

Abstract
The modern horse (Equus caballus) is the product of over 50 million yrs of evolution. The athletic abilities of the horse have been enhanced during the past 6000 yrs under domestication. Therefore, the horse serves as a valuable model to understand the physiology and molecular mechanisms of adaptive responses to exercise. The structure and function of skeletal muscle show remarkable plasticity to the physical and metabolic challenges following exercise. Here, we reveal an evolutionary layer of responsiveness to exercise-stress in the skeletal muscle of the racing horse. We analysed differentially expressed genes and their co-expression networks in a large-scale RNA-sequence dataset comparing expression before and after exercise. By estimating genome-wide dN/dS ratios using six mammalian genomes, and FST and iHS using re-sequencing data derived from 20 horses, we were able to peel back the evolutionary layers of adaptations to exercise-stress in the horse. We found that the oldest and thickest layer (dN/dS) consists of system-wide tissue and organ adaptations. We further find that, during the period of horse domestication, the older layer (FST) is mainly responsible for adaptations to inflammation and energy metabolism, and the most recent layer (iHS) for neurological system process, cell adhesion, and proteolysis.
Keywords: horse, exercise, evolution, RNA sequencing, re-sequencing
1. Introduction
The horse (Equus caballus) is a subspecies of the family Equidae and has evolved over the last 45–55 million yrs, from a small multi-toed mammal into the large single-toed animal it is today.1 Wild horses were domesticated in Central Eurasia ∼5500–6000 yrs ago2 and were primarily bred for their endurance, strength, and speed. This makes the horse not only a useful biological model to study the physiology of exercise, but also to identify the molecular mechanisms of adaptive responses to exercises. The horse is also used as a model for some human disorders, such as infertility, inflammatory disease, and various muscular disorders.3 There are currently >300 breeds of horse, and humans use them in a wide range of activities.4 Among these, the Thoroughbred is a well-known breed of the racing horse. The structure of their skeletal muscle possesses remarkable plasticity, which is able to respond to environmental changes as well as to the physical and metabolic challenges following training and exercise.5 To date, the genetic components of any evolutionary adaptations in skeletal muscle have not yet been identified. Recently, transcriptome analyses of skeletal muscle and blood have been carried out to identify the genes expressed during exercise in horses.6–8 In our study, we show the evolutionary process underlying the response to exercise-induced stress in the skeletal muscle of racehorses by means of gene expression network analysis performed after exercise (AE), and by integrating the genome-wide signatures of selection of three different evolutionary phases.
2. Materials and methods
2.1. RNA-seq data between before and after exercises
We generated RNA-sequence (RNA-seq) data in six horses before exercise (BE) and AE and described elsewhere.9 Briefly, samples of skeletal muscle and blood were taken from six Thoroughbred horses BE and AE. ‘BE’ samples were collected from the triceps brachii of the right leg and from the jugular vein and carotid artery of each horse. After a resting period of several hours, ‘AE’ samples were collected immediately after a 30-min trot from the same tissues of each individual. Thoroughbred horses usually canter for 17–18 min per day. For the purposes of this study, a 30-min trot was taken to be the equivalent to 17–18 min of cantering. Total RNAs from skeletal muscle and blood samples were isolated using TRIzol (Invitrogen) and the RNeasy RNA purification kit with DNase treatment (Qiagen). mRNA was isolated from the total RNA using oligo-dT beads and reverse transcribed into double-stranded cDNA fragments. Construction and sequencing of an RNA-seq library for each sample was carried out based on Illumina HiSeq2000 protocols in order to generate 90 pair-end reads. Twenty-four sets of transcriptome data were generated for muscle and blood from six horses both BE and AE. TopHat (version 1.4.1) was used to map the sequences to a horse reference genome and annotated using the EquCab2 database (http://hgdownload.cse.ucsc.edu/downloads.html#horse).
2.2. Identification of differentially expressed genes
We used the R package edgeR,10 which is based on a negative binomial model, to examine differential expression of replicated count data. This is because RNA-seq data may exhibit more variability than expected in a Poisson distribution, because it is more widely dispersed in the genome. When a negative binomial model is used, the dispersion has to be estimated before a differentially expressed gene (DEG) analysis is carried out. EdgeR provides an estimation of this dispersion using a Cox-Reid profile-adjusted likelihood method. After negative binomial models are fitted and dispersion estimates are obtained, it is possible to proceed with the tests for determining differential expression using a generalized linear model (GLM) likelihood ratio test. GLMs specify probability distributions according to the mean–variance relationship; for example, the quadratic mean–variance relationship for a read count. The GLM likelihood ratio test is based on the idea of fitting negative binomial GLMs with Cox-Reid dispersion estimates. This automatically takes all known sources of variation into account. Significant DEGs were detected with a cut-off value of false discovery rate (FDR) <0.01, based on a paired design between ‘BE’ and ‘AE’. The equine Ensembl gene IDs were converted to official gene symbols by cross-matching with human Ensembl gene IDs and the official gene symbols. The official gene symbols of human homologues of equine genes were used for functional clustering and enrichment analyses using the Database for Annotation, Visualization, and Integrated Discovery (DAVID).11 The representation of functional groups in blood and skeletal muscle relative to the whole genome was investigated using the Expression Analysis Systematic Explorer (EASE) tool12 within the DAVID, of which the EASE is a modified Fisher's exact test used to measure enrichment of gene ontology (GO) terms.13
2.3. Analysis of co-expression gene network
Mapped reads were assembled using Cufflinks (version 1.3.0)14 to estimate the abundance of genes. Fragments per kilobase of exon per million fragments (FPKM) of each sample were calculated to estimate the expression levels of the genes. The FPKM value set for all genes in the muscle and blood tissue samples was normalized using Quantile normalization.15 Connectivity was calculated using the Weighted Gene Co-expression Network Analysis (WGCNA) R-package16 for both up-regulated and down-regulated genes (DRGs) having six points of FPKM for both ‘BE’ and ‘AE’. This connectivity, referred to as the degree of connectivity, is the sum of the connection strengths with other genes or networks. Following calculation of the degree of connectivity, a gene co-expression network was generated and the genes clustered onto a topological overlap matrix (TOM), based on their dissimilarity. Genes with a high connectivity to each other clustered at the same module. Modules were formed based on one of the exercise conditions, and the extent to which the module was preserved was calculated for a different condition in order to identify any reciprocal disruption of gene expression. An Eigengene-based connectivity was calculated for up-regulated genes (URGs) of the modules, AE, in both muscle and blood. The cut-off value, module membership values >0.95, and gene significance (GS) values >20 were applied to identify the genes with a high GS and high intra-modular connectivity in each module.
