

Summary
PTSD symptoms are associated with heightened fear responses in laboratory fear conditioning paradigms. This study examined the effects of dexamethasone administration on hypothalamic–pituitary–adrenal (HPA) function and fear-potentiated startle (FPS) in trauma-exposed individuals with and without PTSD. We used an established fear discrimination procedure, in which one visual stimulus (CS+, danger cue) was paired with aversive airblasts to the throat (unconditioned stimulus, US), and another stimulus (CS−, safety cue) was presented without airblasts. In addition to FPS, the dexamethasone suppression test (DST) was performed. The study sample (N = 100) was recruited from a highly traumatized civilian population in Atlanta, GA. Half of the subjects (n = 54, 16 PTSD, 38 controls) underwent conditioning at baseline and the other half (n = 46, 17 PTSD, 29 controls) after DST, in a cross-sectional design. We found a significant interaction effect of diagnostic group and dexamethasone treatment. Under baseline conditions, subjects with PTSD showed more than twice as much fear-potentiated startle to the danger cue compared to traumatized controls, F(1,53) = 8.08, p = 0.006. However, there was no group difference in subjects tested after dexamethasone suppression. Furthermore, there was a significant treatment effect in PTSD subjects but not in controls, with dexamethasone reducing fear-potentiated startle to the CS+, F(1,32) = 4.00, p = 0.05. There was also a positive correlation between PTSD subjects’ FPS and cortisol levels, r = 0.46, p = 0.01. These results suggest that transient suppression of HPA function via dexamethasone suppression may reduce exaggerated fear in patients with PTSD.
Keywords: PTSD, Trauma, HPA, Cortisol, Dexamethasone, Fear conditioning, Fear-potentiated startle
1. Introduction
Posttraumatic stress disorder (PTSD) is a debilitating psychiatric disorder, marked by symptoms of heightened fear and arousal (APA, 1994) that can develop in individuals who become exposed to extremely traumatic events. While PTSD is frequently observed in soldiers returning from war zones (Hoge et al., 2004), it is also prevalent in survivors of civilian trauma, such as sexual assault (Rothbaum et al., 2001), and natural disasters, such as hurricanes (Galea et al., 2007). A growing number of studies (Alim et al., 2006; Breslau et al., 2004; Schwartz et al., 2005; Switzer et al., 1999) indicate that African Americans living in low income urban environments are at especially high risk for both exposure to traumatic events and PTSD.
Although PTSD is highly prevalent, the incidence is relatively low given the high rates of trauma exposure. The current estimate for PTSD prevalence among civilian populations with high levels of trauma is about 20% (Gillespie et al., 2009). This indicates that there may be genetic and environmental risk factors that may differentially affect vulnerability to develop PTSD following trauma. Recently, genetic markers have been discovered that interact with early childhood abuse to increase risk for PTSD; specifically, polymorphisms of the FKBP5 gene have been associated with PTSD symptoms (Binder et al., 2008; Xie et al., 2010). This gene is involved in regulating cortisol feedback function (Binder, 2009; Binder et al., 2008), the stress hormone mechanism that is one of the most frequently reported neurobiological alterations in PTSD (Baker et al., 1999; de Kloet et al., 2007; Yehuda, 2009; Yehuda et al., 1991a). Heightened cortisol reactivity in response to psychosocial stress has been observed in victims of physical and sexual childhood abuse (Heim et al., 2000). On the other hand, exaggerated suppression of hypothalamic–pituitary–adrenal (HPA) axis activity following administration of dexamethasone, a cortisol analogue, has been a consistent finding in PTSD (Yehuda et al., 2002, 2004a,b), although recent studies indicate that this effect is strongest in individuals with higher genetic risk (Binder et al., 2008). Furthermore, concentrations of corticotropin-releasing hormone (CRH) in the cerebrospinal fluid are higher in PTSD patients (Baker et al., 1999; Bremner et al., 1997). Elevated CRH levels are associated with increased fear response (Kalin and Takahashi, 1990), including the startle response (Keen-Rhinehart et al., 2008; Lee and Davis, 1997; Liang et al., 1992) and enhanced fear conditioning (Roozendaal et al., 2002; Swerdlow et al., 1989). Taken together, these studies suggest that HPA dysregulations are long-term sequelae of trauma (Heim et al., 2008; Yehuda et al., 2002) that are associated with PTSD risk.
Another frequently observed neurobiological alteration in PTSD is heightened amygdala response to emotional stimuli, as evidenced by neuroimaging studies. Functional magnetic resonance imaging (fMRI) and positron emission tomography (PET) data demonstrate that PTSD patients appear to have greater amygdala activation compared to controls (for a recent review, see Liberzon and Sripada, 2008). The amygdala is centrally involved in generating fear responses (Davis, 1992b; Lang et al., 2000; LeDoux, 1998), which can be measured indirectly by using fear conditioning methods, such as fear-potentiated startle (Davis, 1992a; Davis et al., 1993). Fear-potentiated startle (FPS) is the relative increase in the startle response elicited in the presence of a conditioned stimulus (CS+) that was previously paired with an aversive unconditioned stimulus (US). Studies of Vietnam and Gulf war veterans by Grillon et al. (1998) and Morgan et al. (1995) found that PTSD was associated with increased FPS to specific cues as well as threatening contexts. Previous studies in our lab have shown heightened FPS to danger cues (CS+) as well as impaired inhibition of fear to safety cues (CS−) in both combat (Jovanovic et al., 2009) and civilian PTSD (Jovanovic et al., 2010a,b).
To date, very few studies have examined the interaction of cortisol and fear-potentiated startle in humans. One study using fear-potentiated startle in healthy humans found that high cortisol to dehydroepiandrosterone-sulfate (DHEA-S) ratios after fear conditioning were associated with increased startle potentiation (Grillon et al., 2006). We recently examined the relationship between fear-potentiated startle and HPA function in PTSD patients and found a positive correlation between FPS and ACTH levels (Jovanovic et al., 2010b).
Although not widely studied in humans, the animal literature suggests that glucocorticoids in the amygdala enhance fear conditioning (Roozendaal et al., 1992, 2008) and are necessary for neonatal development of fear responses (Sullivan, 2001) and that glucocorticoid receptor (GR) antagonists normalize heightened fear responses (Kohda et al., 2007). A potential mechanism of action of the HPA axis dysregulation is GR hypersensitivity (Yehuda, 2009), which is evident from exaggerated negative feedback (Yehuda et al., 2002), and may be due to increased numbers of GR receptors in PTSD (Yehuda et al., 1991b) or altered receptor binding properties. As described above, the amygdala is part of the fear neurocircuitry that is dysregulated in PTSD. In addition, the amygdala has outputs to the hypothalamus, which is the target of negative feedback by cortisol via GR. Decreased levels of cortisol may differentially activate GR in PTSD subjects compared to healthy controls. Based on these data and our findings of the association of FPS with glucocorticoids, (Jovanovic et al., 2010b), we hypothesized that dexamethasone administration would decrease cortisol levels and reduce fear responses in subjects with PTSD compared to traumatized controls without PTSD.
2. Methods
2.1. Study participants
Participants were recruited as part of a larger study investigating the genetic and environmental factors that contribute to PTSD in a primarily African-American, low socioeconomic, inner-city population (Binder et al., 2008; Gillespie et al., 2009). Exclusion criteria for participation in the study included active psychosis and major medical illnesses as assessed by history and physical examinations conducted by medical professionals. A urine pregnancy screen was used to exclude pregnant women from the dexamethasone test. Participants were also excluded for positive urine toxicology including cocaine and opiates, as well as hearing impairment as assessed by an audiometer (Grason-Stadler, Model GS1710). Prior to their participation, all participants provided written informed consents that were approved by the Emory University Institutional Review Board.
2.2. Psychological assessment
2.2.1. Modified PTSD Symptom Scale
PTSD Symptom Scale (PSS) is a psychometrically valid 17-item self-report scale assessing PTSD symptomatology over the two weeks prior to rating (Falsetti et al., 1993; Foa and Tolin, 2000; Schwartz et al., 2005). The categorical definition of PTSD was determined based on DSM-IV (APA, 1994) A–E criterion responses to the PSS questionnaire.
2.2.2. Childhood Trauma Questionnaire (CTQ)
The CTQ is a self-report inventory assessing childhood physical, sexual and emotional abuse. Studies have established the internal consistency, stability over time, and criterion validity of both the original 70-item CTQ and the current brief version (Bernstein and Fink, 1998; Bernstein et al., 2003). The CTQ yields a total score and subscale scores for each of the types of child abuse.
2.2.3. Traumatic Events Inventory (TEI)
The TEI (Schwartz et al., 2005) assesses lifetime history of trauma exposure and is a measure of both child abuse and non-child abuse traumas. The TEI assesses past experience and frequency of 13 separate types of traumatic events as well as feelings of terror, horror, and helplessness with such events.
