato genome sequence (Fig. 2). The numbers of cSNPs and sSNPs located within the coding regions were 2212 (31.4%) and 2296 (32.5%), respectively. The numbers of uSNPs and dSNPs were 516 (7.3%) and 1311 (18.6%), respectively. There were fewer iSNPs and gSNPs than the other types of SNPs, i.e. 485 (6.9%) and 234 (3.3%), respectively, because the SNPs on the probes of the assays were selected from the transcriptome sequences.9,12 The total percentage of cSNPs, uSNPs, and dSNPs having the capacity to alter gene functions was 57.3%. Most of these SNPs were located on the gene-rich euchromatic regions in each chromosome (Fig. 3). There were no significant differences in the distribution on the genome among the six SNP categories.
Figure 2.

Numbers and percentages of the SNPs classified into six categories.
Figure 3.

Locations of the 7054 SNPs on the tomato genome sequence (SL2.40). The locations of the six groups of SNPs—cSNPs, sSNPs, iSNPs, uSNPs, dSNPs, and gSNPs—are indicated by bars coloured red, blue, sky blue, orange, green, and purple, respectively. The bars below the boxes indicate the regions of the euchromatins (magenta) and heterochromatins (grey).
Among the 2212 cSNP loci, nonsense mutations were found in 15 genes (Table 3 and Supplementary Table S1). Of the 15 nonsense mutations, 3 were found in the alleles of the wild relatives, i.e. Solyc01g106570.2.1 [cystathionine beta-synthase (IPR000644)], Solyc03g083470.2.1 [protein kinase core (IPR000719)], and Solyc01g080340.2.1 [BSD (IPR005607)], and 2 mutations were found on the alleles of the Micro-Tom lines, i.e. Solyc05g025810.2.1 [six-bladed beta-propeller TolB-like (IPR011042)] and Solyc08g076000.2.1 [epidermal growth factor (EGF)-like region conserved site (IPR013032)]. Three mutations on the alleles of Solyc04g014400.2.1 [leucine-rich repeat (IPR001611)], Solyc04g064900.1.1 [glycoside hydrolase family 9 (IPR001701)], and Solyc09g090350.2.1 [long-chain fatty alcohol dehydrogenase (IPR012400)] were distributed in the inbred lines. The other 7 mutations were found across the 40 tomato lines. In ‘Heinz 1706-BG,’ whose genomic sequence has been determined,7 the five nonsense mutations were found on Solyc01g080340.2.1 [BSD (IPR005607)], Solyc01g091770.2.1 [zinc finger RING/FYVE/PHD-type (IPR013083)], Solyc02g050330.1.1 (hypothetical protein), Solyc04g056630.2.1 [cystathionine beta-synthase core (IPR000644)], and Solyc11g071510.1.1 (unnamed protein product). The 15 genes that contained cSNP-caused nonsense mutations are expected to help reveal the divergence and evolution between wild and cultivated tomato lines as well as within cultivated tomato lines.
Table 3.
Annotations of the genes in which nonsense mutations caused by cSNPs were identified
| ITAG gene model | ITAG 2.3 (InterPro) | Annotation (KDRI) |
|---|---|---|
| Solyc01g080340.2.1 | BSD (IPR005607) | Similar to BSD domain-containing protein |
| Solyc01g091770.2.1 | Zinc finger RING/FYVE/PHD-type (IPR013083) | Ring finger protein, putative |
| Solyc01g106570.2.1 | Cystathionine beta-synthase (IPR000644) | Predicted membrane protein contains two CBS domains |
| Solyc01g112110.2.1 | - | Hypothetical protein |
| Solyc02g050330.1.1 | - | Hypothetical protein |
| Solyc02g092220.1.1 | - | Hypothetical protein |
| Solyc03g083470.2.1 | Protein kinase core (IPR000719) | Serine/threonine protein kinase |
| Solyc04g014400.2.1 | Leucine-rich repeat (IPR001611) | Leucine-rich repeat |
| Solyc04g056630.2.1 | Cystathionine beta-synthase core (IPR000644) | Conserved hypothetical protein |
| Solyc04g064900.1.1 | Glycoside hydrolase family 9 (IPR001701) | Hypothetical protein |
| Solyc05g025810.2.1 | Six-bladed beta-propeller TolB-like (IPR011042) | Predicted alkaloid synthase/surface mucin hemomucin |
| Solyc07g008760.2.1 | Tetratricopeptide repeat (IPR019734) | Uncharacterized conserved protein contains tetratricopeptide repeat |
| Solyc08g076000.2.1 | EGF-like region conserved site (IPR013032) | Apoptotic ATPase |
| Solyc09g090350.2.1 | Long-chain fatty alcohol dehydrogenase (IPR012400) | Hypothetical protein |
| Solyc11g071510.1.1 | - | Unnamed protein product |
The ‘Annotation (KDRI)’ indicates the deduced product name based on homology searches against the following databases: NCBI's NR, TAIR10, KOG, and KEGG.
