

Abstract
A novel bis-thiourea BINAM-based catalyst for the asymmetric aza-Henry reaction has been developed. This catalyst promotes the reaction of N-Boc imines with nitroalkanes to afford β-nitroamines with good yields and high enantioselectivities. This catalyst has the advantage that it can be prepared in a single step from commercially available materials. A model is proposed for the catalyst action where the both components of the reaction are activated simultaneously by hydrogen bonding. Regardless of the mechanism, the success of the present catalyst demonstrates the potential of bis-thioureas as an interesting class of relatively unexplored catalysts.
Keywords: Thiourea, Aza-Henry reaction, organocatalyst, asymmetric catalysis, nitroalkanes, BINAM
The nucleophilic addition of nitroalkanes to imines, known as the aza-Henry or nitro-Mannich reaction, is a fundamental C-C bond forming reaction in organic chemistry.1 The resulting 1,2-nitro amines are useful precursors to a variety of nitrogen containing chiral building blocks such as vicinal diamines via reduction of the nitro group2 and α-amino carbonyl compounds by means of the Nef reaction.3 Given the importance of this transformation, considerable effort has been directed towards the development of catalytic asymmetric aza-Henry reaction over the past several years. The first pioneering examples were reported by Shibasaki4 and Jorgensen5 and subsequently other metal-based catalysts have been reported.6 Beyond these metal-catalyzed variants, an increasing interest in organocatalysts in the last few years7 has led to the appearance of the first examples of enantioselective organocatalytic aza-Henry reactions. Takemoto et. al. have reported that the aza-Henry reaction of N-Boc imines with nitroalkanes can be promoted by a bifunctional asymmetric catalyst bearing both a thiourea function and an N,N-dimethylamino group leading to β-nitroamines with good enantioselectivities,8 while shortly thereafter, Yoon and Jacobsen were able to promote the highly stereoselective addition of a range of nitroalkanes to aromatic N-Boc imines using a new thiourea based bifunctional catalyst.9 A third type of thiourea organocatalyst was recently reported for this reaction by Ricci10 and by Schaus11 which involved a thiourea-cinchona alkaloid hybrid. The most recent thiourea catalyst is a hybrid with a chiral oxazoline and is also effective for the aza-Henry reaction.12 Apart from the thioureas, other organocatalysts have been reported for the aza-Henry reaction which are either based on cinchona alkaloids13 or on the triflate salts of bis(amidines).14
It is known that both imines and nitro compounds will interact with thioureas15 and yet a bis-thiourea catalyst has not been reported for the aza-Henry reaction. The first chiral bis-thiourea catalyst to be reported was compound 1 by Nagasawa which was based on trans-cyclohexane-1,2-diamine.16 This catalyst proved effective for the Morita-Baylis-Hillman reaction but not for a number of other reactions.17 Other bis-thiourea catalysts that have been reported include the interesting guanidine derivative 2 which has three double-hydrogen donors and was developed by Nagasawa for the Henry reaction.18 Finally, Berkessel has developed the bis-thiourea catalyst 4 for the Morita-Baylis-Hillman reaction19 and the catalyst 3 for the kinetic resolution of azlactones.20 We report here that the bis-thiourea catalyst 12 based on the 2,2’-diaminobinapthalene (BINAM) chiral scaffold is an effective catalyst for the aza-Henry reaction.21
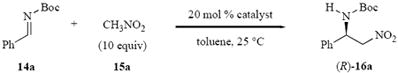
The aza-Henry reaction of the Boc imine 14a and nitromethane were screened with a number of thiourea catalysts and the results are presented in Table 1. This screen includes Nagasawa’s bis-thiourea 1 and the BINAM based thiourea 5 developed by Wang for the Morita-Baylis-Hillman reaction and subsequently found to be effacious for a number of Michael addition reactions.22 The bis-thioureas 6–13 are all preparable in one or two steps from the commercially available (R)-(+)-1,1’-binaphthyl-2,2’-diamine and the details can be found in the supporting information. The bis-thiourea 1 was found to be very effective in promoting the aza-Henry reaction of Boc imine 14a but the asymmetric induction for this reaction was only 8% ee (entry 1). The bifunctional thiourea (R)-5 was not capable of promoting the reaction in the absence of an added base and even in the presence of triethylamine the reaction only went to 50% completion in 12 h and gave 16a in 4% ee. From these results it can be inferred that dimethylamino group in (R)-5 is not sufficiently basic to deprotonate nitromethane. For this reason, the bifunctional catalyst (R)-6 was prepared which has two thiourea functions and two basic functions in the form of imidazoles. As shown by the data in Table 1 this catalyst was capable of promoting the reaction in the absence of triethylamine, but unfortunately, it was not able to provide any significant asymmetric induction (entry 4). The series of six bis-thiourea catalysts 7-12 were then prepared and evaluated and while a few of them were active catalysts only compound (R)-12 with the bis-3,5-trifluoromethylphenyl substituent on the thiourea units was capable of providing significant asymmetric induction (74% ee, entry 10). The macrocyclic bis-thiourea (R,R)-13 was also an active catalyst, but as with most of the catalysts screened for this reaction very low asymmetric induction was observed.