2.2.4. The Beck Depression Inventory (BDI)
The BDI (Beck et al., 1961) was administered to measure depressive symptoms. The BDI consists of a 21-item questionnaire and has been previously validated in the Grady population (Bradley et al., 2008; Jovanovic et al., 2010a). Each of the items measures the presence and severity of depressive symptoms that are rated on a scale from 0 to 3.
2.3. Experimental design
After the participants were consented, screened, and enrolled in the study, they were scheduled for a baseline visit followed by the dexamethasone suppression test (DST) visit approximately one week later. In a cross-sectional treatment design, approximately half of the subjects were assigned to the fear-potentiated startle task on the baseline visit (the no treatment control condition, No-TMT group) and the other half were assigned to the fear-potentiated startle task on the DST visit (the dexamethasone treatment condition, TMT group), see Fig. 1 for a diagram of the experiment.
Figure 1.
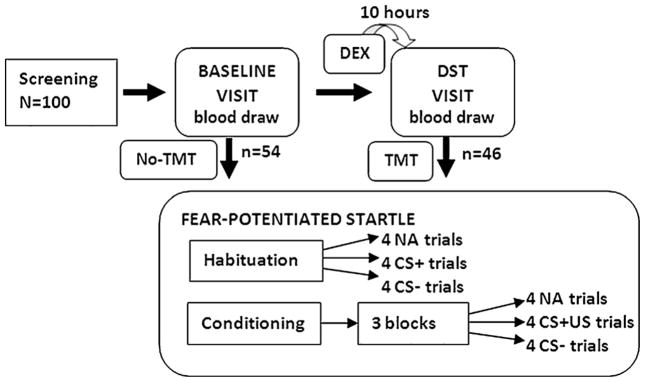
Diagram of the experimental design. Half of the subjects (n = 54) underwent fear conditioning on the first visit (baseline) and the other half (n = 46) underwent fear conditioning on the second visit (DST), in a cross-sectional design. Abbreviations: DEX = dexamethasone (0.5 mg); NA = noise alone startle probe trial; DST = dexamethasone suppression test; CS+ = conditioned stimulus reinforced with the airblast (reinforced); CS− = conditioned stimulus without the airblast (nonreinforced).
The fear-potentiated startle task included two phases: habituation and conditioning. The habituation phase consisted of six startle probes presented alone (noise-alone trials, NA). Immediately following habituation, participants underwent the conditioning phase, which consisted of three blocks, each of which included four trials of each CS type and four NA trials for a total of 12 trials per block. All CS+ trials were reinforced with the US, while the CS− trials were not reinforced. Both conditioned stimuli were different colored shapes presented on a computer monitor and were 6 s in duration. The US was a 250 ms airblast with an intensity of 140 psi directed to the larynx. The airblast was emitted by a compressed air tank attached to the polyethylene tubing and controlled by a solenoid switch. This US has been used in our studies previously (Jovanovic et al., 2005; Norrholm et al., 2006) and produces robust fear-potentiated startle. In all phases of the experiment, inter-trial intervals were of randomized duration ranging from 9 to 22 s.
2.4. HPA hormone assays
Blood was drawn on two separate visits: the baseline visit and the dexamethasone suppression test (DST) visit (see Fig. 1). At the end of the baseline visit, medically cleared subjects were given a 0.5 mg dexamethasone tablet and instructed to take it at 11 PM the night before the next study visit. Subjects were instructed not to take anything by mouth other than water after midnight the night before each study visit. Fasting blood specimens were obtained by venipuncture between 8 AM and 9 AM on the mornings of the baseline and DST visits. Only experienced nurses at the Grady hospital General Clinical Research Center were designated for obtaining blood samples. After collection, samples underwent routine processing and were transferred to a −80 °C freezer for storage until analysis.
Baseline hormone measures included plasma cortisol and adrenocorticotropin hormone (ACTH) levels. DST measures included plasma cortisol and ACTH levels as well as a dexamethasone detection assay to confirm that subjects had taken the dexamethasone tablets as instructed. Cortisol, ACTH, and dexamethasone determinations were completed by the Yerkes Biomarkers Core Laboratory at Emory University. Cortisol and ACTH levels were measured by commercially available radio-immunoassay kits (Cortisol RIA from Diagnostic Systems Laboratories, Webster, TX and ACTH RIA from DiaSorin, Inc., Stillwater, MN). The presence or absence of dexamethasone was ascertained by Elisa using commercially prepared kits from Neogen (Lexington, KY). All intra-assay and inter-assay coefficients of variability were under 10%.
2.5. Startle response measurement
Fear-potentiated startle testing was conducted on the first visit, after the baseline blood draw, for 54 subjects (No-TMT group). For the remaining 46 subjects, startle testing was conducted on the second visit, after the DST blood draw (TMT group), see Fig. 1. Given that the subjects had been fasting for the blood draw, they were given breakfast prior to the startle phase of the study. The startle response data were acquired at a 1000 Hz sampling frequency using the electromyography (EMG) module of the Biopac MP150 for Windows (Biopac Systems, Inc., Aero Camino, CA). The acquired data were filtered, rectified, and smoothed using MindWare software (MindWare Technologies, Ltd., Gahanna, OH) and exported for statistical analyses. The EMG signal was filtered with low- and high-frequency cutoffs at 28 and 500 Hz, respectively. The maximum amplitude of the eyeblink muscle contraction 20–200 ms after presentation of the startle probe was used as a measure of the acoustic startle response.
As previously described (Jovanovic et al., 2005, 2006) the eyeblink component of the acoustic startle response was measured by EMG recordings of the right orbicularis oculi muscle with two 5-mm Ag/AgCl electrodes filled with electrolyte gel. One electrode was positioned 1 cm below the pupil of the right eye and the other was 1 cm below the lateral canthus. Impedance levels were less than 6 kΩ for each participant. The startle probe was a 108-dB (A) SPL, 40 ms burst of broadband noise with near instantaneous rise time, delivered binaurally through headphones.
2.6. Contingency awareness
A response keypad unit (SuperLab, Cedrus Corp., San Pedro, CA) was incorporated into the startle session in order to assess trial-by-trial US expectancy and contingency awareness (Jovanovic et al., 2006). Subjects were instructed to respond on each CS trial, within 3 s of CS onset, by pressing one of three buttons: a button marked ‘+’ when they expected the US, a second button marked ‘−’ when they did not expect the US, and a third button marked ‘0’ when they were uncertain of the contingency. The exact instructions given to the subjects were: “During this experiment you will hear some sudden tones and noises in addition to seeing several colored shapes on the computer monitor. The noises are there to elicit startle and occur every time something happens. However, some of the shapes will be followed by the blast of air while other shapes will not. Throughout the experiment please press the button on the keypad to tell us whether you think a shape will be followed by air (the plus sign), or will not be followed by air (the minus sign). If you do not know, press the 0 sign. You should press a button for each shape.’
2.7. Data analysis
Demographic and clinical data such as age, PTSD symptoms, as well as childhood and adult trauma levels, were compared between the PTSD and No-PTSD trauma control groups, and between the TMT and No-TMT groups within each diagnostic group using 1-way analysis of variance (ANOVA); categorical data, such as sex and race, were analyzed using Chi-square analyses.
Dexamethasone suppression was tested using a repeated measures analysis of variance (RM ANOVA) comparing cortisol and ACTH levels with visit (2 levels: baseline, DST) as the within-subject factor and diagnostic category as the between-group factor, as well as separately within each diagnostic group.
Fear-potentiated startle was calculated using a difference score by subtracting startle magnitude to the noise alone (NA) trials from the startle magnitude on CS+ trials and CS− trials in each conditioning block. These variables were analyzed in a 4-way mixed ANOVA with the within-subject factor of block (3 levels), CS type (2 levels: CS+, CS−), and the between-group factor of diagnosis (2 levels: PTSD, No-PTSD) and treatment (2 levels: TMT, No-TMT). Thus the visit variable refers to the within-subject comparison of HPA data, whereas the treatment variable refers to the between-group comparison of startle data with and without dexamethasone.
Significant interactions were followed up by univariate ANOVAs as well as analyses of covariance (ANCOVAs), with childhood trauma (CTQ) and adult trauma (TEI) levels, as well as depression symptoms (BDI), used as covariates in all analyses involving diagnostic groups. Baseline startle reactivity was measured by comparing average startle magnitude to the noise alone trials between diagnostic and treatment groups using a univariate ANCOVA. Contingency awareness was analyzed by comparing US expectancy using keypad responses to the CS+ and CS− with a RM ANOVA with diagnosis and treatment as between-group factors.
In order to investigate the association between hormones and startle data, we ran bivariate correlations between the cortisol levels (baseline and DST) and the startle potentiation variables (average difference score to CS+). We also examined these correlations separately in the PTSD and No-PTSD groups. All analyses were performed using SPSS 17.0 for Windows (SPSS, Inc., Chicago, IL), with an alpha level of 0.05.