2.4. Analysis of horse DNA re-sequencing data
Whole-blood samples were collected from 14 Thoroughbred racing stallions from the Korean Racing Authority and from four male and two female Jeju domestic ponies (Equus caballus) from the Jeju Provincial Livestock Institute, Korea. Blood (10 ml) was drawn from the carotid artery and was treated with heparin to prevent clotting. A genomic DNA quality check was carried out using fluorescence-based quantification on an agarose gel, a standard electrophoresis on a 0.6% agarose gel, and via a pulsed-field gel, using 200 ng of DNA. Manufacturers' instructions were followed to create a paired library of 500-bp fragments. This consisted of the following: purified genomic DNA fragments of <800 bp, fragments with blunt ends, fragments with 5′ phosphorylated ends, fragments with a 3′-dA overhang, some with adaptor-modified ends, purified ligation product, and a genomic DNA library. Following this, we generated a sequence data using HiSeq 2000 (Illumina, Inc.). Using the Burrows-Wheeler Aligner17 with the default setting, pair-end sequence reads were mapped to the reference horse genome (equCab2). We used the following open-source software packages; Picard Tools, SAMtools,18 and the Genome analysis toolkit,19 for downstream processing and variant calling. Substitution calls were made with GATK UnifiedGenotyper.20 All calls with a Phred-scaled quality of <20 were filtered out. For each chromosome, we inferred the phased haplotype and imputed the missing alleles for the entire set of Thoroughbred populations simultaneously using BEAGLE.21 After phasing, observed heterozygosity and minor allele frequency (MAF) were calculated from autosome set using PLINK (version 1.07).22 Average observed heterozygosities were 0.285 in Thoroughbred horses and 0.345 in Jeju ponies. Average MAFs were 0.207 in Thoroughbred horses and 0.220 in Jeju ponies.
We additionally genotyped 11 Thoroughbred horses using EquineSNP50 Genotyping BeadChip (Illumina, Inc.). Common loci in both SNP chip and DNA re-sequencing data were extracted and compared using VCFtools.20 We identified 98.278–99.572% of genotype concordance (Supplementary Table S2).
2.5. Estimation of iHS and FST value
The iHS23 was calculated using the rehh R package24 and all SNPs from the horse genomes with the same ancestral state as that from the Jeju domestic ponies. Values were accepted when core SNPs were located in genes. We selected a significant iHS at P-value 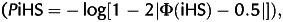 where Φ(x) represents the Gaussian cumulative distribution function of iHS, which is >2,25 and iHS <0. Conventional FST26 values were calculated for genes using Arlequin 3.527 based on pairwise differences between the haplotypes of Thoroughbred racing horses and those of Jeju domestic ponies. The gene region was derived from the phased haplotype of the horse genomes, using genomic information (Ensembl Genes67 and equCab2). The cut-off point for FST is 95% quantile of the empirical distribution of FST.
where Φ(x) represents the Gaussian cumulative distribution function of iHS, which is >2,25 and iHS <0. Conventional FST26 values were calculated for genes using Arlequin 3.527 based on pairwise differences between the haplotypes of Thoroughbred racing horses and those of Jeju domestic ponies. The gene region was derived from the phased haplotype of the horse genomes, using genomic information (Ensembl Genes67 and equCab2). The cut-off point for FST is 95% quantile of the empirical distribution of FST.