Table 1.
Screen of thioureas catalysts with Boc imine 14a.a
| ||||
|---|---|---|---|---|
| entry | catalyst | Et3N (equiv) | % conversion b | % ee c |
| 1 | (1S,2S)-1 | 0.4 | 100 | 8 |
| 2 | (R)-5 | — | 0 | — |
| 3 | (R)-5 | 0.4 | 50 | 4 |
| 4 | (R)-6 | — | 65 | 5 |
| 5 | (R)-7 | 0.4 | 100 | 5 |
| 6 | (R)-8 | 0.4 | 10 | 4 |
| 7 | (R)-9 | 0.4 | 36 | 0 |
| 8 | (R)-10 | 0.4 | 100 | 16 |
| 9 | (R)-11 | 0.4 | 73 | <5 |
| 10 | (R)-12 | 0.4 | 100 | 74 |
| 11 | (R,R)-13 | 0.4 | 100 | 6 |
Reactions conducted with 10 equiv CH3NO2 and 20 mol % catalyst in toluene at 25 °C for 12 h except entry 10 which was 2 h.
Determined by 1H NMR spectrum of crude reaction mixture.
Determined by HPLC on Chiralpak OJ-H column.
A brief survey of the reaction conditions for the reaction of imine 14a with nitromethane catalyzed by the chiral thiourea (R)-12 in the presence of 0.4 equiv of Et3N was conducted and the data in Table 2 reveal the effects of changes in the solvent, temperature and catalyst loading. The least effective solvent was THF which was not unanticipated since the catalyst design was predicated on the assumption that there would be H-bonds involved in the association of the catalyst and substrate. Solvents that are effective H-bond acceptors would be expected to disrupt such an association in the catalyst-substrate complex. Toluene was identifed as the best of the solvents that were examined and lowering the temperature of the reaction lead to an initial significant improvement in the asymmetric induction when the temperature was dropped to -5 °C (82% ee, entry 5) but then only to a small improvement as the temperature was further reduced to −35 °C (86% ee, entry 6). Interestingly, the induction was not increased when the temperature was further lowered to −55 °C and the only effect observed was a decrease in the rate of the reaction (entry 7). Lowering the catalyst loading to 10 mol% at the optimal temperature of −35 °C for the reaction in toluene lead to a dropoff in rate as well as a slight lowering of the asymmetric induction (entries 6 & 11). Thus, the optimal conditions revealed by variations in the conditions for the reaction of imine 14a with nitromethane are those indicated in entry 6 of Table 2.
Table 2.