3.4. Substitution effects on the genes induced by cSNP, uSNP, and dSNP
To estimate the substitution effects on protein and gene functions, the KOG functional categories21 were assigned to the corresponding genes classified into the categories cSNP, sSNP, iSNP, uSNP, and dSNP. Fig. 4 shows the percentage of each of these types of SNPs on the 25 KOG categories. In addition, the standard deviations (s) of the percentages of the 25 KOG categories were calculated in each SNP type (data not shown). Hereafter, KOG categories that showed more or <1.5 s (s: sigma distribution) in the percentages were considered as significantly different KOGs in the distribution of each type of SNP. In the case of cSNPs, large ratios were observed for KOG L (replication, recombination, and repair, 41.0%) and KOG Q (secondary metabolites biosynthesis, transport, and catabolism, 40.7%), whereas a smaller ratio was identified for KOG F (nucleotide transport and metabolism, 18.8%). In the case of uSNP, significantly higher distributions were observed for KOG W (extracellular structures, 12.0%) and KOG H (coenzyme transport and metabolism, 12.5%), whereas lower distributions were identified for KOG J (translation, ribosomal structure, and biogenesis, 3.4%), KOG L (replication, recombination, and repair, 3.6%) and KOG Q (secondary metabolites biosynthesis, transport, and catabolism, 2.5%). In the case of dSNP, high and low ratios were observed for KOG Z (cytoskeleton, 35.7%), and KOG Y (nuclear structure, 11.1%), respectively. If promoters or terminators were located in the 1 Kb regions upstream or downstream from the start or stop codons of the genes, uSNP and dSNP might affect their gene expressions. For a more substantial estimation of the substitution effects on uSNP and dSNP, it is necessary to detect the promoter and terminator regions on each corresponding gene.
Figure 4.

Percentages of the 5 types of SNPs on each of the 25 KOG categories. The percentages of the observed cSNPs, sSNPs, uSNPs, dSNPs, and iSNPs are indicated by white, grey, dots, thick dots, and slated bars, respectively.
Most of the substitutions involving changes in the charges or polarity of the amino acid residues were related to alterations in the physicochemical properties of proteins. Therefore, these alterations in the physicochemical properties of the amino acid residues by substitutions were investigated for 2212 cSNP loci. The amino acid properties were not altered by the substitutions at 926 (41.9%) of the 2212 cSNP loci (grey bars in Fig. 5), whereas they were altered to non-polar (slanted bars) or polar (dotted bars) in 861 (38.9%) and 346 (15.6%) of the 2212 SNP loci, respectively. Amino acid residues whose properties were altered on a change in polarity were found in 64 (2.9%) SNP loci (white bars). In total, 1271 (57.5%) cSNP loci were predicted to alter the properties of amino acid residues.
Figure 5.

Substitution patterns of the properties of amino acid residues caused by cSNPs. The substitutions that did not alter the properties of amino acid residues are indicated by grey bars. The substitutions related to changes in polarity, e.g. to polar and to non-polar, are shown by dotted and slated bars, respectively. The substitutions related to changes in charges or amino acid residues, i.e. negatively charged (acidic) or positively charged (basic), are shown by white bars. The substitutions related to nonsense mutations are shown by black bars.