2.6. Estimation of the dN/dS value
We downloaded the protein and reference mRNA sequences for humans, mouse, dog, pig, cow, and horse from ENSEMBL.28 The 1 : 1 orthologs of all six species were made using Mestortho.29 Following this, 9000 1 : 1 orthologs were found and used to estimate the synonymous and non-synonymous substitution rates in mammals. Phylogenetic trees were obtained using Timetree.30 Orthologous gene sets were aligned using PRANK31 with the default settings, and poorly aligned sites were eliminated using Gblocks.32 The maximum likelihood method (codeml of PAML 4)33 was used to estimate the dN (the rate of non-synonymous substitutions), dS (the rate of synonymous substitutions), and ω (the ratio of non-synonymous substitutions to the rate of synonymous substitutions), with F3 × 4 codon frequencies under the branch model (model = 2, NSsites = 0) and the basic mode l (model = 0, NSsites = 0). Orthologs with dS > 3 or ω > 5 were filtered.34 After this process, 8417 orthologs remained. A log-likelihood ratio test was performed to compare these models, and we applied an FDR correction.35
2.7. Gene network analysis of positively selected genes of the three evolutionary layers
To ascertain the FPKM values of the URGs in skeletal muscle tissue, signed correlation values [adjacency values: (0.5 × (1 + correlation))soft-thresholding power] were calculated using WGCNA, and a weighted network adjacency matrix was drawn up. We applied a cut-off value of >0.6 to the adjacency, which signifies 0.86 of R2 and 0.92 of the correlation to a linear line fitting with in a power-law distribution. This resulted in a scale free topology of the network. Those genes directly connected to the module core genes and that were associated with the dN/dS, FST, and iHS statistics, which were selected to form the single-depth connection network for the core genes. The correlation network plot was generated using the network visualization tool, Cytoscape (version 2.83).36 A KEGG pathway enrichment test was performed using EASE, with a cut-off value of <0.1 for the selected and core genes associated with dN/dS, FST, and iHS.
3. Results
3.1. DEGs between BE and AE
We generated 24 RNA-seq datasets from muscle and blood tissue from six horses BE and AE. We identified 1822 URGs in muscle tissue, 222 URGs in blood tissue, 930 DRGs in muscle tissue, and 200 DRGs in blood tissue AE (Fig. 1 and Supplementary Fig. S1). Muscle tissue contains a higher proportion of DEGs than blood (10.2 compared with 1.6%). Since the DEGs in the muscle are considerably biased towards URGs, we performed further analysis of the URGs and used an arbitrary cut-off of a >64-fold, which is log2(FC)>6. Enriched pathways of the genes showed a nucleotide-binding oligomerization domain-like receptor (NLR) signalling pathway, a JAK-STAT signalling pathway, a MAPK signalling pathway, and a Toll-like receptor (TLR) signalling pathway (Fig. 1). Notably, interleukin (IL)-6 and IL-8 were the top URGs and were widely involved in the pathways. Skeletal muscle produces and releases cytokines called myokines37 as part of the extracellular signalling pathway in response to factors, such as exercise. The exercise-induced myokines include IL-6 and IL-8,38 and many studies have demonstrated that IL-6 production is associated with muscle contraction.39 Many of the URGs and DRGs in each tissue are covered by the enriched terms present in the Gene Ontology, Biological Processes (GO-BP; Supplementary Figs S2–S6). Representative terms include an intracellular signalling cascade, the response to organic substances, the immune response, regulation of cell proliferation, and apoptosis (Supplementary Fig. S2). Exercise-induced muscle damage is a well-known phenomenon40,41 that elicits an inflammatory response.42 We found that many of the enriched GO-BP terms in URGs are related to the inflammatory response. A further analysis of enriched KEGG pathways (Fig. 2) revealed that many are activated in muscle and blood tissues as a result of exercise-induced stress. Among them, TLR and NLR signalling pathways are commonly found in the URGs of both of these tissues. TLRs and NLRs are two major types of innate immune sensors that provide an immediate response to pathogens or tissue damage.43 The JAK-STAT signalling pathway is also up-regulated in URGs in muscle tissue. The JAK-STAT signalling cascade is a crucial factor in myogenesis17,44,45 and has a role in the inflammatory response.46 We also found a significant up-regulation of focal adhesion, apoptosis, the p53 signalling pathway, and an increased regulation of the actin cytoskeleton (Fig. 2). The p53 protein has a known role in apoptosis of skeletal muscle,47 and the actin cytoskeleton is also involved in the regulation of apoptotic signalling.48
Figure 1.

Plot of the log value for fold change against the log value of the abundance of each gene, using Cox-Reid common dispersion, in skeletal muscle. The x-axis shows the log2-concentration (log2Conc.); i.e. the abundance of each gene. The y-axis shows the log2 fold change of ‘AE’ over ‘BE’. The red circles show the significantly DEGs in skeletal muscle. The symbol for the genes whose expression was changed by over 6-fold (dotted line) AE is shown. The gene-enriched KEGG pathways as well as all included genes are shown on the upper right-hand side.
Figure 2.

Enriched KEGG pathways associated with DEGs in skeletal muscle tissue and blood. For each set of up-regulated and down-regulated DEG in skeletal muscle and blood, a KEGG pathway enrichment analysis was performed. Starting from the left, the figure shows: tissue type, status of regulation AE, KEGG pathway terms, higher-level KEGG pathway terms, and the highest level of KEGG pathway terms. The number of genes associated with the KEGG pathway is given in brackets on the right.