Optimization of the reaction of imine 14a with thioureas (R)-12.a
| entry | solvent | cat load (mol %) | temp (°C) | % conversion b | % ee c |
|---|---|---|---|---|---|
| 1 | CH2Cl2 | 20 | 25 | 82 | 59 |
| 2 | THF | 20 | 25 | 52 | 28 |
| 3 | benzene | 20 | 25 | 100 | 62 |
| 4 | toluene | 20 | 25 | 100 | 74 |
| 5 | toluene | 20 | −5 | 100 | 82 |
| 6 | toluene | 20 | −35 | 83 | 86 |
| 7 | toluene | 20 | −55 | 50 | 84 |
| 8 | mesitylene | 20 | −35 | 86 | 84 |
| 9 | chlorobenzene | 20 | −35 | 93 | 84 |
| 10 | toluene | 10 | −10 | 100 | 80 |
| 11 | toluene | 10 | −35 | 53 | 78 |
| 12 | mesitylene | 10 | −10 | 90 | 82 |
Reactions conducted with 10 equiv CH3NO2 in 0.2 M solution in imine 14a with x mol% (R)-12 and 0.4 equiv Et3N. Reaction time at 25 °C was 24 h and all other temperatures was 36 h.
Determined by 1H NMR spectrum of crude reaction mixture with Ph3CH as internal standard.
% ee of (R)-16a determined by HPLC on Chiralpak OJ-H column.
With the preparation and screen of the eleven catalysts indicated in Table 1 and the survey of the reaction conditions summarized in Table 2 it was time to review the substrate compatiblity of the reaction with the set of ten different imines and three nitroalkanes presented in Table 3 which serves to bring the reaction scope into focus. The asymmetric induction for these reactions does not appear to be particularly sensitive to the presence of either electron donating or electron withdrawing substituents on the phenyl group of the imine. It was also of note to observe that the reactions of the methoxy substituted imines did not appear to be slower than the imine from benzaldehyde, however, the imines with electron withdrawing groups were more reactive and were complete in shorter reactions times. It was also of interest to find that the reaction will tolerate the presence of a pyridine ring in the imine substrate (entry 10). A decrease in induction was noted for the ortho substituted aryl imines 14d and 14g (entries 4 and 7) and an explanation for this is not exactly clear especially upon consideration that the 1-naphthyl imine 14i does not give an induction that is suppressed relative to that of imine 14a (entries 1 vs. 9). Finally, the asymmetric induction drops from 86% ee for nitromethane to 70% ee for nitroethane with the same imine (entries 1 vs. 11). Interestingly, the drop is much smaller for nitropropane (entry 12). In the last two entries of the Table, the major diastereomer is the syn adduct 16 with a syn:anti ratio of approximately 4:1.
Table 3.
Scope of the aza-Henry reaction of the N-Boc imine 14 catalyzed by the bis-thiourea (R)-12.a
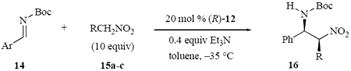
| |||||||
|---|---|---|---|---|---|---|---|
| entry | Imine | Ar | time (h) | R | adduct | % yield b | % ee c |
| 1 | 14a | C6H5 | 36 | H | 16a | 55 | 86 |
| 2 | 14b | 4-ClC6H4 | 15 | H | 16b | 62 | 85 |
| 3 | 14c | 3-ClC6H4 | 15 | H | 16c | 53 | 91 |
| 4 | 14d | 2-ClC6H4 | 15 | H | 16d | 61 | 74 |
| 5 | 14e | 4-BrC6H4 | 24 | H | 16e | 50 | 78 |
| 6 | 14f | 4-MeOC6H4 | 36 | H | 16f | 50 | 89 |
| 7 | 14g | 2-MeOC6H4 | 36 | H | 16g | 40 | 65 |
| 8 | 14h | 4-MeC6H4 | 36 | H | 16h | 48 | 86 |
| 9 | 14i | 1-napthyl | 36 | H | 16i | 65 | 85 |
| 10 | 14j | 3-pyridyl | 22 | H | 16j | 63 | 81 |
| 11 | 14a | C6H5 | 36 | Me | 16k | 59 d | 70 e |
| 12 | 14a | C6H5 | 36 | Et | 16l | 63 d | 80 f |
Reactions conducted with 10 equiv CH3NO2 with 0.2 M 14 in toluene at −35 °C with 0.4 equiv Et3N and 20 mol % (R)-12.