3. Results
3.1. Subject characteristics
One hundred and thirteen participants were recruited; of those, 5 (1 PTSD, 4 No-PTSD) were excluded due to incomplete trauma histories, 2 (1 PTSD, 1 No-PTSD) were excluded as outliers as their DST ACTH values were more than 3 standard deviations above the mean, and 6 (all controls) were excluded due to a negative assay for dexamethasone on the DST visit, indicating non-compliance. This resulted in a final sample of 100 participants: 54 who were fear conditioned prior to taking the dexamethasone treatment (No-TMT group) and 46 who were fear conditioned 10 h after taking the dexamethasone treatment (TMT group), see Fig. 1. Table 1 shows the demographic and clinical information of the subjects across the different groups, as well as their trauma history. The PTSD and No-PTSD groups were matched on sex, age and race. Furthermore, the treatment groups (TMT, No-TMT) did not differ on any demographic or clinical variables (see Table 1).
Table 1.
Demographic, trauma history, and PTSD symptom data for the study sample. The participants in the two treatment conditions (TMT, No-TMT) do not differ on any of these variables; the two diagnostic groups (PTSD, No-PTSD control) are matched on demographic variables but differ in the degree of traumatization and symptom severity. Childhood trauma was assessed using the Childhood Trauma Questionnaire (CTQ); adult trauma was assessed using the Traumatic Events Interview (TEI); PTSD symptoms were assessed using the PTSD Symptom Scale (PSS); depression symptoms were assessed using the Beck Depression Inventory (BDI).
| PTSD (n = 33)
|
No-PTSD (n = 67)
|
PTSD vs. No-PTSD | |||||
|---|---|---|---|---|---|---|---|
| TMT (n = 17) | No-TMT (n = 16) | TMT vs. No-TMT | TMT (n = 29) | No-TMT (n = 38) | TMT vs. No-TMT | ||
| Demographics | |||||||
| Sex (% female) | 70.6 | 56.3 | χ2 = 0.35, p = 0.56 | 51.7 | 57.9 | χ2 = 0.25, p = 0.62 | χ2 = 0.64, p = 0.42 |
| Race (% AA) | 94.1 | 93.8 | χ2 = 0.31, p = 0.58 | 96.6 | 92.1 | χ2 = 0.92, p = 0.63 | χ2 = 0.60, p = 0.74 |
| Age (M, SD) | 41.8 (7.3) | 36.3 (12.3) | F = 1.37, p = 0.25 | 39.6 (12.9) | 41.0 (12.9) | F = 0.20, p = 0.66 | F = 0.14, p = 0.71 |
| Trauma history | |||||||
| Childhood trauma (M, SD) | 56.9 (26.9) | 55.4 (22.5) | F = 0.03, p = 0.87 | 38.5 (15.3) | 36.2 (12.7) | F = 0.43, p = 0.51 | F = 24.61, p < 0.0001 |
| Adult trauma (M, SD) | 5.1 (2.6) | 4.6 (1.9) | F = 0.44, p = 0.51 | 2.6 (2.1) | 3.0 (2.5) | F = 0.53, p = 0.47 | F = 17.91, p < 0.0001 |
| PTSD symptoms | |||||||
| Total score (M, SD) | 25.0 (8.3) | 28.1 (10.2) | F = 0.94, p = 0.34 | 7.0 (7.2) | 7.2 (5.9) | F = 0.01, p = 0.92 | F = 145.33, p < 0.0001 |
| Re-experiencing (M, SD) | 6.1 (3.5) | 6.8 (4.3) | F = 0.22, p = 0.65 | 2.0 (2.5) | 1.5 (2.0) | F = 0.79, p = 0.38 | F = 60.31, p < 0.0001 |
| Avoidance (M, SD) | 10.3 (3.7) | 11.8 (4.6) | F = 1.00, p = 0.32 | 2.4 (2.7) | 2.8 (3.3) | F = 0.21, p = 0.65 | F = 129.89, p < 0.0001 |
| Hyper-arousal (M, SD) | 8.4 (3.0) | 9.4 (3.6) | F = 0.79, p = 0.38 | 2.6 (3.7) | 2.9 (2.9) | F = 0.17, p = 0.69 | F = 77.37, p < 0.0001 |
| Depression symptoms | |||||||
| Total score (M, SD) | 21.8 (15.2) | 23.4 (8.2) | F = 0.14, p = 0.71 | 9.8 (7.8) | 12.1 (8.8) | F = 1.18, p = 0.28 | F = 28.17, p < 0.001 |
We did not exclude subjects who were taking psychotropic medication. Of the 100 participants, 3 (1 PTSD, 2 No-PTSD) used benzodiazepines (χ2 = 0.01, ns), 11 (6 PTSD, 5 No-PTSD) used selective serotonin reuptake inhibitor (SSRI) antidepressants (χ2 = 2.60, ns), and 6 (4 PTSD, 2 No-PTSD) used anti-psychotics (χ2 = 3.72, p = 0.07). None of the medications had an effect on either baseline or fear-potentiated startle.
3.2. Clinical assessments
As shown in Table 1, all PTSD subjects had significantly greater levels of childhood (F(1,99) = 24.61, p < 0.0001) and adult (F(1,99) = 17.91, p < 0.0001) trauma compared to the traumatized No-PTSD controls. In addition, PTSD subjects had significantly higher symptoms of depression compared to No-PTSD subjects, F(1,93) = 28.17, p < 0.001. Given the potential confounding effects of these variables on the study results, these variables were used as covariates in analyses between diagnostic PTSD and No-PTSD groups. As expected, the subjects who met current criteria for PTSD had significantly higher total PSS scores, F(1,99) = 145.33, p < 0.0001, than traumatized controls without PTSD. Furthermore, the PTSD subjects had significantly greater symptom severity in each of the three PTSD symptom clusters, i.e., re-experiencing (F(1,99) = 60.31, p < 0.0001), avoidance (F(1,99) = 129.89, p < 0.0001), and hyper-arousal (F(1,99) = 77.37, p < 0.0001). It is important to note, however, that neither trauma history, depression, nor PTSD symptoms differed between dexamethasone treatment groups.
3.3. Dexamethasone suppression
Of the 100 participants with startle and clinical data, baseline cortisol and ACTH levels were available for 90 participants (30 PTSD, 60 No-PTSD), and DST cortisol and ACTH levels were available for 71 participants (27 PTSD, 44 No-PTSD). Missing data were primarily due to: (1) participants not showing up for follow-up appointments, (2) difficulties in drawing blood from participants, despite extensive experience by the nurses, or (3) errors in the hormone assays. Fig. 2 shows the baseline and DST cortisol levels in each group. A RM ANOVA of baseline and DST cortisol levels found a significant main effect of visit, F(1,68) = 160.71, p < 0.001, but no effect of diagnosis and no interaction effect of DST by diagnostic group. Because we wanted to confirm significant dexamethasone suppression of cortisol in each group, we repeated the analysis separately in each group, and found significant effects of visit in PTSD subjects, F(1,25) = 98.36, p < 0.001, and No-PTSD subjects, F(1,43) = 91.92, p < 0.001. However, there were no group differences in baseline cortisol, F(1,89) = 0.30, ns, DST cortisol levels, F(1,70) = 1.52, ns, or change scores from baseline to DST cortisol, F(1,70) = 0.04, ns. These data suggest that we did not find hypersuppression of cortisol in the PTSD group in this sample.
Figure 2.

Cortisol levels across visits (baseline and DST) and diagnostic groups. Dexamethasone effectively suppressed cortisol levels in both groups.
A RM ANOVA of ACTH levels also showed a significant main effect of visit, F(1,64) = 9.04, p < 0.01, but no significant effect of diagnostic group, and no interaction effect of group by DST visit. A RM ANOVA performed within each diagnostic group separately found a significant reduction in ACTH levels in the No-PTSD subjects, F(1,39) = 6.89, p = 0.01, but only a trend for a reduction in PTSD subjects, F(1,25) = 2.99, p < 0.1. Between-group analyses of baseline and DST ACTH did not show significant group differences, F(1,85) = 0.18, ns, and F(1,66) = 0.05, ns, respectively. The change score from baseline to DST ACTH levels also did not differ between groups, F(1,66) = 0.03, ns. As in the case of cortisol, this study did not find ACTH hypersuppression in PTSD.
