

Abstract
High throughput phenotypic screening of large commercially available libraries through two NIH programs has produced thousands of potentially interesting hits for further development as antitubercular agents. Unfortunately, these screens do not supply target information, and further follow up target identification is required to allow optimal rational design and development of highly active and selective clinical candidates. Cheminformatic analysis of the quinoline and quinazoline hits from these HTS screens suggested a hypothesis that certain compounds in these two classes may target the mycobacterial tubulin homolog, FtsZ. In this brief communication, activity of a lead quinoline against the target FtsZ from M. tuberculosis (Mtb) is confirmed as well as good in vitro whole cell antibacterial activity against Mtb H37Rv. The identification of a putative target of this highly tractable pharmacophore should help medicinal chemists interested in targeting FtsZ and cell division develop a rational design program to optimize this activity towards a novel drug candidate.
Keywords: tuberculosis, FtsZ, quinoline
1. Introduction
There is an urgent demand for new antitubercular drugs possessing novel mechanisms of action with the goal of developing more effective combination treatments against drug resistant forms of M. tuberculosis.1–2 As part of the NIH strategy to encourage the discovery and development of new drugs against Mtb, the Tuberculosis Antimicrobial Acquisition and Coordinating Facility (TAACF) and the Molecular Libraries Screening Center Network (MLSCN) have made high throughput screening data against Mtb H37Rv for over 300,000 drug-like small molecules available via the PubChem database.3–5 Through this effort, over one thousand interesting compounds appear to show excellent lead antibacterial activity as well as selectivity relative to a mammalian cell toxicity counter screen. Unfortunately, continued progression of these leads is hindered by a lack of target information that would greatly facilitate a modern rational drug design program to produce highly potent and selective second generation analogs for clinical development. As part of our ongoing effort to advance leads from the TAACF and MLSCN programs, we have continued to mine the active samples from these whole cell phenotypic screens as they are an excellent basis set for the development of new antitubercular drugs due to the fact that these samples may serve as privileged scaffolds active versus whole bacteria for further target identification and chemical optimization. Such an approach may circumvent reported issues from pharmaceutical target-based infectious disease HTS campaigns.6 In this regard, we have had notable success pursuing whole cell active samples for target identification and chemical optimization through cheminformatic approaches as exemplified by our work with mycobacterial DHFR, IMPDH, and FtsZ.7–9
Among the reported actives from the government HTS programs, a small number of interesting quinolines and quinazolines were reported to have significant antitubercular activity, and one sample (1) showed excellent potency and good selectivity as measured relative to growth inhibition of a mammalian cell, Vero cells.
A further cheminformatic search of highly related quinoline scaffolds noted significant and diverse antibacterial activity of this class, and small molecule quinoline-based compounds show a variety of interesting target activities including inhibition of Mtb.10–13 In fact a diarylquinoline, Bedaquiline (TMC207 or R207910), is a recently approved FDA antituberculosis drug.13 Beyond the broad antitubercular activity of the two classes, however, there was a large body of useful data concerning the mechanism of action of highly related compounds in the area of anticancer drug discovery. Published work from Sirisoma et al. demonstrated potent activity of structurally related quinazolines (see Figure 1 structure 2) against mammalian tubulin along with a thorough structure-activity analysis for the putative colchicine binding site in tubulin.14–16 Our work with FtsZ and the reported activity of colchicine against Mtb FtsZ suggested a hypothesis that at least one target of these quinolines that are highly homologous to the quinazolines reported by Sirisoma et al. may be a potential colchicine like binding site in Mtb FtsZ.9 The tubulin homolog FtsZ (Filamenting temperature-sensitive mutant Z) is essential for cell division in most bacteria, including Mtb, and it is an ideal target for novel antimicrobials that has recently received considerable attention in the discovery of novel anti-TB drugs.17–28
Figure 1.