Isolated yield after purification by chromatography on silica gel.
Determined by HPLC on Chiralpak OJ-H column.
Combined yield of syn and anti adducts.
%ee of syn isomer; syn:anti = 77:23.
%ee of syn isomer; syn:anti = 80:20.
The absolute stereochemistry of the adducts 16 was determined by comparing the HPLC data and optical rotations of the products with the data reported for those derivatives of 16 that have been previously reported and then the remaining derivatives of 16 were assigned by correlation and the details can be found in the Supporing Information. To account for the absolute sense of the enantioselectivity of the reactions with the bis-thiourea catalyst 12 we propose structure 17 as a model for the catalyst-substrate complex. The proposed structure 17 shown in Figure 2 is generated from (S)-12 and would give the (S)-enantiomer of the adduct 16a. On the right side of structure 17 one of the thiourea groups is interacting with the N-Boc imine via two hydrogen bonds with the nitrogen and oxygen atoms of the imine. On the left side of structure 17 the other thiourea group is interacting with the anion of nitromethane via hydrogen bonds to each of the oxygens of the nitronate anion. This would lead to re-face addition to the imine and the formation of the (S)-enantiomer of 16a which is in accord with the experimental observation that the (R)-enantiomer of 12 gives rise to the (R)-enantiomer of 16a.
Figure 2.
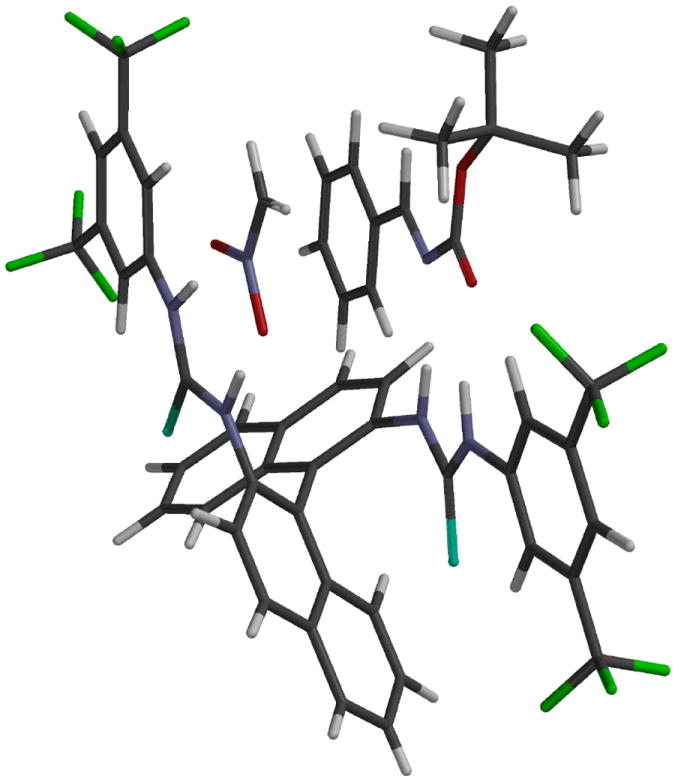
Catalyst-substrate complex 17 as a proposed model for the reaction with catalyst (S)-12 as it is hydrogen bonded to imine 14a and to the anion of CH3NO2.