3.4. Fear-potentiated startle (FPS)
Table 2 shows startle magnitude to the noise alone, CS+, and CS− trials across each block and dexamethasone treatment condition for each diagnostic group. Within each block there was a significant effect of trial type (block 1, F(2,192) = 30.41, p < 0.001; block 2, F(2,192) = 35.16, p < 0.001; block 3, F(2,192) = 28.45, p < 0.001). In the first two blocks there were no other significant main effects or interaction effects. In the third block, there was a significant 3-way interaction, F(2,192) = 4.51, p = 0.01. Follow-up analyses indicated that PTSD subjects had significantly higher startle magnitude than the controls on the CS+ and CS− trials, but not the NA trials. In all blocks, regardless of diagnostic or treatment group, all subjects showed significant fear conditioning to the CS+ compared to NA (see Table 2). In order to reduce the impact of individual differences in baseline (NA) startle magnitude, we calculated a value for fear-potentiated startle; FPS was assessed using the difference score between baseline startle and startle in the presence of the conditioned stimuli. Although there were no significant differences in baseline startle between groups, given that the NA startle was higher on average in the PTSD group than the control group (see Table 2), we performed a bivariate correlation between NA startle and the difference score (i.e., FPS) for the CS+ and CS− in the PTSD group. Neither score was correlated with NA startle (CS+, r = 0.12, ns; CS−, r = −0.08, ns).
Table 2.
Startle magnitude across groups and dexamethasone (DEX) treatment conditions across the three blocks of conditioning.
| Startle magnitude (μV) | PTSD (n = 33)
|
No-PTSD (n = 67)
|
||
|---|---|---|---|---|
| No DEX TMT (n = 16) | DEX TMT (n = 17) | No DEX TMT (n = 38) | DEX TMT (n = 29) | |
| Block 1 | ||||
| Noise alone (M, SE) | 131.71 (27.0) | 69.82 (26.2)‡ | 72.43 (17.5) | 74.05 (20.1) |
| CS+ (M, SE) | 164.84 (31.2)† | 100.39 (30.3)† | 113.83 (20.3)† | 107.08 (23.2)† |
| CS− (M, SE) | 190.54 (32.0) | 104.45 (31.1) | 107.65 (20.8) | 116.85 (23.8) |
| Block 2 | ||||
| Noise alone (M, SE) | 104.34 (21.1) | 59.89 (20.5) | 58.52 (13.7) | 65.31 (15.7) |
| CS+ (M, SE) | 182.53 (30.4)† | 96.32 (29.5)†,‡ | 112.70 (19.7)† | 102.29 (22.6)† |
| CS− (M, SE) | 171.70 (30.1) | 94.59 (29.2) | 83.39 (19.5)* | 91.93 (22.4) |
| Block 3 | ||||
| Noise alone (M, SE) | 99.41 (23.0) | 51.75 (22.3) | 51.62 (14.9) | 46.05 (17.1) |
| CS+ (M, SE) | 183.92 (30.4)† | 79.90 (29.8)†,‡ | 80.36 (19.9)†,* | 91.81 (22.8)† |
| CS− (M, SE) | 138.31 (23.3) | 72.85 (22.6) | 58.11 (15.1)* | 58.81 (17.3) |
Diagnostic group effect, p < 0.05.
Trial type effect (CS+ vs. NA), p < 0.05.
DEX treatment effect, p < 0.05.
Fear-potentiated startle data were analyzed across 3 blocks of conditioning using a 4-way mixed ANOVA with block (3 levels) × trial type (CS+, CS−) × group (PTSD, No-PTSD) × dexamethasone treatment (TMT, No-TMT) and childhood and adult trauma history as covariates. This analysis revealed a significant 4-way interaction, F(2,192) = 4.79, p = 0.009. Follow-up analyses within each block of conditioning indicated a significant 3-way interaction of trial type -× group × treatment in the 3rd block of conditioning, F(21,94) = 3.91, p = 0.05. There were no significant interactions or main effects of group or dexamethasone treatment in the earlier conditioning blocks. In order to assess group differences in differential conditioning (i.e., FPS to CS+ vs. CS−) without the dexamethasone treatment, we examined the effect of trial type in the last block of conditioning with a 2-way mixed ANOVA with trial type (CS+, CS−) × group (PTSD, No-PTSD). This analysis revealed significantly higher FPS to the CS+ than the CS− across both groups, F(1,52) = 13.25, p = 0.001. There was no interaction effect of trial type and group; however, there was a significant main effect of group, with PTSD having higher FPS to both trial types, F(1,52) = 8.19, p = 0.006. We repeated the same 2-way ANOVA of trial type (CS+, CS−) × group (PTSD, No-PTSD) with dexamethasone treatment and found that there was still a significant, albeit not as strong, effect of trial type, F(1,44) = 5.32, p = 0.03. Again, there was no interaction of trial type and group, and there was also no longer a significant main effect of group, F(1,44) = 0.11, ns.
3.5. Dexamethasone effect on FPS
In order to examine the effect of dexamethasone treatment on each trial type, we performed a univariate analysis of FPS to the CS+ in the 3rd block of conditioning, with diagnostic group (PTSD, No-PTSD) and dexamethasone treatment (TMT, No-TMT) as between-group variables, with and without trauma history and depression as covariates. The analysis was repeated with the CS− trial type. The analysis of the CS+ trial type revealed a 2-way interaction of diagnosis by treatment, F(1,99) = 6.83, p = 0.01. Adding trauma history as covariates to the model increased the interaction effect, F(1,99) = 7.54, p = 0.007, which was further strengthened by adding depression to the ANCOVA, F(1,93) = 8.07, p = 0.006. This interaction was followed by comparing diagnostic groups separately for each treatment, and by comparing treatment conditions within each diagnostic category. The first analysis found significantly greater fear potentiation to the CS+ in PTSD subjects compared to No-PTSD subjects (ANOVA F(1,53) = 8.08, p = 0.006; ANCOVA with trauma, F(1,53) = 7.52, p = 0.008; ANCOVA with trauma and depression, F(1,50) = 4.33, p = 0.04) when subjects were tested at baseline cortisol levels (within the No-TMT condition, see Fig. 3A). However, in the TMT condition, the group difference was no longer present (ANOVA F(1,45) = 0.77, ns; ANCOVA with trauma, F(1,45) = 0.28, ns; ANCOVA with trauma and depression, F(1,42) = 0.39, ns). When we compared the dexamethasone treatment conditions within each diagnostic group, there was a significant treatment effect in PTSD subjects, with dexamethasone reducing fear-potentiated startle to the CS+ (F(1,32) = 4.00, p = 0.05). However, there was no significant treatment effect in the No-PTSD control group (F(1,66) = 1.94, ns).
Figure 3.

(A) Fear-potentiated startle to the CS+ and CS− in the last block of conditioning across treatment conditions and diagnostic groups. The significant group difference in the no dexamethasone condition was eliminated in the dexamethasone condition. Abbreviations: CS+ = conditioned stimulus reinforced with the airblast (reinforced); CS− = conditioned stimulus without the airblast (nonreinforced). (B) Fear-potentiated startle to the CS+ and CS− in the last block of conditioning across treatment conditions and diagnostic groups converted to Tscores. Tscores were calculated based on the individual’s average noise alone startle magnitude set as the mean, i.e., T = 50. The significant group difference in the no dexamethasone condition was eliminated in the dexamethasone condition. Abbreviations: CS+ = conditioned stimulus reinforced with the airblast (reinforced); CS− = conditioned stimulus without the airblast (nonreinforced).
In an effort to standardize the startle data, we converted the raw startle magnitude data to T scores according to the methods used in the NIMH Center for Emotion and Attention (e.g. McTeague et al., 2010). Fig. 3B shows the above data presented as T scores, with the individual’s average NA startle magnitude set as the mean, i.e., T = 50. Using the T scores in the same analyses as above, we replicated the effect of dexamethasone treatment on the group differences in fear potentiation to CS+ trials. There was significantly greater fear potentiation to the CS+ in PTSD subjects compared to No-PTSD subjects, F(1,53) = 4.98, p = 0.03 (covariates did not change the results), when No-TMT subjects were tested (no dexamethasone condition, see Fig. 3B). However, in the TMT condition, the group difference was no longer present, F(1,45) = 0.40, ns. Furthermore, when we compared the dexamethasone treatment conditions within each diagnostic group, the analyses of T scores did not reveal a significant effect of dexamethasone in either group.
The analysis of the CS− trial type showed a significant main effect of diagnostic group, with higher fear-potentiated startle in the PTSD subjects compared to No-PTSD subjects (ANOVA F(1,99) = 3.99, p = 0.05; ANCOVA with trauma, F(1,99) = 4.35, p = 0.04). However, this was not the case when depression was added to the model (ANCOVA with trauma and depression, F(1,93) = 2.23, ns). There was no effect of treatment and no interaction effect of group by treatment. The analysis was repeated with T scores of the startle magnitude to the CS− trials; there were no significant effects of group or treatment.