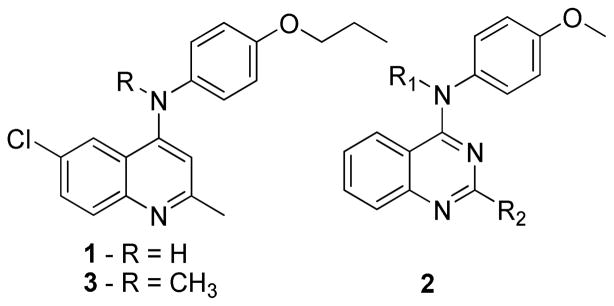
Structures of quinoline compounds 1 and 3 and generalized quinazolines (2) reported by Sirisoma et al. where R1 = H or Me and R2 = Cl or Me.
Herein we report further target-based studies relating to the initially reported quinoline (1) as well as the preparation and screening of a new analog (3) with the goal of identifying a potential target in Mtb that will help researchers in the area of Mtb drug discovery advance this excellent and synthetically tractable pharmacophore.
2. Results and Discussion
The TAACF active 6-Chloro-2-methyl-N-(4-propoxyphenyl)quinolin-4-amine (1) was repurchased as a dry powder from ChemBridge as its hydrochloride salt and was used as supplied without further analysis. Based on the reported SAR work of Sirisoma et al. suggesting that the 4-NMe group reduced free rotation of the 4-substituent and possible off target kinase binding, we decided to prepare and screen the related derivative 3. The synthetic route for compound 3 is outlined in Scheme 1 following reported methods.16,29,30
Scheme 1.
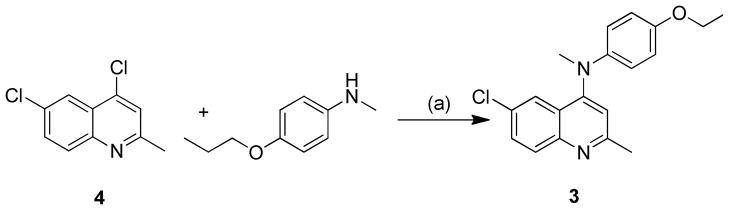
Synthetic pathway to compound 3. N-methyl-4-propoxyaniline is available via reported methods.29 Reagents and conditions: a) Conc. HCl, iso-propanol, rt.30
Both compounds 1 and 3 were screened for inhibition of: 1) polymerization of Mtb FtsZ, and its mammalian homolog, tubulin, by reported methods9; 2) in vitro growth of Mtb H37Rv to determine the IC90 (the concentration that inhibits 90% of growth) by reported methods4; and 3) in vitro growth of Vero cells for cytotoxicity (CC50).4 Screening results are given in Table 1. Compound 1 was only modestly less active than its N-methyl analog 3 against H37Rv growth in vitro but showed good activity for FtsZ. The N-Me compound was inactive against FtsZ and showed good activity in the tubulin polymerization assay.
Table 1.
Biological data for compounds 1 and 3.a
In summary, through a thorough cheminformatics analysis of active quinolines and quinazolines reported under the auspices of the TAACF and MLSCN programs, a hypothesis was developed that the active quinoline 1 may target the putative colchicine binding site in Mtb FtsZ. It is notable that this hypothesis derived from related quinazolines reported as highly active anticancer compounds that were shown to bind in the colchicine binding site of tubulin. Mammalian target data were also extremely useful in identifying potential targets of the N,N′-diphenylurea class in our earlier Mtb IMPDH work, and it is highly recommended that other researchers assessing antitubercular HTS activity data not ignore this vast body of important data as appropriate as it not only helps address potential toxic effects, but it may also help elucidate possible antibacterial targets for screening and medicinal chemistry development. Beyond pinpointing a potential target of this class in Mtb, the vast body of anticancer/tubulin information and the resulting SAR clearly suggested optimal approaches that would help clarify and separate the anticancer/tubulin and antibacterial/FtsZ activity. Among these, Sirisoma et al. clearly demonstrated the value of N-methylation to enhance target anticancer activity and selectivity. Interestingly, the -NMe (3) analog of the lead antitubercular agent 1, while it retained strong antitubercular activity in vitro, was inactive versus FtsZ, was highly toxic to Vero cells, and was a good inhibitor of tubulin polymerization. The fact that compound 3 still retained activity versus Mtb H37Rv in vitro while losing FtsZ activity suggests that these agents may target more than FtsZ in Mtb H37Rv. Surprisingly, the finding that a methylamino substitution is required for activity against tubulin and completely abolishes activity against FtsZ is a useful finding for selectivity against mycobacterial FtsZ. Furthermore, the toxicity of 3 is possibly due to inhibition of tubulin polymerization.