3.2. Co-expression network analysis of the DEGs
We conducted a co-expression network analysis using the WGCNA R software package.16 We found a higher number of modules in muscle tissue ‘AE’ compared with ‘BE’, indicating that the exercise-induced stress has a dramatic effect on gene expression in muscle (Fig. 3). We identified 321 core genes AE in muscle tissues (Fig. 3b), but only one in the blood (Supplementary Fig. S7). Pathways that are significantly enriched in these 321 genes include: the MAPK signalling pathway, the NLR signalling pathway, the TLR signalling pathway, the JAK-STAT signalling pathway, the p53 signalling pathway, cell adhesion molecules, and apoptosis (Fig. 3c). Thorough analysis of the DEGs and core genes of the co-expression network analysis consistently indicate the following: (i) after a single bout of exercise, gene expression in muscle tissue is more disrupted than in blood, and (ii) pathways involved in exercise-induced stress are related to those involved in inflammation and apoptosis in skeletal muscle.
Figure 3.

Dendrogram of the weighted gene co-expression network, the network modules BE (a) and AE (b) in URGs of skeletal muscle and enriched KEGG pathways of module genes (c). The genes are clustered on a TOM, based on dissimilarity, and are presented as a dendrogram. Where genes have a high connectivity, they are clustered in the same module. The upper modules are based on the gene network formed BE, and the lower modules are based on that formed AE. The grey module contains uncharacterized genes. The preservation score of the condition ‘BE’ is shown in the box at the bottom of the modules. For each module, the number of the genes included in the enriched KEGG pathway is listed in the box.
3.3. Positive selection in horse lineage: dN/dS analysis
In this study, we aimed to detect the selection signatures of the three evolutionary phases of E. caballus, using dN/dS, FST, and iHS (Fig. 4). The first phase, calculated by dN/dS, shows the positive selection that occurred during ∼82.5 million yrs of evolution of the horse species. The second phase, calculated by FST, indicates the relatively later selection events that occurred during the domestication of the horse, ∼6000 yrs ago. The final phase, calculated by means of iHS, shows the most recent selective sweep. To analyse the dN/dS phase, we identified 9000 genes from six mammals (human, mouse, dog, horse, cow, and pig), which were orthologous with a 1 : 1 ratio. Using a maximum likelihood method (PAML 4),33 the ratio of non-synonymous to synonymous substitutions (dN/dS) was estimated using a branch model. Of the orthologous genes, 495 were significantly enhanced in the horse lineage (Supplementary Fig. S8). This gene enhancement indicates that the horse lineage is well adapted for basic functions relating to their athletic performance, such as ion transport, cell motility, and cellular response to stress (Fig. 4). Muscle structure and respiratory systems have also evolved in horses over time (Fig. 4). We also found a very strong correlation between dN/dS and expression levels, both in blood and in muscle tissues (Fig. 5). It is known that highly expressed genes evolve more slowly.49 Since expression levels from BE and AE are more disrupted in skeletal muscle, a consistently stronger correlation in muscle tissue indicates that it may be more sensitive to the deleterious side effects caused by highly expressed genes.
Figure 4.

Signatures of selection from the dN/dS, FST, and iHS evolutionary phases of the horse. The horse lineage (red line) in the evolutionary tree showing six species, showing the branching point between the Thoroughbred and the Jeju pony. Tables show enrichment GO terms from the significant genes for dN/dS, FST, and iHS, and the HGNC symbols indicate the core genes. The number of genes involved in the pathway is shown in brackets.
Figure 5.

Scatter plots showing expression values (FPKM) and dN/dS values; in skeletal muscle tissue BE (a), AE (b), and in blood BE (c), and AE (d). After sorting into non-overlapping bins of a 20-gene interval, the log10 average values of FPKM against the average values of dN/dS (x-axis) were plotted. A simple linear regression line is shown in red, and ρ represents Spearman's rank correlation coefficient.
3.4. Positive selection during the domestication period: FST and iHS analyses
To detect a selection signal during the domestication of the horse, we performed a comparative genome-wide FST26 and iHS50 statistical analysis based on the protein-coding genes of 14 Thoroughbreds and 6 Jeju ponies using 10-fold re-sequencing data. The Jeju pony was used as a comparative group because this breed displays pronounced phenotypic differences with the Thoroughbreds, especially with regard to body shape and racing performance (Supplementary Table S1). FST statistics are generally more sensitive to older selection events of an intermediate to high frequency, while the iHS test is used to detect evidence of a recent, stronger, positive selection.50 Using an empirical p–value of <0.05 of FST, we identified 1199 significantly differentiated protein-coding genes (Supplementary Fig. S9). Using p-values of iHS <0.01, and iHS <0, we identified 2182 SNPs and 756 genes, which have undergone recent selective sweep events (Supplementary Fig. S10). The FST phase shows enhanced gene differentiation mainly in the inflammatory response, while the most recent selection events indicated by the iHS value involve the genes of the neurological system process, cell adhesion, and proteolysis. Although dN/dS values strongly correlate with expression values in both types of tissue, we found no correlation between expression levels and FST or iHS values (Supplementary Fig. S11). This indicates that gene expression levels of this biological system have been modulated by long-term evolutionary forces. We also found no correlation between any pairs of dN/dS, FST, and iHS (Supplementary Fig. S12). These results are as expected, because the three statistical methods indicate different phases of evolution separated by time.