3.5. Inference of direct substitution effects on protein functions
The amino acid residues directly altering protein functions were inferred according to the locations of substitutions on the functional sites of protein structures. A total of 1271 cSNP loci that altered the charges or polarities on amino acid residues were identified in 1108 genes. Of these 1108 genes, the three-dimensional structures of 843 corresponding proteins were constructed by homology modelling using Modeller v9.10 software.18 Furthermore, the candidate functional sites were predicted by FPocket software.19
These analyses together revealed 215 cSNP loci that were candidate functional sites on 200 proteins (Supplementary Table S1). The genes whose cSNPs were located on candidate functional sites have the potential to be applied as functional markers because they could directly affect the protein functions. Therefore, differences in the cSNP distribution on the 25 KOG categories were investigated according to the cSNP positions on functional or other sites (Fig. 6). Significantly different distributions were observed on KOG O (posttranslational modification, protein turnover, and chaperones), KOG C (energy production and conversion), KOG G (carbohydrate transport and metabolism), KOG E (amino acid transport and metabolism), and KOG Q (secondary metabolites biosynthesis, transport, and catabolism).
Figure 6.

Distribution of the annotated KOGs in the genes with identified cSNPs inside or outside of functional sites. The numbers of annotated genes with cSNP located inside or outside of the functional sites are shown by white and grey bars, respectively.
Solyc09g010080.2.1, also known as brix9-2-5,29,30 controls fruit sugar content and is involved in KOG G (carbohydrate transport and metabolism). It is also annotated as beta-fructofuranosidase. The three-dimensional structure of Solyc09g010080 was built by homology modelling based on the crystal structure of a cell-wall invertase from Arabidopsis thaliana (PDB id: 2ac1) (Fig. 7). The identity of the amino acid sequences between Solyc09g010080 and 2ac1 was 51.0% in 92.6% length coverage. In the constructed structure, 28 pockets were found as candidate functional sites by the FPocket program.19 This program usually detects several candidates for functional sites, which are called pockets, in a protein structure because there are many small or large clefts on the surface of proteins. In a previous study,29,30 three amino acid substitutions (Gln297, Asn315, and Leu322) caused by cSNPs were found in brix9-2-5 between S. lycopersicum and S. pennellii. Asn315, which is one of the three amino acid residues, was identified on pocket 8 that was a relatively larger pocket that included 15 amino acid residues (Asn315, Leu316, Ser317, Lys319, Ser430, Tyr431, Lys432, Ile433, Val459, Asp460, Val461, Asp462, Leu463, Asp465, and Lys488). The amino acid residue at the 315th position was altered from Asn to Asp, and the amino acid property was changed from ‘polar’ to ‘negatively charged (acidic).’ The amino acid residue at the 315th position was Asp in the wild tomatoes and S. lycopersicum var cerasiforme (WIR25/30 and TOMJPF00009), whereas it was Asn in the other tomatoes. Aspartic acid residues play important roles in the catalytic sites of proteins, and, therefore, the activities of Solyc09g010080 might differ between the wild and cultivated tomatoes.
Figure 7.
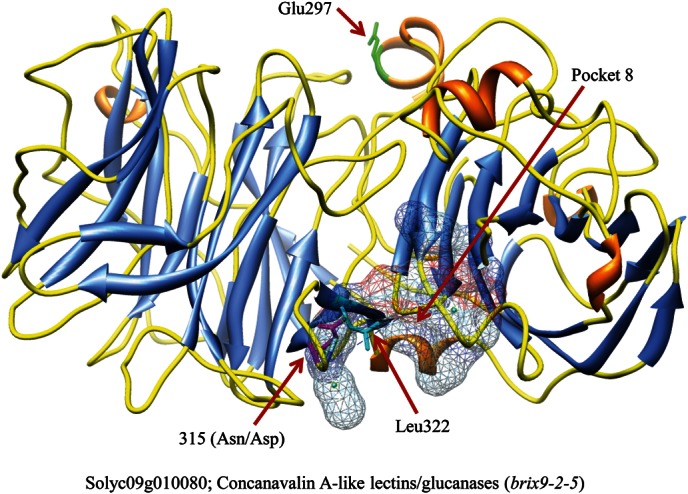
Locations of the amino acid residues substituted by a cSNP on Solyc09g010080. The amino acid residue (number 315) substituted by a cSNP was located on pocket 8. The α-helices, β-sheets, and turns are shown in orange, blue, and yellow, respectively. Pocket 8 is displayed with a mesh model. In the mesh model, the carbon, oxygen, and nitrogen atoms are coloured grey, red, and blue, respectively.