While the numerous actives from the NIH phenotypic Mtb screens are an excellent resource, it has historically been extremely difficult to develop potent and selective compounds without specific target information. Based on our analysis and screens, we would propose that one possible target of the lead quinoline 1 is the crucial cell division protein FtsZ. Furthermore, while there is significant interest in this target in bacteria and Mtb, there are very few compounds targeting mycobacterial FtsZ, and several of the reported scaffolds suffer from issues of ease of synthesis as well as problems with toxicity and medicinal chemistry profiles. We would suggest that the quinolines are an established “privileged” scaffold with broad based activities that include bioavailability and antitubercular activity. As such, with the information provided herein, the class and target is an excellent starting point for the development of a rational medicinal chemistry program to develop new antitubercular agents with a novel mechanism of action. Overall, these data as well as the ease of preparing new analogs suggest that these drug-like 4-substituted quinolines may be a good lead series for further scaffold expansion in order to develop new antitubercular agents targeting a novel and essential protein in Mtb, FtsZ.
Acknowledgments
Funding: This work was supported in part by grants from the National Institutes of Health NCI 1R01CA131378 (RCR PI). R.C.R. acknowledges the American Reinvestment and Recovery Act Grant 1RC1AI086677-01 that provided support for the presented study (National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (NIAID)) – “Targeting MDR-Tuberculosis.”
Footnotes
Competing interests: None declared.
Ethical approval: Not required.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Bini Mathew, Email: mathew@southernresearch.org.
Larry Ross, Email: ross@southernresearch.org.
Robert C. Reynolds, Email: rcrreynolds@gmail.com.
References and Notes
- 1.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. The challenge of new drug discovery for tuberculosis. Nature. 2011;469:483–90. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 2.Goldman RC, Laughon BE. Discovery and validation of new antitubercular compounds as potential drug leads and probes. Tuberculosis (Edinb) 2009;89:331–3. doi: 10.1016/j.tube.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maddry JA, Ananthan S, Goldman RC, Hobrath JV, Kwong CD, Maddox C, Rasmussen L, Reynolds RC, Secrist JA, III, Sosa MI, White EL, Zhang W. Antituberculosis activity of the molecular libraries screening center network library. Tuberculosis (Edinb) 2009;89:354–63. doi: 10.1016/j.tube.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ananthan S, Faaleolea ER, Goldman RC, Hobrath JV, Kwong CD, Laughon BE, Maddry JA, Mehta A, Rasmussen L, Reynolds RC, Secrist JA, III, Shindo N, Showe DN, Sosa MI, Suling WJ, White EL. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb) 2009;89:334–53. doi: 10.1016/j.tube.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reynolds RC, Ananthan S, Faaleolea E, Hobrath JV, Kwong CD, Maddox C, Rasmussen L, Sosa MI, Thammasuvimol E, White EL, Zhang W, Secrist JA., III High throughput screening of a library based on kinase inhibitor scaffolds against Mycobacterium tuberculosis H37Rv. Tuberculosis (Edinb) 2012;92:72–83. doi: 10.1016/j.tube.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 7.Barrow EW, Suling WJ, Seitz LE, Reynolds RC, Barrow WW. New antifolate inhibitors for Mycobacterium avium. Med Chem. 2006;2:505–10. doi: 10.2174/157340606778250225. [DOI] [PubMed] [Google Scholar]