3.5. Integration of gene expression network and selection signature in the three layers
We integrated both the gene expression and the evolutionary phases. We describe a positively selected core gene (PSCG) as a gene identified in gene expression networks and which has a signature indicative of positive selection. Among the 321 core genes of the URGs in skeletal muscle tissue, we found 10 expressed in the dN/dS phase, 14 in the FST phase, and 8 PSCGs in the iHS phase (Fig. 4). We suggest that these genes are the major components of the evolutionary adaptation found in the skeletal muscle of the racing horse to exercise-induced stress. Furthermore, using a cut-off value of >0.6 to a weighted network adjacency, we identified genes directly connected by the PSCGs (Fig. 6). All except for two PSCGs connected into a large single network consisting of 626 genes. Genes involved in the inflammatory and apoptosis-related pathways are over-represented in the network (Fig. 6). Gene networks for each evolutionary phase (Supplementary Figs S13–S15) also show significant over-representations of the inflammatory and apoptosis-related pathways. To conclude, we suggest that the skeletal muscle of the racing horse has evolved not only for muscle strength, but also more importantly for the production of an efficient exercise-induced stress response. We believe that our integrative analysis can be used to reveal multiple evolutionary phases in response to certain biological conditions. This approach enables us to study the molecular adaptive evolution of many unique biological populations.
Figure 6.

Single-depth correlation network of the core genes associated with dN/dS, FST, and iHS. A correlation network was constructed to show expression values AE. The blue, orange, and green circles show the core genes associated with dN/dS, FST, and iHS.
4. Discussion
Skeletal muscle produces and releases cytokines called myokines,37 and the tissue cell secretes myokines as part of the extracellular signalling pathway in response to factors such as exercise, and the secreted factors can participate in nutrient generation, mediating angiogenesis, and regulating myogenesis37,51 in which exercise-induced myokines include IL-6, IL-8, IL-15, fibroblast growth factor (FGF) 21, and brain-derived neurotrophic factor (BDNF).38 Concordantly, we found many up-regulated myokines and their receptors AE in the muscle, including IL-6, IL-8, IL-15 receptor, FGF7, FGF receptor 1, FGF receptor 3, and BDNF. Numerous studies have demonstrated that IL-6 production is associated with muscle contraction.39 Concordantly, IL-6 is the most extremely up-regulated in muscle tissue, but it is not significantly up-regulated in the blood. IL-6 is inactive in resting muscles, but is rapidly activated by muscle contraction, and its release from muscle during exercise may be related to free radical metabolism, especially with reactive oxygen species generation.52 IL-8 is a paracrine mediator secreted from contracting skeletal muscle. IL-8 is a chemokine that acts as an attractor for neutrophils and as an angiogenic factor.38
Since physical exercise provides a challenge to homeostasis throughout the body, the immune system displays substantial perturbations in response to a single bout of exercise. Inflammation represents a series of events, which is usually initiated by tissue trauma and is terminated with tissue repair. Exercise-induced muscle damage is a well-known phenomenon40,41 that elicits an inflammatory response.42 Exercise-induced muscle trauma induces an acute inflammatory response characterized by an initial removal of necrotic tissue or cellular debris and a subsequent repair of injured muscle, nerve fibres, blood vessels, as well as extracellular matrix. We found that the enriched GO terms in URGs in both the tissues are directly related to such system-wide response to exercise (Fig. 2), especially one related to inflammatory response.
We found that there are a wide scope of pathways that are elicited by exercise-stress in muscle and blood tissues, which includes pathways belong to cellular processes, environmental information processing, and organismal systems. Among the pathways associated with immune system, TLR, and NLR signalling pathways are commonly found in the URGs of each tissue. TLRs and NLRs are two major forms of innate immune sensors, which provide immediate responses against pathogenic invasion or tissue injury. Activation of these sensors induces the recruitment of innate immune cells, such as macrophages and neutrophils, initiates tissue repair processes, and results in adaptive immune activation.43 The JAK-STAT signalling pathway is enriched in up-regulated in muscle. The JAK/STAT signalling cascade has been identified as a crucial factor for myogenesis17,44,45 and could also have a role in inflammation.46 Efficient utilization of myogenesis allows for the formation of mature muscle fibres for muscle repair/regeneration.53 Additional inflammatory related pathways are also enriched in muscle URGs: haematopoietic cell lineage, phagocytosis, B-cell receptor signalling pathway, and chemokine receptor signalling pathway. We also found a significant enrichment for genes in cellular processes: focal adhesion, apoptosis, p53 signalling pathway, and regulation of actin cytoskeleton. Genes in the focal adhesion are important for muscle integrity because of its role in the protection against mechanically induced damage.54 Concordantly, a genome scan for positive selection in Thoroughbred horses revealed that positively selected genomic regions contain significant over-representation of focal adhesion pathway.55 The p53 protein is known for its role in apoptosis in skeletal muscle,47 and actin cytoskeleton are also involved in the regulation of apoptotic signalling.48 Furthermore, using mice experiment, it was recently shown that exercise-conditioned serum is capable of inducing apoptosis.56 Although we did not find apoptosis-related KEGG pathway enrichment of the URGs in blood, apoptosis-related genes seem to be highly enriched in those of blood tissues on the basis of the GO enrichment analysis result.
We conducted gene co-expression network analysis using WGCNA R package.16 The network analysis package has been extensively used for microarray expression data and recently evaluated for RNA-seq data network inference showing greater sensitivity and dynamic range than that of microarray data.57 For URGs AE for each of the tissue, gene co-expression network was constructed and clustered as modules. The modules are based on one condition BE or AE and their module preservation scores in another condition are calculated. We found higher number of modules in AE (18 modules) compared with BE (9 modules) in muscle tissue. It indicates that the exercise-stress cause dramatic perturbation of gene expression in muscle in response to the stress in the tissue. We found only one gene was identified as a core gene in AE condition of blood samples (Supplementary Fig. S7), whereas 321 genes in that of muscle samples (Fig. 3b).