In this study, we predicted the functions of 7054 SNPs identified by array assays. Although this was the total number of SNPs we investigated, we expect that greater numbers of SNPs exist on the tomato genome. The advance of NGS technologies has enabled the discovery of genome-wide SNPs by the re-sequencing of multiple tomato lines. The approach demonstrated in this study is expected to be applied to larger-scale functional analysis of SNPs identified by re-sequencing.
4. Conclusion
A total of 7054 SNP loci were genotyped in 40 tomato lines, including S. lycopersicum, S. lycopersicum var. cerasiforme, S. pimpinellifolium, S. pennellii, S. peruvianum, and S. chilense. Based on the SNP genotypes, these 40 lines were classified into 4 clusters, and the LDs were detected on the 40 lines and each cluster. The SNP effects on gene expression were inferred by the sequences within 1 Kb upstream and downstream from the start codons because there was no information about the promoter and terminator regions in the annotations on ITAG. To investigate the effects of SNP more precisely, promoter and terminator regions should be identified by computational and experimental procedures. On the other hand, the SNP effects on protein functions were inferred by modelled structures because the molecular modelling method has been found useful for estimating the effects of the substitution of amino acid residues on protein function.6,31 Therefore, homology modelling was applied to the tomato genes with cSNPs. As a result, it was predicted that the protein functions (or catalytic activities) of the 200 genes might be altered by the substitutions because the SNPs were located on the candidate functional sites. The uSNPs, dSNPs, and cSNPs have the capacity to affect gene expressions and protein functions, so they might be related to the differences in phenotypes among tomato lines. These SNPs are expected to be applied to marker-assisted selection because they could be considered functional markers. Recently, the large-scale genotyping of SNPs has been achieved using the NGS or array platform. The identification of the SNPs that directly affect protein and gene functions is quite important for molecular breeding and enzymology.
4.1. Availability
The genotyping data, annotations, and modelled protein structures with cSNPs are available at the Kazusa Tomato Genomics DataBase (http://plant1.kazusa.or.jp/tomato/).
Supplementary data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the Kazusa DNA Research Institute Foundation and the Ministry of Agriculture, Forestry, and Fisheries of Japan (Genomics for Agricultural Innovation Foundation, DD-4010 and SGE-1001), and MEXT KAKENHI Grant number 24510286, Grant-in-Aid for Scientific Research (C).
Supplementary Material
Acknowledgements
We are grateful to Dr Ariizumi (National BioResource Project, Japan), Dr Saito (NARO Institute of Vegetable and Tea Sciences, Japan), Dr Tam (Tomato Genetic Resource Center, USA), and Dr Tanksley (Cornell University, USA) for providing plant materials. The SNP BeadChip was developed by Illumina's technology and the USDA-NIFA Agriculture and Food Research Initiative (AFRI) sponsored Solanaceae Coordinated Agricultural Project.