- 8.Usha V, Hobrath JV, Gurcha SS, Reynolds RC, Besra GS. Identification of novel Mt-Guab2 inhibitor series active against M. tuberculosis. PLOS One. 2012;7:e33886. doi: 10.1371/journal.pone.0033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White EL, Suling WJ, Ross LJ, Seitz LE, Reynolds RC. 2-Alkoxycarbonylpyridines: inhibitors of Mycobacterium tuberculosis FtsZ. J Antimicrob Chemother. 2002;50:111–4. doi: 10.1093/jac/dkf075. [DOI] [PubMed] [Google Scholar]
- 10.Lilienkampf A, Mao J, Wan B, Wang Y, Franzblau SG, Kozikowski AP. Structure-activity relationships for a series of quinoline-based compounds active against replicating and nonreplicating Mycobacterium tuberculosis. J Med Chem. 2009;52:2109–18. doi: 10.1021/jm900003c. [DOI] [PubMed] [Google Scholar]
- 11.Mao J, Wang Y, Wan B, Kozikowski AP, Franzblau SG. Design, synthesis, and pharmacological evaluation of mefloquine-based ligands as novel antituberculosis agents. ChemMedChem. 2007;2:1624–30. doi: 10.1002/cmdc.200700112. [DOI] [PubMed] [Google Scholar]
- 12.Jayaprakash S, Iso Y, Wan B, Franzblau SG, Kozikowski AP. Design, synthesis, and SAR studies of mefloquine-based ligands as potential antituberculosis agents. ChemMedChem. 2006;1:593–7. doi: 10.1002/cmdc.200600010. [DOI] [PubMed] [Google Scholar]
- 13.Andries K, Verhasselt P, Guillemont J, Goehlmann HWH, Neefs J-M, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier VA. Diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–7. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 14.Sirisoma N, Pervin A, Zhang H, Jiang S, Adam Willardsen J, Anderson MB, Mather G, Pleiman CM, Kasibhatla S, Tseng B, Drewe J, Cai SX. Discovery of N-methyl-4-(4-methoxyanilino)quinazolines as potent apoptosis inducers. Structure-activity relationship of the quinazoline ring. Bioorg Med Chem Lett. 2010;20:2330–4. doi: 10.1016/j.bmcl.2010.01.155. [DOI] [PubMed] [Google Scholar]
- 15.Sirisoma N, Pervin A, Zhang H, Jiang S, Adam Willardsen J, Anderson MB, Mather G, Pleiman CM, Kasibhatla S, Tseng B, Drewe J, Cai SX. Discovery of N-(4-methoxyphenyl)-N,2-dimethylquinazolin-4-amine, a potent apoptosis inducer and efficacious anticancer agent with high blood brain barrier penetration. J Med Chem. 2009;52:2341–51. doi: 10.1021/jm801315b. [DOI] [PubMed] [Google Scholar]
- 16.Sirisoma N, Kasibhatla S, Pervi A, Zhang H, Jiang S, Willardsen JA, Anderson MB, Baichwal V, Mather GG, Jessing K, Hussain R, Hoang K, Pleiman CM, Tseng B, Drewe J, Cai SX. Discovery of 2-chloro-N-(4-methoxyphenyl)-N-methylquinazolin-4-amine (EP128265, MPI-0441138) as a potent inducer of apoptosis with high in vivo activity. J Med Chem. 2008;51:4771–9. doi: 10.1021/jm8003653. [DOI] [PubMed] [Google Scholar]
- 17.Ma S, Ma S. The development of FtsZ inhibitors as potential antibacterial agents. Chem Med Chem. 2012;7:1161–72. doi: 10.1002/cmdc.201200156. [DOI] [PubMed] [Google Scholar]
- 18.Stokes NR, Baker N, Bennett JM, Berry J, Collins I, Czaplewski LG, Logan A, Macdonald R, Macleod L, Peasley H, Mitchell JP, Nayal N, Yadav A, Srivastava A, Haydon DJ. An improved small-molecule inhibitor of FtsZ with superior in vitro potency, drug-like properties, and in vivo efficacy. Antimicrob Agents Chemother. 2013;57:317–25. doi: 10.1128/AAC.01580-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar K, Awasthi D, Lee S-Y, Zanardi I, Ruzsicska B, Knudson S, Tonge PJ, Slayden RA, Ojima I. Novel trisubstituted benzimidazoles, targeting Mtb FtsZ, as a new class of antitubercular agents. J Med Chem. 2011;54:374–81. doi: 10.1021/jm1012006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science. 2008;321:1673–5. doi: 10.1126/science.1159961. [DOI] [PubMed] [Google Scholar]