The imine could have been hydrogen-bonded to the catalyst in two different ways. The alternative to the binding mode for the imine shown in Figure 2 is to have the imine rotated 180° such that the oxygen and nitrogen atoms of the imine switch hydrogens on the thiourea. Our preference for the binding mode of the imine shown in Figure 2 is based on the following reasoning. The conformation of the 3,5-bistrifluoromethyl substituted arene ring is known to be co-planar with the thiourea as a result of hydrogen bonding of one of the ortho-hydrogens of the arene with the sulfur of the thiourea.23 The consequence of this is that there would not be any obvious non-covalent positive interactions between the phenyl of the imine and the bis-3,5-trifluoromethyl phenyl group of the catalyst in the alternative binding mode. In contrast, the binding mode of the imine that is shown in Figure 2 could benefit from CH-π interactions between the edge of the phenyl group of the imine and the face of one of the naphthalene rings of the catalyst. In addition it is likely that the nitromethane is deprotonated after it is hydrogen bonded to the catalyst. Triethylamine is not sufficiently basic enough to completely deprotonate nitromethane since its pKa in DMSO is 17 and in water it is 10. The pKa of nitromethane should be lowered upon binding to the catalyst via two hydrogen bonds. This is consistent with the observation that at −35 °C the reaction only goes to 9% completion for the formation of 16a without catalyst under the conditions in entry 1 in Table 3. Therefore, the catalyst does have a significant effect on the rate of the reaction which may be the result of many factors. The coordination of the imine by the catalyst is supported by 1H NMR experiments. The signal for the HC=N proton of the amine is shifted from 8.88 ppm in the uncomplexed imine to 8.65 ppm in the presence of two equivalents of the catalyst. The interpretation of this shift is not clear at this point and this will be one of many subjects of future efforts to gain additional experimental evidence towards the understanding of the mechanism of this catalyst in the aza-Henry reaction.
Experimental Section
General procedure for the asymmetric aza-Henry reaction (Table 3)
A flame-dried round bottom flask was loaded with 0.2 equiv. of catalyst 12 (0.2 mmol, 160 mg) and 1 equiv of imine 14a-j (1 mmol). The solid mixture was dissolved in 4 mL of toluene and then RCH2NO2 (10 equiv., 0.52 mL) was added at −35 °C. After 5 minutes, Et3N (0.4 equiv, 56 μL) was added and then the mixture was stirred at −35 °C for 17-36 hours. The volatiles were evaporated and the crude product was purified by column chromatography on silica gel (20% acetone in hexanes) to afford products 16a – 16l.
For all the optimization studies of the aza-Henry reaction detailed in Tables 1 and 2, the experimental procedure is the same with slight modifications that are indicated in each Table. Whenever a % conversion is mentioned, this means the product was not purified, and the % conversion was determined from the 1H NMR spectrum of the crude reaction mixture by integration of product peaks versus the C(=N)H proton if the imine. The only species observed in the crude 1H NMR spectrum of the crude reaction mixture was the catalyst, the desired product and the starting imine.
Supplementary Material
Figure 1.
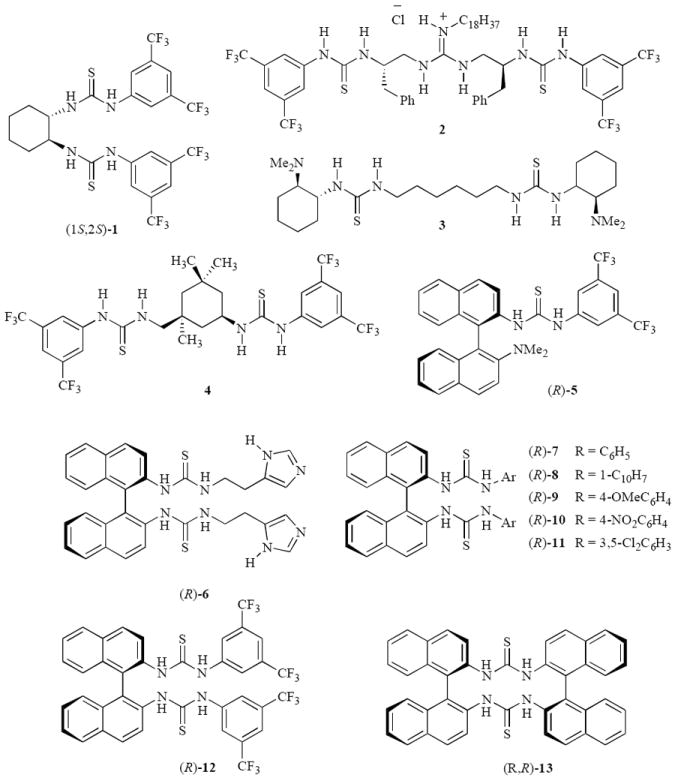
Chiral thioureas and bis-thiourea organocatalysts.