On the basis of evolutionary tree of the six species, the accelerated genes have been evolved during 82.5 million yrs, therefore, representing the most ancient time of gene evolution among our evolutionary statistics. We found a very strong correlation between dN/dS and expression level in both the blood and muscle samples (Fig. 5). Interestingly, muscle samples contain stronger correlations between them than those of blood samples. It has been known that highly expressed genes evolve slowly.49 Therefore, the best predictor of a protein's evolutionary rate is its expression level from bacteria to mammals.58–61 Interestingly, muscle samples also contain stronger correlations between dN/dS and expression level compared with those of blood samples. It was suggested that selection against the toxicity of misfolded proteins generated by ribosome errors suffices to create a negative correlation between the evolution rate and expression level.61 Thus, highly expressed genes tend to be under purifying selection. Our results indicate that muscle tissue might be more susceptible to the toxicity of misfolded proteins than blood tissue. Considering that the expression levels of BE and AE are more perturbed in the skeletal muscle (Fig. 3a and b; Supplementary Fig. S7), consistently stronger correlation in the muscle tissue indicate that the skeletal muscles have been evolved to adapt the evolutionary susceptibility. Previously tissues composed of neurones were shown to have strongest trends in fly, mouse, and human.61 However, to our limited knowledge, there has been no report of strong correlation in a skeletal muscle tissue.
Our limited sample size may have resulted in a minor fraction of false positive error and, thus, a very large sample size is required, which is unable to be delivered in this study. However, we believe that our analyses, not confounded by tenuous population demographic history, may be considered as preliminary foundation for further replication studies in harbouring genes that underlie phenotypic variation in the racing horse.
Generally, dN/dS ratio detects, at least, more than a million years of evolutionary time62 and, therefore, tends to fail to detect recent or within-species evolutionary process. In order to detect recent and within-species evolutionary forces related to exercise responsive genes in the horse, we further analysed FST and iHS statistics of genome-wide protein-coding genes. The FST statistics are generally more sensitive to older selection events that have reached a intermediate to high frequency, while the iHS test is most powerful for detecting evidence of a recent, strong, positive selection.50 However, there is no clear cut between the FST and iHS evolutionary layers. Therefore, it was our intention to demonstrate older domestication process using FST and relatively recent process using iHS, there must be a significantly overlapping period between the two layers.
Supplementary data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by a grant from the Next-Generation BioGreen 21 Program (PJ008106 and PJ008191), Rural Development Administration, Republic of Korea, and this work was also supported by Korea Racing Authority (KRA) for the project of thoroughbred horse (No. 0569-20110008).
Supplementary Material
Acknowledgements
The RNA-sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE37870. The DNA re-sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA) database under accession number SRA053569 and SRA054885.
Footnotes
Edited by Dr Mikita Suyama
References
- 1.van de Goor L.H.P., van Haeringen W.A., Lenstra J.A. Population studies of 17 equine STR for forensic and phylogenetic analysis. Anim. Genet. 2011;42:627–33. doi: 10.1111/j.1365-2052.2011.02194.x. doi:10.1111/j.1365-2052.2011.02194.x. [DOI] [PubMed] [Google Scholar]
- 2.Outram A.K., Stear N.A., Bendrey R., et al. The earliest horse harnessing and milking. Science. 2009;323:1332–5. doi: 10.1126/science.1168594. doi:10.1126/science.1168594. [DOI] [PubMed] [Google Scholar]
- 3.Wade C.M., Giulotto E., Sigurdsson S., et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326:865–7. doi: 10.1126/science.1178158. doi:10.1126/science.1178158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ling Y.H., Ma Y.H., Guan W.J., et al. Evaluation of the genetic diversity and population structure of Chinese indigenous horse breeds using 27 microsatellite markers. Anim. Genet. 2011;42:56–65. doi: 10.1111/j.1365-2052.2010.02067.x. doi:10.1111/j.1365-2052.2010.02067.x. [DOI] [PubMed] [Google Scholar]
- 5.Marini M., Veicsteinas A. The exercised skeletal muscle: a review. Eur. J. Transl. Myol. – Myol. Rev. 2010;20:105–20. [Google Scholar]
- 6.Barrey E., Mucher E., Robert C., Amiot F.,, Gidrol X. Gene expression profiling in blood cells of endurance horses completing competition or disqualified due to metabolic disorder. Equine. Vet. J. 2006;38:43–9. doi: 10.1111/j.2042-3306.2006.tb05511.x. doi:10.1111/j.2042-3306.2006.tb05511.x. [DOI] [PubMed] [Google Scholar]
- 7.McGivney B.A., Eivers S.S., MacHugh D.E., et al. Transcriptional adaptations following exercise in thoroughbred horse skeletal muscle highlights molecular mechanisms that lead to muscle hypertrophy. BMC Genomics. 2009;10:638. doi: 10.1186/1471-2164-10-638. doi:10.1186/1471-2164-10-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGivney B.A., McGettigan P.A., Browne J.A., et al. Characterization of the equine skeletal muscle transcriptome identifies novel functional responses to exercise training. BMC Genomics. 2010;11:398. doi: 10.1186/1471-2164-11-398. doi:10.1186/1471-2164-11-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park K.-D., Park J., Ko J., et al. Whole transcriptome analyses of six thoroughbred horses before and after exercise using RNA-Seq. BMC Genomics. 2012;13:473. doi: 10.1186/1471-2164-13-473. doi:10.1186/1471-2164-13-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. doi:10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dennis G., Jr, Sherman B.T., Hosack D.A., et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. doi:10.1186/gb-2003-4-5-p3. [PubMed] [Google Scholar]
- 12.Hosack D.A., Dennis G., Jr, Sherman B.T., Lane H.C., Lempicki R.A. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. doi:10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alterovitz G., Ramoni M.F. Knowledge Based Bioinformatics. Wiley Online Library; 2010. [Google Scholar]
- 14.Trapnell C., Williams B.A., Pertea G., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. doi:10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolstad B. Preprocesscore: a collection of pre-processing functions, R package version 1.18.0.