3.6. Contingency awareness
Response pad data were available for 59 participants (19 PTSD, 40 controls, the reduction in sample size was due to computer error); these data were analyzed with a RM ANOVA comparing US expectancy responses to CS+ and CS− across blocks of conditioning, with diagnosis and treatment as between-group factors. This analysis revealed a significant main effect of trial type, F(1,53) = 130.27, p < 0.0001, but no main effects of dexamethasone treatment or diagnostic group. There was also a significant interaction effect of block and trial type, F(2,106) = 15.23, p < 0.001, showing that awareness increased over conditioning blocks. In order to better compare awareness with the startle data, we analyzed US expectancy specifically in the 3rd block of conditioning, with and without trauma history and depression as covariates. The results of the response keypad data showed that, across both groups, subjects understood the experimental contingencies demonstrating significant differential conditioning between the CS+ and CS− (Fig. 4) (RM ANOVA F(1,55) = 117.88, p < 0.0001; RM ANOVA with trauma covariates F(1,53) = 9.39, p = 0.003; RM ANOVA with trauma and depression covariates F(1,49) = 4.88, p = 0.03). Furthermore, contingency awareness was not affected by dexamethasone. Analysis of US expectancy did not show main effects of diagnostic group, treatment, or interaction effects of the two factors. Although most participants demonstrated awareness, there were a few who did not (2 PTSD, 7 No-PTSD). Therefore, we analyzed the main findings in only the aware participants (defined as having a greater US expectancy on the CS+ than CS− on the final conditioning block). The results did not change, i.e., without dexamethasone PTSD subjects had exaggerated FPS to the CS+ compared to No-PTSD subjects, F(1,28) = 8.39, p = 0.007, while with dexamethasone there was no difference between groups, F(1,20) = 2.37, ns. These data suggest that PTSD symptoms did not affect cognitive awareness of the CS contingency and that awareness was also unaffected by dexamethasone.
Figure 4.
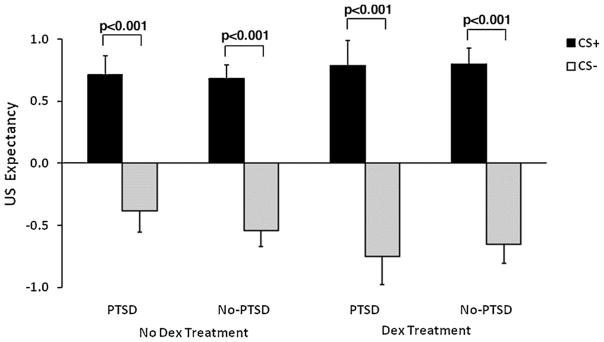
Response keypad data for the CS+ and CS− trials in the last block of conditioning across dexamethasone treatment conditions and diagnostic groups. Expectancy of the US was significantly higher to the CS+ than CS− in every condition, but did not differ by treatment condition or group. Abbreviations: CS+ = conditioned stimulus reinforced with the airblast (reinforced); CS− = conditioned stimulus without the airblast (nonreinforced); US = unconditioned stimulus, i.e., airblast.
3.7. Cortisol levels and FPS
Given the significant suppression of cortisol with dexamethasone, we wanted to see whether cortisol levels were associated with FPS to the CS+. We performed correlations on baseline and DST cortisol levels in the total sample as well as separately for each diagnostic group. In the total sample, we did not find a correlation between baseline cortisol and FPS (r = 0.09, ns), or DST cortisol and FPS (r = −0.16, ns). However, when we examined each diagnostic group separately, we found a significant positive correlation in the PTSD group between baseline cortisol and FPS to the CS+ (r = 0.46, p = 0.01), see Fig. 5; but not between DST cortisol and FPS (r = −0.01, ns). Neither association was significant in the No-PTSD group. These data suggest that HPA axis activity, represented by baseline cortisol, is associated with heightened fear in PTSD, and that the suppression of HPA function in the above studies may be correlated with normalization of fear responses.
Figure 5.

Scatter plot of fear-potentiated startle responses to the CS+ and baseline cortisol levels in the two diagnostic groups. A positive correlation was found only in the PTSD group. Abbreviation: CS+ = conditioned stimulus reinforced with the airblast (reinforced).
4. Discussion
This is the first study to examine fear-potentiated startle in PTSD subjects undergoing the dexamethasone suppression test. We found that diagnostic differences in fear responses were normalized under conditions of suppressed cortisol levels. This finding suggests that glucocorticoids may be involved in pathological fear that is observed in PTSD patients (Jovanovic et al., 2009, 2010b; Lissek et al., 2005; Shin et al., 2005). A recent meta-analysis of 15 studies using fear conditioning found that patients with anxiety disorders showed higher levels of fear responses compared to healthy controls (Lissek et al., 2005). These data suggest that the fear response is over-active, while the inhibition of fear is deficient in PTSD, which has led researchers to use fear conditioning models to examine some of the core PTSD symptoms. An early study (Grillon and Morgan, 1999) using a FPS paradigm with Gulf war veterans diagnosed with PTSD found equivalent levels of fear potentiation to the CS+ in the PTSD and control groups during conditioning, but heightened FPS to the CS+ in the PTSD patients during later phases of the experiment, and greater startle to the CS− during conditioning. On the other hand, a study by Orr et al. (2000) that examined fear conditioning in PTSD subjects using skin conductance found that PTSD subjects discriminated between the danger and safety cues better than controls. However, even in this study, the larger discrimination was due to increased skin conductance responses to the CS+ in the PTSD group. A prospective study of police academy cadets found that greater skin conductance responses to threatening stimuli and slower habituation prior to trauma exposure were predictive of PTSD symptom severity after trauma exposure (Pole et al., 2009). These studies underscore the consistency of heightened fear conditioned responses to the CS+ in PTSD.
Given that fear acquisition requires learning of the association between the CS and the US, the fear expression increases over repeated trials, so that fear-potentiated startle is greatest in the final block of conditioning. However, healthy participants may acquire the association quickly and begin to habituate to the US, a phenomenon originally observed by Rescorla (1973) so that the greatest fear expression is in the second block, and diminishes by the third block. PTSD patients, on the other hand, maintain high levels of fear which results in the greatest group differences being observed in the third block. This effect of dysregulated fear responses (i.e., greater fear after many acquisition trials) is precisely what is reduced by dexamethasone.
In the current study, both PTSD subjects and No-PTSD trauma controls exhibited significant cortisol and ACTH suppression following dexamethasone. Interestingly, we did not find a PTSD effect on either baseline or DST cortisol or ACTH levels; i.e., we did not observe exaggerated dexamethasone suppression (Newport et al., 2004; Yehuda et al., 2002, 2004a), possibly because we did not exclude patients with comorbid depression, which frequently yields a non-suppression effect (Yehuda et al., 2004b). Another potential reason for the lack of a group effect is that both groups were traumatized. de Kloet et al. (2007) found that combat veterans with PTSD had similar levels of DST cortisol to trauma controls, and both had reduced levels compared to non-trauma controls. Additionally, as mentioned above, dexamethasone hypersuppression may be associated with genetic risk for PTSD, rather than PTSD per se (Binder et al., 2008).
Data on baseline cortisol levels in PTSD have been somewhat inconsistent (see recent review by Yehuda, 2009), with several studies reporting low levels in PTSD (Yehuda et al., 1990, 2000; Young et al., 2004) and others finding no diagnostic differences (Shalev et al., 2008; Wheler et al., 2006). A recent meta-analysis concluded that cortisol levels were only reduced if serum or plasma levels were measured (Meewisse et al., 2007). On the other hand, emotional stimuli, such as trauma reminders have been found to increase salivary cortisol levels in PTSD patients (Elzinga et al., 2003). Given the lack of group differences in baseline cortisol in the present study, it does not appear to be cortisol per se that is enhancing fear-potentiated startle in the PTSD group. Rather, it may be the sensitivity of glucocorticoid receptors (GR) to endogenous cortisol in limbic structures that is accounting for the observed effect. Preclinical studies using animal models of repeated stress have found GR sensitization to fear stimuli (Cook, 2002). Thus, even equivalent amounts of cortisol may have different effects in populations at risk for psychopathology. GR hypersensitivity has been shown in many studies with PTSD patients (Yehuda, 2009) and is consistent with studies showing increased genetic risk for PTSD in individuals carrying FKBP5 polymorphisms coding for alterations in GR sensitivity (Binder et al., 2008; Xie et al., 2010) as well as reduced FKBP5 gene expression in PTSD (Yehuda et al., 2009a).
In our previous studies using a conditional discrimination paradigm (Jovanovic et al., 2005), we have consistently found that PTSD is associated with impaired inhibition of fear (Jovanovic et al., 2009, 2010a). In this paradigm we also found increased FPS to the CS− in subjects with PTSD; however, we did not find any effects of dexamethasone on the CS−. On the other hand, we found exaggerated FPS to the CS+, which was more than two-fold greater than that of the traumatized No-PTSD controls, and positively correlated to baseline cortisol levels. In our previous studies, we have found that fear expression to the CS+ (danger signal) and inhibition of fear to the CS− (safety signal) appear to be different processes, likely mediated by different neurocircuitry. While fear responses may only involve subcortical areas of the brain located primarily in the limbic circuitry, safety signals may require a cognitive, cortical component (Bremner et al., 2005; Weike et al., 2008). For example, a recent study by Weike et al. (2008) examined the temporal domain of fear conditioning with a danger and safety signal and found that safety signal processing was slower than danger processing. Thus it would seem that the effects of dexamethasone may be limited to reducing amygdala hyper-activation, thereby not affecting safety signal processing.