Acknowledgments
This work was supported by a grant from the NIH (GM 63019).
Footnotes
Supporting Information Available: Experimental protocols, characterization procedures, spectral data for all compounds and X-ray data for 13.
References
- 1.(a) Baer HH, Urbas L. In: The Chemistry of the Nitro and Nitroso Groups. Part 2. Patai S, editor. Interscience; New York: 1970. p. 117. [Google Scholar]; (b) Ono N. The Nitro Group in Organic Synthesis. 1. Wiley; New York: 2001. [Google Scholar]; (c) Westermann B. Angew Chem. 2003;115:161–163. [Google Scholar]; Angew Chem Int Ed. 2003;42:151–153. [Google Scholar]; (d) Palomo C, Oiarbide M, Mielgo A. Angew Chem. 2004;116:5558–5560. doi: 10.1002/anie.200460506. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:5442–5444. doi: 10.1002/anie.200460506. [DOI] [PubMed] [Google Scholar]; (e) Anderson JC, Blake AJ, Howell GP, Wilson C. J Org Chem. 2005;70:549–555. doi: 10.1021/jo048304h. [DOI] [PubMed] [Google Scholar]
- 2.a) Lloyd DH, Nichols DE. J Org Chem. 1986;51:4294–4295. [Google Scholar]; b) Barrett AGM, Spilling CD. Tetrahedron Lett. 1988;29:5733–5734. [Google Scholar]; c) Kende AS, Mendoza JS. Tetrahedron Lett. 1991;32:1699–1702. [Google Scholar]; d) Sturgess MS, Yarberry DJ. Tetrahedron Lett. 1993;34:4743–4746. [Google Scholar]; e) Adams H, Anderson JC, Peace S, Pennell AMK. J Org Chem. 1998;63:9932–9934. [Google Scholar]
- 3.a) Pinnick HW. Org React. 1990;38:655–792. [Google Scholar]; b) Matt C, Wagner A, Moiskowski C. J Org Chem. 1997;62:234–235. doi: 10.1021/jo962110n. [DOI] [PubMed] [Google Scholar]; c) Ballini R, Petrini M. Tetrahedron. 2004;60:1017–1047. [Google Scholar]
- 4.a) Yamada K, Harwood SJ, Groger H, Shibasaki M. Angew Chem Int Ed Engl. 1999;38:3504–3506. doi: 10.1002/(sici)1521-3773(19991203)38:23<3504::aid-anie3504>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; b) Yamada K, Moll G, Shibasaki M. Synlett. 2001:980–982. [Google Scholar]; c) Tsuritani N, Yamada K, Yoshikawa N, Shibasaki M. Chem Lett. 2002:276–277. [Google Scholar]
- 5.a) Knudsen KR, Risgaard T, Nishiwaki N, Gothelf KV, Jørgensen KA. J Am Chem Soc. 2001;123:5843–5844. doi: 10.1021/ja010588p. [DOI] [PubMed] [Google Scholar]; b) Nishiwaki N, Knudsen KR, Gothelf KV, Jørgensen KA. Angew Chem Int Ed. 2001;40:2992–2995. doi: 10.1002/1521-3773(20010817)40:16<2992::AID-ANIE2992>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; c) Knudsen KR, Jorgensen KA. Org Biomol Chem. 2005;3:1362–1364. doi: 10.1039/b500618j. [DOI] [PubMed] [Google Scholar]
- 6.a) Anderson JC, Howell GP, Lawrence RM, Wilson CS. J Org Chem. 2005;70:5665–5670. doi: 10.1021/jo050762i. [DOI] [PubMed] [Google Scholar]; b) Gao F, Zhu J, Tang Y, Deng M, Qian C. Chirality. 2006;18:741–745. doi: 10.1002/chir.20315. [DOI] [PubMed] [Google Scholar]; c) Palomo C, Liarbide M, Halder R, Laso A, Lopez R. Angew Chem Int Ed. 2006;45:117–120. doi: 10.1002/anie.200502674. [DOI] [PubMed] [Google Scholar]; d) Handa S, Gnanadesikan V, Matsunaga S, Masakatsu S. J Am Chem Soc. 2007;129:4900–4901. doi: 10.1021/ja0701560. [DOI] [PubMed] [Google Scholar]; e) Trost BM, Lupton DW. Org Lett. 2007;9:2023–2026. doi: 10.1021/ol070618e. [DOI] [PubMed] [Google Scholar]
- 7.For general reviews on asymmetric organocatalysis, see: Dalko PI, Moisan L. Angew Chem Int Ed. 2001;40:3726–3748. doi: 10.1002/1521-3773(20011015)40:20<3726::aid-anie3726>3.0.co;2-d.. Dalko PI, Moisan L. Angew Chem Int Ed. 2004;43:5138–5175. doi: 10.1002/anie.200400650.. Seayad J, List B. Org Biomol Chem. 2005;3:719–724. doi: 10.1039/b415217b..