- 16.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. doi:10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y.P., Xu Y., Li W., et al. STAT3 induces muscle stem cell differentiation by interaction with myoD. Cytokine. 2009;46:137–41. doi: 10.1016/j.cyto.2008.12.015. doi:10.1016/j.cyto.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 18.Li H., Handsaker B., Wysoker A., et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–79. doi: 10.1093/bioinformatics/btp352. doi:10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A., Hanna M., Banks E., et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. doi:10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DePristo M.A., Banks E., Poplin R., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–98. doi: 10.1038/ng.806. doi:10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browning B.L., Yu Z. Simultaneous genotype calling and haplotype phasing improves genotype accuracy and reduces false-positive associations for genome-wide association studies. Am. J. Hum. Genet. 2009;85:847–61. doi: 10.1016/j.ajhg.2009.11.004. doi:10.1016/j.ajhg.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S., Neale B., Todd-Brown K., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–75. doi: 10.1086/519795. doi:10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. doi:10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gautier M., Vitalis R. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics. 2012;28:1176–77. doi: 10.1093/bioinformatics/bts115. doi:10.1093/bioinformatics/bts115. [DOI] [PubMed] [Google Scholar]
- 25.Gautier M., Naves M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol. Ecol. 2011;20:3128–43. doi: 10.1111/j.1365-294X.2011.05163.x. doi:10.1111/j.1365-294X.2011.05163.x. [DOI] [PubMed] [Google Scholar]
- 26.Wright S. The genetical structure of populations. Ann. Hum. Genet. 1951;15:323–54. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 27.Excoffier L.,, Lischer H.E.L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010;10:564–67. doi: 10.1111/j.1755-0998.2010.02847.x. doi:10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 28.Hubbard T., Barker D., Birney E., et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. doi:10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim K.M., Sung S., Caetano-Anollés G., Han J.Y., Kim H. An approach of orthology detection from homologous sequences under minimum evolution. Nucleic Acids Res. 2008;36:e110. doi: 10.1093/nar/gkn485. doi:10.1093/nar/gkn485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hedges S.B., Dudley J., Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–72. doi: 10.1093/bioinformatics/btl505. doi:10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- 31.Loytynoja A., Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl Acad. Sci. USA. 2005;102:10557–62. doi: 10.1073/pnas.0409137102. doi:10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talavera G., Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007;56:564–77. doi: 10.1080/10635150701472164. doi:10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z.H. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. doi:10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 34.Castillo-Davis C., Kondrashov F., Hartl D., Kulathinal R. The functional genomic distribution of protein divergence in two animal phyla: coevolution, genomic conflict, and constraint. Genome Res. 2004;14:802–11. doi: 10.1101/gr.2195604. doi:10.1101/gr.2195604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benjamini Y., Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001;29:1165–88. doi:10.1214/aos/1013699998. [Google Scholar]
- 36.Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–32. doi: 10.1093/bioinformatics/btq675. doi:10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pedersen B.K., Febbraio M.A. Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol. Rev. 2008;88:1379–406. doi: 10.1152/physrev.90100.2007. doi:10.1152/physrev.90100.2007. [DOI] [PubMed] [Google Scholar]
- 38.Pedersen B.K. Edward F. Adolph Distinguished Lecture: muscle as an endocrine organ: IL-6 and other myokines. J. Appl. Physiol. 2009;107:1006–14. doi: 10.1152/japplphysiol.00734.2009. doi:10.1152/japplphysiol.00734.2009. [DOI] [PubMed] [Google Scholar]
- 39.Starkie R.L., Arkinstall M.J., Koukoulas I., Hawley J.A., Febbraio M.A. Carbohydrate ingestion attenuates the increase in plasma interleukin-6, but not skeletal muscle interleukin-6 mRNA, during exercise in humans. J. Physiol. Lond. 2001;533:585–91. doi: 10.1111/j.1469-7793.2001.0585a.x. doi:10.1111/j.1469-7793.2001.0585a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cannon J.G., St Pierre B.A. Cytokines in exertion-induced skeletal muscle injury. Mol. Cell. Biochem. 1998;179:159–67. doi: 10.1023/a:1006828425418. doi:10.1023/A:1006828425418. [DOI] [PubMed] [Google Scholar]