- 21.Kumar K, Awasthi D, Berger WT, Tonge PJ, Slayden RA, Ojima I. Discovery of anti-TB agents that target the cell-division protein FtsZ. Future Medicinal Chemistry. 2010;2:1305–23. doi: 10.4155/fmc.10.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urgaonkar G, Pierre SLH, Meir I, Lund H, Chaudhuri DR, Shaw JT. Synthesis of antimicrobial natural products targeting FtsZ: (+/−)-dichamanetin and (+/−)-2‴-hydroxy-5″-benzylisouvarinol-B. Org Lett. 2005;7:5609–12. doi: 10.1021/ol052269z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Galgoci A, Kodali S, Hearth KB, Jayasuriya H, Dorso K, Vicente F, Gonzalez A, Cully D, Bramhill D, Singh S. Discovery of a small molecule that inhibits cell division by blocking FtsZ, a novel therapeutic target of antibiotics. J Biol Chem. 2003;278:44424–8. doi: 10.1074/jbc.M307625200. [DOI] [PubMed] [Google Scholar]
- 24.Margailt DN, Romberg L, Mets RB, Hebert AM, Mitchinson TJ, Krischner MW, Chaudhuri DR. Targeting cell division: small-molecule inhibitors of FtsZ GTPase perturb cytokinetic ring assembly and induce bacterial lethality. Proc Natl Acad Sci USA. 2004;101:11821–6. doi: 10.1073/pnas.0404439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erickson HP. FtsZ, a prokaryotic homolog of tubulin. Cell. 1995;80:367–70. doi: 10.1016/0092-8674(95)90486-7. [DOI] [PubMed] [Google Scholar]
- 26.Mathew B, Srivastava S, Ross LJ, Suling WJ, White EL, Woolhiser LK, Lenaerts AJ, Reynolds RC. Novel pyridopyrazine and pyrimidothiazine derivatives as FtsZ inhibitors. Bioorg Med Chem. 2011;19:7120–8. doi: 10.1016/j.bmc.2011.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds RC, Srivastava S, Ross LJ, Suling WJ, White EL. A new 2-carbamoyl pteridine that inhibits mycobacterial FtsZ. Bioorg Med Chem Lett. 2004;14:3161–4. doi: 10.1016/j.bmcl.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 28.Temple C, Rose JD, Elliot RD, Montgomery JA. Synthesis of potential antimalarial agents. II. 6,8-Disubstituted pyrido[2,3-b]pyrazines. J Med Chem. 1968;11:1216–8. doi: 10.1021/jm00312a024. [DOI] [PubMed] [Google Scholar]
- 29.Kadin SB. Monomethylation of aromatic amines via sodium borohydride mediated carbon-nitrogen bond cleavage. J Org Chem. 1973;38:1348–50. [Google Scholar]
- 30.6-Chloro-N,2-dimethyl-N-(4-propoxyphenyl)quinolin-4-amine(3) Concentrated HCl (1 drop) was added to a solution of 4,6-dichloro-2-methylquinoline 4 (175 mg, 0.825 mmol) and N-methyl-4-propoxyaniline29 (150 mg, 0.91 mmol) in 10 mL isopropanol while stirring at room temperature. The reaction mixture was stirred at room temperature under argon atmosphere for 18 hours. The reaction mixture was diluted with CH2Cl2 and washed with aq. NaHCO3. The aqueous layer was washed with CH2Cl2 (2 × 20 mL). The combined organic layer was dried over anhydrous Na2SO4 and the solvent was removed under vacuum. The product was purified by flash column chromatography to afford the title compound 3 (86 mg, 31%, LCMS Purity: 100%) as a white solid. mp 83–84 °C. 1H NMR (CDCl3, 300 MHz): δ 7.86 (d, 1H, J = 9.0 Hz, 8-H), 7.56 (d, 1H, J = 2.4 Hz, 5-H), 7.46 (dd, 1H, J = 2.4 Hz, 9.0 Hz, 7-H), 6.90–6.80 (m, 5H, 2′-H, 3′-H, 5′-H, 6′-H, 3-H), 3.91 (t, 2H, J = 6.6 Hz, -OCH2), 3.38 (s, 3H, -NCH3), 2.67 (s, 3H, 2-CH3), 1.85–1.73 (m, 2H, -CH2-CH3), 1.05 (t, 3H, J = 7.5 Hz, -CH2-CH3). HRMS calcd for [C20H21ClN2O+H]+: 341.14152, Found: 341.14182.