- 8.a) Okino T, Nakamura S, Furukawa T, Takemoto Y. Org Lett. 2004;6:625–627. doi: 10.1021/ol0364531. [DOI] [PubMed] [Google Scholar]; b) Xu X, Furukawa T, Okino T, Miyabe H, Takemoto Y. Chem Eur J. 2006;12:466–476. doi: 10.1002/chem.200500735. [DOI] [PubMed] [Google Scholar]
- 9.Yoon TP, Jacobsen EN. Angew Chem. 2005;117:470–472. doi: 10.1002/anie.200461814. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:466–468. doi: 10.1002/anie.200461814. [DOI] [PubMed] [Google Scholar]
- 10.Bernardi L, Fini F, Herrera RP, Ricci A, Sgarzani V. Tetrahedron. 2006;62:375–380. [Google Scholar]
- 11.Bode CM, Ting A, Schaus SE. Tetrahedron. 2006;62:11499–11505. [Google Scholar]
- 12.Chang Y-W, Yang J-J, Dang J-N, Xue Y-X. Synlett. 2007:2283–2285. [Google Scholar]
- 13.a) Fini F, Sgarzani V, Pettersen D, Herrera RP, Bernardi L, Ricci A. Angew Chem Int Ed. 2005;44:7975–7978. doi: 10.1002/anie.200502646. [DOI] [PubMed] [Google Scholar]; b) Polomo C, Oiarbide M, Laso A, Lopez R. J Am Chem Soc. 2005;127:17622–17623. doi: 10.1021/ja056594t. [DOI] [PubMed] [Google Scholar]
- 14.a) Nugent BM, Yoder RA, Johnston JN. J Am Chem Soc. 2004;126:3418–3419. doi: 10.1021/ja031906i. [DOI] [PubMed] [Google Scholar]; b) Singh AS, Yoder RA, Shen B, Johnston JN. J Am Chem Soc. 2007;129:3466–3467. doi: 10.1021/ja068073r. [DOI] [PubMed] [Google Scholar]
- 15.a) Connon SJ. Chem Eur J. 2006;12:5418–5427. doi: 10.1002/chem.200501076. [DOI] [PubMed] [Google Scholar]; b) Taylor MS, Jacobsen EN. Angew Chem Int Ed. 2006;45:1520–1543. doi: 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]
- 16.Sohtome Y, Tanatani A, Hashimoto Y, Nagasawa K. Tetrahedron Lett. 2004;45:5589–5592. [Google Scholar]
- 17.a) Herrera RP, Sgarzani V, Bernardi L, Ricci A. Angew Chem Int Ed. 2005;44:6576–6579. doi: 10.1002/anie.200500227. [DOI] [PubMed] [Google Scholar]; b) Procuranti B, Connon SJ. Chem Commun. 2007:1421–1423. doi: 10.1039/b618792g. [DOI] [PubMed] [Google Scholar]; c) Herrera RP, Monge D, Martin-Zamora E, Ferandez R, Lassaletta JM. Org Lett. 2007;9:3303. doi: 10.1021/ol071292c. [DOI] [PubMed] [Google Scholar]; d) Martin NJA, Ozores L, List B. J Am Chem Soc. 2007;129:8976–8977. doi: 10.1021/ja074045c. [DOI] [PubMed] [Google Scholar]