- 41.Clarkson P.M., Sayers S.P. Etiology of exercise-induced muscle damage. Can. J. Appl. Physiol. 1999;24:234–48. doi: 10.1139/h99-020. doi:10.1139/h99-020. [DOI] [PubMed] [Google Scholar]
- 42.Tidball J.G. Inflammatory cell response to acute muscle injury. Med. Sci. Sport. Exerc. 1995;27:1022–32. doi: 10.1249/00005768-199507000-00011. doi:10.1249/00005768-199507000-00011. [DOI] [PubMed] [Google Scholar]
- 43.Fukata M., Vamadevan A.S., Abreu M.T. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin. Immunol. 2009;21:242–53. doi: 10.1016/j.smim.2009.06.005. doi:10.1016/j.smim.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 44.Sun L.G., Ma K.W., Wang H.X., et al. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J. Cell. Biol. 2007;179:129–38. doi: 10.1083/jcb.200703184. doi:10.1083/jcb.200703184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K.P., Wang C.H., Xiao F., Wang H.X., Wu Z.G. JAK2/STAT2/STAT3 are required for myogenic differentiation. J. Biol. Chem. 2008;283:34029–36. doi: 10.1074/jbc.M803012200. doi:10.1074/jbc.M803012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Shea J.J., Pesu M., Borie D.C., Changelian P.S. A new modality for immunosuppression: targeting the JAK/STAT pathway. Nat. Rev. Drug. Discov. 2004;3:555–64. doi: 10.1038/nrd1441. doi:10.1038/nrd1441. [DOI] [PubMed] [Google Scholar]
- 47.Saleem A., Adhihetty P.J., Hood D.A. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genomics. 2009;37:58–66. doi: 10.1152/physiolgenomics.90346.2008. doi:10.1152/physiolgenomics.90346.2008. [DOI] [PubMed] [Google Scholar]
- 48.Gourlay C.W., Ayscough K.R. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat. Rev. Mol. Cell. Biol. 2005;6:583–5. doi: 10.1038/nrm1682. doi:10.1038/nrm1682. [DOI] [PubMed] [Google Scholar]
- 49.Drummond D.A., Bloom J.D., Adami C., Wilke C.O., Arnold F.H. Why highly expressed proteins evolve slowly. Proc. Natl Acad. Sci. USA. 2005;102:14338–43. doi: 10.1073/pnas.0504070102. doi:10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voight B.F., Kudaravalli S., Wen X.Q., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:446–58. doi: 10.1371/journal.pbio.0040072. doi:10.1371/journal.pbio.0040446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pedersen B.K., Akerstrom T.C.A. Role of myokines in exercise and metabolism. J. Appl. Physiol. 2007;103:1093–98. doi: 10.1152/japplphysiol.00080.2007. doi:10.1152/japplphysiol.00080.2007. [DOI] [PubMed] [Google Scholar]
- 52.Donges C.E., Duffield R., Drinkwater E.J. Effects of resistance or aerobic exercise training on interleukin-6, C-reactive protein, and body composition. Med. Sci. Sports Exerc. 2010;42:304–13. doi: 10.1249/MSS.0b013e3181b117ca. [DOI] [PubMed] [Google Scholar]
- 53.Charge S.B.P., Rudnicki M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004;84:209–38. doi: 10.1152/physrev.00019.2003. doi:10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 54.Fluck M., Mund S.I., Schittny J.C., Klossner S., Durieux A.C., Giraud M.N. Mechano-regulated Tenascin-C orchestrates muscle repair. Proc. Natl Acad. Sci. USA. 2008;105:13662–67. doi: 10.1073/pnas.0805365105. doi:10.1073/pnas.0805365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gu J.J., Orr N., Park S.D., et al. A genome scan for positive selection in thoroughbred horses. PLoS One. 2009;4:e5767. doi: 10.1371/journal.pone.0005767. doi:10.1371/journal.pone.0005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hojman P., Dethlefsen C., Brandt C., Hansen J., Pedersen L., Pedersen B.K. Exercise-induced muscle-derived cytokines inhibit mammary cancer cell growth. Am. J. Physiol. Endocrinol. Metab. 2011;301:E504–10. doi: 10.1152/ajpendo.00520.2010. [DOI] [PubMed] [Google Scholar]
- 57.Iancu O.D., Kawane S., Bottomly D., Searles R., Hitzemann R., McWeeney S. Utilizing RNA-Seq data for de novo coexpression network inference. Bioinformatics. 2012;28:1592–97. doi: 10.1093/bioinformatics/bts245. doi:10.1093/bioinformatics/bts245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pal C., Papp B., Hurst L.D. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–31. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krylov D.M., Wolf Y.I., Rogozin I.B., Koonin E.V. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res. 2003;13:2229–35. doi: 10.1101/gr.1589103. doi:10.1101/gr.1589103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rocha E.P.C., Danchin A. An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol. Biol. Evol. 2004;21:108–16. doi: 10.1093/molbev/msh004. doi:10.1093/molbev/msh004. [DOI] [PubMed] [Google Scholar]
- 61.Drummond D.A., Wilke C.O. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–52. doi: 10.1016/j.cell.2008.05.042. doi:10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oleksyk T.K., Smith M.W., O'Brien S.J. Genome-wide scans for footprints of natural selection. Philos. Trans. R. Soc. B. 2010;365:185–205. doi: 10.1098/rstb.2009.0219. doi:10.1098/rstb.2009.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.