Enhanced fear conditioned responses with glucocorticoids have been demonstrated in several studies using animal models (Roozendaal et al., 2002, 2008). It has been shown that GR agonists increase memory formation during fear conditioning (Roozendaal et al., 2009), and GR antagonists reduce fear responses (Kohda et al., 2007). It appears that cortisol in the amygdala is a necessary pre-requisite for the development of fear conditioning in neonates (Sullivan, 2001). Although this is an understudied area in the human clinical literature, studies of hippocampally mediated learning processes in PTSD found that conditioning was impaired by large doses of hydrocortisone given systemically 6 h prior to conditioning (Vythilingam et al., 2005). Furthermore, a recent PET neuroimaging study examined glucose metabolism in several brain areas after administering a high dose of hydrocortisone, resulting in increased amygdala activation in PTSD patients (Yehuda et al., 2009b). Taken together, these studies suggest that vulnerability to develop PTSD in the aftermath of extreme traumatic experiences may be, in part, a combination of genes coding for GR hyper-sensitivity and an overactive amygdala that is primed to respond to endogenous glucocorticoids. Whether the amygdala is sensitized by early trauma (environmental effect) or is overactive due to other genetic or epigenetic pre-trauma risk factors is still unclear at this time. However, these two factors could result in an enhanced fear conditioning phenotype which may partially underlie PTSD psychopathology.
The limitations of the study include the use of self-report data for trauma histories and PTSD symptoms and the lack of a placebo control group. With regard to the self-report measures, these have been used in our previous studies with this population (Binder et al., 2008; Gillespie et al., 2009; Jovanovic et al., 2010a,b) and shown validity with other measures (Binder et al., 2008). The present study was based on a naturalistic between-group comparison of dexamethasone; future studies will need to replicate the findings using a placebo-controlled cross-over design.
In the present study, artificially reducing endogenous cortisol levels led to the elimination of diagnostic differences in fear conditioned responses to danger cues. These data have important implications for treatment of PTSD, especially concerning acute states of fear or arousal. Importantly, neither cognitive learning nor motor responses were affected by dexamethasone administration as evidenced by normal use of the response keypad. There have been few studies that have examined glucocorticoids as treatment options for PTSD, and they have yielded mixed results. Acute administration of stress-levels of cortisol impairs memory retrieval in healthy controls (Tollenaar et al., 2007), and appears to reduce traumatic memory retrieval in PTSD (de Quervain, 2008). On the other hand, dexamethasone suppression of cortisol does not appear to affect memory in PTSD (Bremner et al., 2004). Interestingly, chronic daily administration of low dose cortisol appears to ameliorate PTSD symptoms (Aerni et al., 2004), while case studies of acute dexamethasone administration have also indicated improvement in symptoms (Driscoll, 2009). Future studies should focus on GR system targets as a possible adjunct therapy to other treatment strategies in patients with PTSD.
Acknowledgments
Role of the funding sources
Funding for this study was provided by NIMH grants (MH071537 and MH47840), NIH National Centers for Research Resources (M01 RR00039), and the Burroughs Wellcome Fund; these funding agencies had no further role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
This work was primarily supported by National Institutes of Mental Health (MH071537). Support was also Emory and Grady Memorial Hospital General Clinical Research Center, NIH National Centers for Research Resources (M01 RR00039), and the Burroughs Wellcome Fund. We thank Allen Graham, BA, Daniel Crain, BS, Lamya Khoury, BS, Asante Kamkwalala, BS, Lauren Sands, BA, and India Karapanou, BA, as well as the nurses and staff of the Grady GCRC for their assistance with data collection and support.
Footnotes
Conflicts of interest
Dr. Jovanovic has research support from NIMH (F32 MH070129). Ms. Phifer, Ms. Sicking, and Dr. Weiss received past departmental fellowship funding from AstraZeneca. Dr. Norrholm has research support from the National Alliance for Research on Schizophrenia and Depression (NARSAD), the Department of Defense (DOD)/Congressionally Directed Medical Research Program (CDMRP, award #W81XWH-08-2-0170), and the Emory University Research Committee. Dr. Bradley reported research support from American Foundation for Suicide Prevention. Dr. Ressler reported research support from NIMH (MH071537); NIH National Centers for Research Resources (M01 RR00039); NIDA; NARSAD; Burroughs Wellcome Foundation; and is cofounder of Extinction Pharmaceuticals for the development of NMDA-based therapeutics to enhance extinction.
References
- Aerni A, Traber R, Hock C, Roozendaal B, Schelling G, Papassotiropoulos A, Nitsch RM, Schnyder U, de Quervain DJF. Low-dose cortisol for symptoms of posttraumatic stress disorder. American Journal of Psychiatry. 2004;161:1488–1490. doi: 10.1176/appi.ajp.161.8.1488. [DOI] [PubMed] [Google Scholar]
- Alim TN, Graves E, Mellman TA, Aigbogun N, Gray E, Lawson W, Charney DS. Trauma exposure, posttraumatic stress disorder and depression in an African-American primary care population. Journal of the National Medical Association. 2006;98:1630–1636. [PMC free article] [PubMed] [Google Scholar]
- APA. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- Baker DB, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. American Journal of Psychiatry. 1999;156:585–588. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- Beck AT, Ward CH, Mendelsohn M, Mock J, Erbaugh J. An inventory for measuring depression. Archives of General Psychiatry. 1961;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- Bernstein DP, Fink L. Childhood Trauma Questionnaire: A Retrospective Self-Report Manual. The Psychological Corporation; San Antonio, TX: 1998. [Google Scholar]
- Bernstein DP, Stein JA, Newcomb MD, Walker E, Pogge D, Ahluvalia T, Stokes J, Handelsman L, Medrano M, Desmond D, Zule W. Development and validation of a brief screening version of the Childhood Trauma Questionnaire. Child Abuse & Neglect. 2003;27:169–190. doi: 10.1016/s0145-2134(02)00541-0. [DOI] [PubMed] [Google Scholar]
- Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34:S186–S195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. Journal of the American Medical Association. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RG, Binder EB, Epstein MP, Tang Y, Nair HP, Liu W, Gillespie CF, Berg T, Evces M, Newport DJ, Stowe ZN, Heim CM, Nemeroff CB, Schwartz A, Cubells JF, Ressler KJ. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Archives of General Psychiatry. 2008;65:190–200. doi: 10.1001/archgenpsychiatry.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, Nemeroff CB, Charney DS. Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. American Journal of Psychiatry. 1997;154:624–629. doi: 10.1176/ajp.154.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Vermetten E, Schmahl C, Vaccarino V, Vythilingam M, Afzal N, Grillon C, Charney DS. Positron emission tomographic imaging of neural correlates of a fear acquisition and extinction paradigm in women with childhood sexual-abuse-related post-traumatic stress disorder. Psychological Medicine. 2005;35:791–806. doi: 10.1017/s0033291704003290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Vythilingam M, Vermetten E, Afzal N, Nazeer A, Newcomer JW, Charney DS. Effects of dexamethasone on declarative memory function in posttraumatic stress disorder. Psychiatry Research. 2004;129:1–10. doi: 10.1016/j.psychres.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Breslau N, Wilcox HC, Storr CL, Lucia VC, Anthony JC. Trauma exposure and posttraumatic stress disorder: a study of youths in urban America. Journal of Urban Health. 2004;81:530–544. doi: 10.1093/jurban/jth138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook CJ. Glucocorticoid feedback increases the sensitivity of the limbic system to stress. Physiology & Behavior. 2002;75:455–464. doi: 10.1016/s0031-9384(02)00650-9. [DOI] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in fear-potentiated startle: implications for animal models of anxiety. Trends in Pharmacological Sciences. 1992a;13:35–41. doi: 10.1016/0165-6147(92)90014-w. [DOI] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in fear and anxiety. Annual Review of Neuroscience. 1992b;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- Davis M, Falls WA, Campeau S, Kim M. Fear-potentiated startle: a neural and pharmacological analysis. Behavioral Brain Research. 1993;58:175–198. doi: 10.1016/0166-4328(93)90102-v. [DOI] [PubMed] [Google Scholar]