- 18.a) Sohtome Y, Hashimoto Y, Nagasawa K. Adv Synth Catal. 2005;347:1643–1648. [Google Scholar]; b) Sohtome Y, Takemura N, Iguchi T, Hashimoto Y, Nagasawa K. Synlett. 2006:144–146. [Google Scholar]; c) Sohtome Y, Hashimoto Y, Nagasawa K. Eur J Org Chem. 2006:2894–2897. [Google Scholar]; d) Sohtome Y, Takemura N, Takada K, Takagi R, Iguchi T, Nagasawa K. Chem Asian J. 2007;2:1150–1160. doi: 10.1002/asia.200700145. [DOI] [PubMed] [Google Scholar]
- 19.Berkessel A, Roland K, Neudörfl JM. Org Lett. 2006;8:4195–4198. doi: 10.1021/ol061298m. [DOI] [PubMed] [Google Scholar]
- 20.Berkessel A, Mukherjee S, Müller TN, Cleemann F, Roland K, Brandenburg M, Neuförfl JM, Lex J. Org Biomol Chem. 2006;4:4319–4330. doi: 10.1039/b607574f. [DOI] [PubMed] [Google Scholar]
- 21.While this work was in progress the bis-thiourea 12 was reported as a catalyst for the Friedel-Crafts reaction of nitroalkenes which gave 12-50% ee: Fleming EM, McCabe T, Connon SJ. Tetrahedron Lett. 2006;47:7037–7042.
- 22.a) Wang J, Li H, Yu X, Zu L, Wang W. Org Lett. 2005;7:4293–4296. doi: 10.1021/ol051822+. [DOI] [PubMed] [Google Scholar]; b) Wang J, Li H, Duan W, Zu L, Wang W. Org Lett. 2005;7:4713–4716. doi: 10.1021/ol0519137. [DOI] [PubMed] [Google Scholar]; c) Wang J, Li H, Zu L, Wang W. Org Lett. 2006;8:1391–1394. doi: 10.1021/ol0601794. [DOI] [PubMed] [Google Scholar]; d) Li H, Zu L, Wang J, Wang W. Tetrahedron Lett. 2006;47:3145–3148. [Google Scholar]; e) Wang J, Li H, Zu L, Jiang W, Wang W. Adv Synth Catal. 2006;348:2047–2050. [Google Scholar]; f) Berkessel A, Mukherjee S, Müller TN, Cleemann F, Roland K, Brandenburg M, Neudörfl JM, Lex J. Org Biomol Chem. 2006;4:4319–4330. doi: 10.1039/b607574f. [DOI] [PubMed] [Google Scholar]; g) Wang J, Li H, Zu L, Jiang W, Xie H, Duan W, Wang W. J Am Chem Soc. 2006;128:12652–12653. doi: 10.1021/ja065187u. [DOI] [PubMed] [Google Scholar]; h) Bartoli G, Bosco M, Carlone A, Locatelli M, Mazzanti A, Sambri L, Melchiorre P. Chem Commun. 2007:722–724. doi: 10.1039/b613477g. [DOI] [PubMed] [Google Scholar]; i) Zu L, Wang J, Li H, Xie H, Jiang W, Wang W. J Am Chem Soc. 2007;129:1036. doi: 10.1021/ja067781+. [DOI] [PubMed] [Google Scholar]; j) Zu L, Xie H, Li H, Wang J, Jiang W, Wang W. Adv Synth Catal. 2007;349:1882–1886. [Google Scholar]; k) Wang J, Zu L, Li H, Xie H, Wang W. Synthesis. 2007:2576–2580. [Google Scholar]
- 23.Wittkopp A, Schreiner PR. Chem Eur J. 2003;9:407. doi: 10.1002/chem.200390042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.