Footnotes
Edited by Prof. Hiroyuki Toh
References
- 1.Varshney R.K., Nayak S.N., May G.D., Jackson S.A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009;27:522–30. doi: 10.1016/j.tibtech.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Cao J., Schneeberger K., Ossowski S., et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 2011;43:956–63. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- 3.Chagné D., Crowhurst R.N., Troggio M., et al. Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS One. 2012;7:e31745. doi: 10.1371/journal.pone.0031745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee P.H., Shatkay H. F-SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 2008;36:D820–4. doi: 10.1093/nar/gkm904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yue P., Melamud E., Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kharabian A. An efficient computational method for screening functional SNPs in plants. J. Theor. Biol. 2010;265:55–62. doi: 10.1016/j.jtbi.2010.04.017. [DOI] [PubMed] [Google Scholar]
- 7.Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–41. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamilton J.P., Sim S.C., Stoffel K., et al. Single nucleotide polymorphism discovery in cultivated tomato via sequencing by synthesis. The Plant Genome. 2012;5:17–29. [Google Scholar]
- 9.Sim S.C., Van Deynze A., Stoffel K., et al. High-density SNP genotyping of tomato (Solanum lycopersicum L.) reveals patterns of genetic variation due to breeding. PLoS One. 2012;7:e45520. doi: 10.1371/journal.pone.0045520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller L.A., Tanksley S.D., Giovannoni J.J., et al. The Tomato Sequencing Project, the first cornerstone of the International Solanaceae Project (SOL) Comp. Funct. Genomics. 2005;6:153–8. doi: 10.1002/cfg.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yano K., Watanabe M., Yamamoto N., et al. MiBASE: a database of a miniature tomato cultivar Micro-Tom. Plant Biotechnol. J. 2006;23:195–8. [Google Scholar]
- 12.Shirasawa K., Isobe S., Hirakawa H., et al. SNP discovery and linkage map construction in cultivated tomato. DNA Res. 2010;17:381–91. doi: 10.1093/dnares/dsq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pilcher C.D., Wong J.K., Pillai S.K. Inferring HIV transmission dynamics from phylogenetic sequence relationships. PLoS Med. 2008;5:e69. doi: 10.1371/journal.pmed.0050069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradbury P.J., Zhang Z., Kroon D.E., Casstevens T.M., Ramdoss Y., Buckler E.S. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 16.Altschul S.F., Madden T.L., Schäffer A.A., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearson W.R., Lipman D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA. 1988;85:2444–8. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eswar N., Webb B., Marti-Renom M.A., et al. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. 2006 doi: 10.1002/0471250953.bi0506s15. Chapter 2: Unit 2.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Guilloux V., Schmidtke P., Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Hernandez M., Berardini T.Z., Chen G., et al. TAIR: a resource for integrated Arabidopsis data. Funct. Integr. Genomics. 2002;2:239–53. doi: 10.1007/s10142-002-0077-z. [DOI] [PubMed] [Google Scholar]
- 21.Tatusov R.L., Fedorova N.D., Jackson J.D., et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41–54. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Punta M., Coggill P.C., Eberhardt R.Y., et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eddy S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito A., Matsunaga H., Yoshida T., et al. ‘Tomato Chuukanbohon Nou 11’, a tomato parental line with a short-internode trait. Bull. Natl. Inst. Veg. Tea Sci. 2007;6:65–76. [Google Scholar]
- 26.Scott J.W., Harbaugh B.K. Micro-Tom A miniature dwarf tomato. Florida Agr. Expt. Sta. Circ. 1989;370:1–6. [Google Scholar]
- 27.Meissner R., Chague V., Zhu Q., Emmanuel E., Elkind Y., Levy A.A. A high throughput system for transposon tagging and promoter trapping in tomato. Plant J. 2000;22:265–74. doi: 10.1046/j.1365-313x.2000.00735.x. [DOI] [PubMed] [Google Scholar]
- 28.Laterrot H. Combined use of male sterility and resistance to tobacco mosaic virus in tomato. Ann. Amelio. Plant. 1976;26:517–21. [Google Scholar]
- 29.Fridman E., Pleban T., Zamir D. A recombination hotspot delimits a wild-species quantitative trait locus for tomato sugar content to 484 bp within an invertase gene. Proc. Natl. Acad. Sci. USA. 2000;97:4718–23. doi: 10.1073/pnas.97.9.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fridman E., Carrari F., Liu Y.S., Fernie A.R., Zamir D. Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science. 2004;305:1786–9. doi: 10.1126/science.1101666. [DOI] [PubMed] [Google Scholar]
- 31.Kumar R., Kumar S., Sangwan S., Yadav I.S., Yadav R. Protein modeling and active site binding mode interactions of myrosinase-sinigrin in Brassica juncea–an in silico approach. J. Mol. Graph. Model. 2011;29:740–6. doi: 10.1016/j.jmgm.2010.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.