- de Kloet CS, Vermetten E, Heijnen CJ, Geuze E, Lentjes EG, Westenberg HG. Enhanced cortisol suppression in response to dexamethasone administration in traumatized veterans with and without posttraumatic stress disorder. Psychoneuroendocrinology. 2007;32:215–226. doi: 10.1016/j.psyneuen.2006.12.009. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ. Glucocorticoid-induced reduction of traumatic memories: implications for the treatment of PTSD. Progress in Brain Research. 2008;167:239–247. doi: 10.1016/S0079-6123(07)67017-4. [DOI] [PubMed] [Google Scholar]
- Driscoll H. Dexamethasone in Clinical Treatment of Acute Exacerbation of Chronic PTSD. International Society of Traumatic Stress Studies; Atlanta, GA: 2009. [Google Scholar]
- Elzinga BM, Schmahl CS, Vermetten E, van Dyck R, Bremner JD. Higher cortisol levels following exposure to traumatic reminders in abuse-related PTSD. Neuropsychopharmacology. 2003;28:1656–1665. doi: 10.1038/sj.npp.1300226. [DOI] [PubMed] [Google Scholar]
- Falsetti S, Resnick H, Resick P, Kilpatrick D. The modified PTSD Symptom Scale: a brief self-report measure of posttraumatic stress disorder. The Behavior Therapist. 1993;16:161–162. [Google Scholar]
- Foa EB, Tolin DF. Comparison of the PTSD symptom scale-interview version and the clinician-administered PTSD scale. Journal of Traumatic Stress. 2000;13:181–191. doi: 10.1023/A:1007781909213. [DOI] [PubMed] [Google Scholar]
- Galea S, Brewin CR, Gruber M, Jones RT, King DW, King LA, McNally RJ, Ursano RJ, Petukhova M, Kessler RC. Exposure to hurricane-related stressors and mental illness after Hurricane Katrina. Archives of General Psychiatry. 2007;64:1427–1434. doi: 10.1001/archpsyc.64.12.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie CF, Bradley RG, Mercer K, Smith AK, Conneely K, Gapen M, Weiss T, Schwartz AC, Cubells JF, Ressler KJ. Trauma exposure and stress-related disorders in inner city primary care patients. General Hospital Psychiatry. 2009;31:505–514. doi: 10.1016/j.genhosppsych.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Morgan CA., 3rd Fear-potentiated startle conditioning to explicit and contextual cues in Gulf War veterans with posttraumatic stress disorder. Journal of Abnormal Psychology. 1999;108:134–142. doi: 10.1037//0021-843x.108.1.134. [DOI] [PubMed] [Google Scholar]
- Grillon C, Morgan CA, 3rd, Davis M, Southwick SM. Effects of experimental context and explicit threat cues on acoustic startle in Vietnam veterans with posttraumatic stress disorder. Biological Psychiatry. 1998;44:1027–1036. doi: 10.1016/s0006-3223(98)00034-1. [DOI] [PubMed] [Google Scholar]
- Grillon C, Pine DS, Baas JM, Lawley M, Ellis V, Charney DS. Cortisol and DHEA-S are associated with startle potentiation during aversive conditioning in humans. Psychopharmacology. 2006;186:434–441. doi: 10.1007/s00213-005-0124-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, Miller AH, Nemeroff CB. Pituitary–adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. Journal of the American Medical Association. 2000;284:592–597. doi: 10.1001/jama.284.5.592. [DOI] [PubMed] [Google Scholar]
- Heim C, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology. 2008;33:693–710. doi: 10.1016/j.psyneuen.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Hoge CW, Castro CA, Messer SC, McGurk D, Cotting DI, Koffman RL. Combat duty in Iraq and Afghanistan, mental health problems, and barriers to care. New England Journal of Medicine. 2004;351:13–22. doi: 10.1056/NEJMoa040603. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, Keyes M, Fiallos A, Myers KM, Davis M, Duncan EJ. Fear potentiation and fear inhibition in a human fear-potentiated startle paradigm. Biological Psychiatry. 2005;57:1559–1564. doi: 10.1016/j.biopsych.2005.02.025. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, Blanding NQ, Davis M, Duncan E, Bradley B, Ressler KJ. Impaired fear inhibition is a biomarker of PTSD but not depression. Depression and Anxiety. 2010a;27:244–251. doi: 10.1002/da.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, Blanding NQ, Phifer JE, Weiss T, Davis M, Duncan E, Bradley B, Ressler K. Fear potentiation is associated with hypothalamic–pituitary–adrenal axis function in PTSD. Psychoneuroendocrinology. 2010b;35:846–857. doi: 10.1016/j.psyneuen.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, Fennell JE, Keyes M, Fiallos AM, Myers KM, Davis M, Duncan EJ. Posttraumatic stress disorder may be associated with impaired fear inhibition: relation to symptom severity. Psychiatry Research. 2009;167:151–160. doi: 10.1016/j.psychres.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic T, Norrholm SD, Keyes M, Fiallos A, Jovanovic S, Myers KM, Davis M, Duncan EJ. Contingency awareness and fear inhibition in a human fear-potentiated startle paradigm. Behavioral Neuroscience. 2006;120:995–1004. doi: 10.1037/0735-7044.120.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalin NH, Takahashi LK. Fear-motivated behavior induced by prior shock experience is mediated by corticotropin-releasing hormone systems. Brain Research. 1990;509:80–84. doi: 10.1016/0006-8993(90)90311-x. [DOI] [PubMed] [Google Scholar]
- Keen-Rhinehart E, Michopoulos V, Toufexis DJ, Martin EI, Nair H, Ressler KJ, Davis M, Owens MJ, Nemeroff CB, Wilson ME. Continuous expression of corticotropin-releasing factor in the central nucleus of the amygdala emulates the dysregulation of the stress and reproductive axes. Molecular Psychiatry. 2008;14:37–50. doi: 10.1038/mp.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohda K, Harada K, Kato K, Hoshino A, Motohashi J, Yamaji T, Morinobu S, Matsuoka N, Kato N. Glucocorticoid receptor activation is involved in producing abnormal phenotypes of single-prolonged stress rats: a putative post-traumatic stress disorder model. Neuroscience. 2007;148:22–33. doi: 10.1016/j.neuroscience.2007.05.041. [DOI] [PubMed] [Google Scholar]
- Lang PJ, Davis M, Ohman A. Fear and anxiety: animal models and human cognitive psychophysiology. Journal of Affective Disorders. 2000;61:137–159. doi: 10.1016/s0165-0327(00)00343-8. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Fear and the brain: where have we been, and where are we going? Biological Psychiatry. 1998;44:1229–1238. doi: 10.1016/s0006-3223(98)00282-0. [DOI] [PubMed] [Google Scholar]
- Lee Y, Davis M. Role of the hippocampus, the bed nucleus of the stria terminalis, and the amygdala in the excitatory effect of corticotropin-releasing hormone on the acoustic startle reflex. Journal of Neuroscience. 1997;17:6434–6446. doi: 10.1523/JNEUROSCI.17-16-06434.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KC, Melia KR, Miserendino MJD, Falls WA, Campeau S, Davis M. Corticotropin-releasing factor: long-lasting facilitation of the acoustic startle reflex. Journal of Neuroscience. 1992;12:2303–2312. doi: 10.1523/JNEUROSCI.12-06-02303.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon I, Sripada CS. The functional neuroanatomy of PTSD: a critical review. Progress in Brain Research. 2008;167:151–169. doi: 10.1016/S0079-6123(07)67011-3. [DOI] [PubMed] [Google Scholar]
- Lissek S, Powers AS, McClure EB, Phelps EA, Woldehawariat G, Grillon C, Pine DS. Classical fear conditioning in the anxiety disorders: a meta-analysis. Behaviour Research & Therapy. 2005;43:1391–1424. doi: 10.1016/j.brat.2004.10.007. [DOI] [PubMed] [Google Scholar]
- McTeague LM, Lang PJ, Laplante MC, Cuthbert BN, Shumen JR, Bradley MM. Aversive imagery in posttraumatic stress disorder: trauma recurrence, comorbidity, and physiological reactivity. Biological Psychiatry. 2010;67:346–356. doi: 10.1016/j.biopsych.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meewisse ML, Reitsma JB, De Vries GJ, Gersons BPR, Olff M. Cortisol and post-traumatic stress disorder in adults: systematic review and meta-analysis. The British Journal of Psychiatry. 2007;191:387–392. doi: 10.1192/bjp.bp.106.024877. [DOI] [PubMed] [Google Scholar]
- Morgan CA, 3rd, Grillon C, Southwick SM, Davis M, Charney DS. Fear-potentiated startle in posttraumatic stress disorder. Biological Psychiatry. 1995;38:378–385. doi: 10.1016/0006-3223(94)00321-S. [DOI] [PubMed] [Google Scholar]
- Newport DJ, Heim C, Bonsall R, Miller AH, Nemeroff CB. Pituitary–adrenal responses to standard and low-dose dexamethasone suppression tests in adult survivors of child abuse. Biological Psychiatry. 2004;55:10–20. doi: 10.1016/s0006-3223(03)00692-9. [DOI] [PubMed] [Google Scholar]
- Norrholm SD, Jovanovic T, Vervliet B, Myers KM, Davis M, Rothbaum BO, Duncan EJ. Conditioned fear extinction and reinstatement in a human fear-potentiated startle paradigm. Learning & Memory. 2006;13:681–685. doi: 10.1101/lm.393906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr SP, Metzger LJ, Lasko NB, Macklin ML, Peri T, Pitman RK. De novo conditioning in trauma-exposed individuals with and without posttraumatic stress disorder. Journal of Abnormal Psychology. 2000;109:290–298. [PubMed] [Google Scholar]
- Pole N, Neylan TC, Otte C, Henn-Hasse C, Metzler TJ, Marmar CR. Prospective prediction of posttraumatic stress disorder symptoms using fear potentiated auditory startle responses. Biological Psychiatry. 2009;65:235–240. doi: 10.1016/j.biopsych.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA. Effect of US habituation following conditioning. Journal of Comparative Physiology and Psychology. 1973;82:137–143. doi: 10.1037/h0033815. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Barsegyan A, Lee S. Adrenal stress hormones, amygdala activation, and memory for emotionally arousing experiences. Progress in Brain Research. 2008;167:79–97. doi: 10.1016/S0079-6123(07)67006-X. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Brunson KL, Holloway BL, McGaugh JL, Baram TZ. Involvement of stress-released corticotropin-releasing hormone in the basolateral amygdala in regulating memory consolidation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13908–13913. doi: 10.1073/pnas.212504599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Koolhaas JM, Bohus B. Central amygdaloid involvement in neuroendocrine correlates of conditioned stress responses. Journal of Neuroendocrinology. 1992;4:483–489. doi: 10.1111/j.1365-2826.1992.tb00196.x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McReynolds JR, Van der Zee EA, Lee S, McGaugh JL, McIntyre CK. Glucocorticoid effects on memory consolidation depend on functional interactions between the medial prefrontal cortex and basolateral amygdala. Journal of Neuroscience. 2009;29:14299–14308. doi: 10.1523/JNEUROSCI.3626-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbaum BO, Kozak MJ, Foa EB, Whitaker DJ. Post-traumatic stress disorder in rape victims: autonomic habituation to auditory stimuli. Journal of Traumatic Stress. 2001;14:283–293. doi: 10.1023/A:1011160800958. [DOI] [PubMed] [Google Scholar]
- Schwartz AC, Bradley RL, Sexton M, Sherry A, Ressler KJ. Posttraumatic stress disorder among African Americans in an inner city mental health clinic. Psychiatric Services. 2005;56:212–215. doi: 10.1176/appi.ps.56.2.212. [DOI] [PubMed] [Google Scholar]
- Shalev AY, Videlock EJ, Peleg T, Segman R, Pitman RK, Yehuda R. Stress hormones and post-traumatic stress disorder in civilian trauma victims: a longitudinal study. Part I. HPA axis responses. The International Journal of Neuropsychopharmacology. 2008;11:365–372. doi: 10.1017/S1461145707008127. [DOI] [PubMed] [Google Scholar]
- Shin LM, Wright CI, Cannistraro PA, Wedig MM, McMullin K, Martis B, Macklin ML, Lasko NB, Cavanagh SR, Krangel TS, Orr SP, Pitman RK, Whalen PJ, Rauch SL. A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Archives of General Psychiatry. 2005;62:273–281. doi: 10.1001/archpsyc.62.3.273. [DOI] [PubMed] [Google Scholar]
- Sullivan RM. Unique characteristics of neonatal classical conditioning: the role of the amygdala and locus coeruleus. Integrative Physiological & Behavioral Science. 2001;36:293. doi: 10.1007/bf02688797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Britton KT, Koob GF. Potentiation of acoustic startle by corticotropin-releasing factor (CRF) and by fear are both reversed by alphahelical CRF (9–41) Neuropsychopharmacology. 1989;2:285–292. doi: 10.1016/0893-133x(89)90033-x. [DOI] [PubMed] [Google Scholar]
- Switzer GE, Dew MA, Thompson K, Goycoolea JM, Derricott T, Mullins SD. Posttraumatic stress disorder and service utilization among urban mental health center clients. Journal of Traumatic Stress. 1999;12:25–39. doi: 10.1023/A:1024738114428. [DOI] [PubMed] [Google Scholar]
- Tollenaar MS, Elzinga BM, Spinhoven P, Everaerd WA. The effects of cortisol increase on long-term memory retrieval during and after acute psychosocial stress. Acta Psychologica. 2007 doi: 10.1016/j.actpsy.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Vythilingam M, Lawley M, Collin C, Bonne O, Agarwal R, Hadd K, Charney DS, Grillon C. Hydrocortisone impairs hippocampal-dependent trace eyeblink conditioning in post-traumatic stress disorder. Neuropsychopharmacology. 2005;31:182–188. doi: 10.1038/sj.npp.1300843. [DOI] [PubMed] [Google Scholar]
- Weike AI, Schupp HT, Hamm AO. In dubio pro defensio: initial activation of conditioned fear is not cue specific. Behavioral Neuroscience. 2008;122:685–696. doi: 10.1037/0735-7044.122.3.685. [DOI] [PubMed] [Google Scholar]
- Wheler GHT, Brandon D, Clemons A, Riley C, Kendall J, Loriaux DL, Kinzie JD. Cortisol production rate in posttraumatic stress disorder. Journal of Clinical Endocrinology and Metabolism. 2006;91:3486–3489. doi: 10.1210/jc.2006-0061. [DOI] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, Farrer LA, Gelernter J. Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder. Neuropsychopharmacology. 2010;35 (8):1684–1692. doi: 10.1038/npp.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Annals of the New York Academy of Sciences. 2009;1179:56–69. doi: 10.1111/j.1749-6632.2009.04979.x. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Bierer LM, Schmeidler J, Aferiat DH, Breslau N, Dolan S. Low cortisol and risk for PTSD in adult offspring of holocaust survivors. American Journal of Psychiatry. 2000;157:1252–1259. doi: 10.1176/appi.ajp.157.8.1252. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Cai G, Golier JA, Sarapas C, Galea S, Ising M, Rein T, Schmeidler J, Müller-Myhsok B, Holsboer F, Buxbaum JD. Gene expression patterns associated with posttraumatic stress disorder following exposure to the world trade center attacks. Biological Psychiatry. 2009a;66:708–711. doi: 10.1016/j.biopsych.2009.02.034. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Giller EL, Southwick SM, Lowy MT, et al. Hypothalamic–pituitary–adrenal dysfunction in posttraumatic stress disorder. Biological Psychiatry. 1991a;30:1031–1048. doi: 10.1016/0006-3223(91)90123-4. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Golier JA, Halligan SL, Meaney M, Bierer LM. The ACTH response to dexamethasone in PTSD. American Journal of Psychiatry. 2004a;161:1397–1403. doi: 10.1176/appi.ajp.161.8.1397. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Halligan SL, Golier JA, Grossman R, Bierer LM. Effects of trauma exposure on the cortisol response to dexamethasone administration in PTSD and major depressive disorder. Psychoneuroendocrinology. 2004b;29:389–404. doi: 10.1016/s0306-4530(03)00052-0. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Halligan SL, Grossman R, Golier JA, Wong C. The cortisol and glucocorticoid receptor response to low dose dexamethasone administration in aging combat veterans and holocaust survivors with and without posttraumatic stress disorder. Biological Psychiatry. 2002;52:393–403. doi: 10.1016/s0006-3223(02)01357-4. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Harvey PD, Golier JA, Newmark RE, Bowie CR, Wohltmann JJ, Grossman RA, Schmeidler J, Hazlett EA, Buchsbaum MS. Changes in relative glucose metabolic rate following cortisol administration in aging veterans with posttraumatic stress disorder: an FDG-PET neuroimaging study. Journal of Neuropsychiatry and Clinical Neurosciences. 2009b;21:132–143. doi: 10.1176/jnp.2009.21.2.132. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Lowry MT, Southwick SM, Mason JW, Giller EL. Increased number of glucocorticoid receptors in posttraumatic stress disorder. American Journal of Psychiatry. 1991b;148:499–504. doi: 10.1176/ajp.148.4.499. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Southwick SM, Nussbaum G, Wahby V, Giller EL, Jr, Mason JW. Low urinary cortisol excretion in patients with posttraumatic stress disorder. The Journal of Nervous and Mental Disease. 1990;178:366–369. doi: 10.1097/00005053-199006000-00004. [DOI] [PubMed] [Google Scholar]
- Young EA, Tolman R, Witkowski K, Kaplan G. Salivary cortisol and posttraumatic stress disorder in a low-income community sample of women. Biological Psychiatry. 2004;55:621–626. doi: 10.1016/j.biopsych.2003.09.009. [DOI] [PubMed] [Google Scholar]