

Abstract
Alzheimer’s disease (AD) is a major cause of dementia in the elderly with no effective treatment. Accumulation of amyloid-β peptide (Aβ) in the brain, one of the pathological features of AD, is considered to be a central disease-causing and disease-promoting event in AD. In this study, we showed that feeding male AβPP/PS1 transgenic mice, a well established mouse model of AD, with a diet containing phenolic antioxidant tert-butylhydroquinone (TBHQ) dramatically reduced brain Aβ load with no significant effect on the amounts of alpha- and beta-C-terminal fragments or full-length AβPP. Further studies showed that TBHQ diet inhibited the expression of plasminogen activator inhibitor-1 (PAI-1), a protease inhibitor which plays a critical role in brain Aβ accumulation in AD, accompanied by increases in the activities of tissue type and urokinase type plasminogen activators (tPA and uPA) as well as plasmin. Moreover, we showed that TBHQ diet increased the expression of low density lipoprotein related protein-1, a multi ligand endocytotic receptor involved in transporting Aβ out of the brain, and plasma Aβ40 and Aβ42 levels. We also showed that TBHQ diet increased the concentration of glutathione, an important antioxidant, and suppressed the expression of NADPH oxidase 2 as well as lipid peroxidation. Collectively, our data suggest that TBHQ may have therapeutic potential for AD by increasing brain antioxidant capacity/reducing oxidative stress level and by stimulating Aβ degradation/clearance pathways.
Keywords: Alzheimer’s disease, amyloid-β degradation, amyloid-β efflux, antioxidant
INTRODUCTION
Alzheimer’s disease (AD), a major course of dementia in the elderly, is a progressive neurodegenerative disease that gradually damages the neocortex and hippocampus. Despite extensive studies, there is no effective treatment for this devastating disease due to an incomplete understanding of the etiology and pathophysiology of the disease. Formation of extracellular senile plaques, which are abnormal aggregates of amyloid-β peptide (Aβ), is a pathological feature of AD. Accumulation of both soluble and insoluble Aβ in the brain has been suggested to be a disease-causing and disease-promoting event in AD. Consequently, developing the strategies to reduce brain Aβ levels has been a focus of many studies including several clinical trials. Nonetheless, although several studies have shown that reduction of brain Aβ burden is associated with improvement of learning/memory function in animals and in AD patients [1–5], no drugs have been developed yet due to potential safety concerns or adverse effects.
Aβ production is increased in early-onset (familial) AD due to mutations in the amyloid-β protein precursor (AβPP) gene or the presenilin 1 and presenilin 2 (PS1 and PS2) genes. However, the mechanism underlying increased Aβ accumulation in the brain of late-onset (sporadic) AD, which account for 95% of the cases, remains largely unknown. The brain is a site with high metabolic activity, high concentrations of prooxidants such as iron, and low antioxidant capacity, and therefore is highly sensitive to oxidative damage. It has been well documented that oxidative stress increases with age and in AD. It has also been shown that oxidative stress contributes importantly to AD pathology including neuronal cell death and Aβ accumulation while antioxidants, including lipoic acid and N-acetylcysteine (NAC), reduce brain Aβ burden and/or improve cognitive function in animal models of AD [6–9]. These observations suggest therapeutic potential of antioxidants for the treatment of AD.
The phenolic compound tert-butyl hydroquinone (TBHQ) is a highly effective preservative used in a wide range of food and cosmetic products at the concentrations as high as 1% w/w with relative low toxicity (WHO Food Additives Series 4). In addition to directly scavenging free radicals [10], studies from this and other labs have shown that TBHQ increases the expression of the enzymes involved in the synthesis of glutathione (GSH), the most abundant intracellular free thiol and an important antioxidant, in various types of cells and in animal models [11–15]. TBHQ has also been shown to induce the expression of several other enzymes involved in detoxification and antioxidant defense through activation of nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor controlling a pleiotropic cellular defense [14, 16, 17]. Most importantly, it has been reported that TBHQ protects neurons from oxidative damage induced by different toxicants or ischemia-reperfusion in vitro and in vivo [14, 15, 18–23]. Whether TBHQ can ameliorate pathological changes in AD, a disease with many pathological features of oxidative stress, however, has not been reported.
In this study, we explored the therapeutic potential of TBHQ for AD using AβPP/PS1 mice, a well-established animal model of AD. Our results showed that feeding AβPP/PS1 mice with TBHQ-containing diet significantly reduced brain Aβ load. Further studies suggest that TBHQ does so probably by increasing antioxidant capacity/reducing oxidative stress and stimulating Aβ degradation and/or efflux from the brain. The results from this study suggest that TBHQ may have therapeutic value for AD.
MATERIALS AND METHODS
Animals and TBHQ feeding
AβPP/PS1 double transgenic mice, purchased from JAXMICE, were generated by co-injection of human AβPP and PS1 transgene constructs containing AD mutations (a mutant human presenilin 1 (DeltaE9) and a chimeric mouse/human amyloid-β protein precursor) and were maintained on a C57BL/6 genetic background. Two and a half month old male AβPP/PS1 mice were fed with either a control diet or 1% TBHQ-containing diet for 6 weeks. All the mice were maintained on a 12-h light/dark cycle at 22°C with free access to water and food. Body weight and food intake were recorded weekly in order to monitor potential toxicity of TBHQ. By the end of feeding, mice were euthanized and blood withdrawn from the heart, followed by transcardial perfusion with cold PBS as we have described before [24]. Brain was dissected sagittally into right and left hemispheres with the right hemisphere fixed in 10% PBS buffered formalin for immunostaining analysis and the left hemisphere dissected and hippocampus and cerebral cortex frozen in liquid nitrogen immediately for biochemistry analyses. All procedures involving animals were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham.
ELISA analyses of Aβ and PAI-1 proteins in the brain
For the measurement of soluble and insoluble Aβ, mouse brain tissues were prepared as we have described previously [24]. Briefly, the brain tissues were homogenized in a Aβ extraction buffer containing 20 mM Tris–HCl (pH 7.6), 137 mM NaCl, 1% Triton X-100, 2% SDS, and protease inhibitors (complete protease inhibitor cocktail, Boehringer Mannheim, Mannheim, Germany), and centrifuged at 100,000 × g for 1 h. Supernatant was collected (SDS soluble) and stored at −80°C until analysis. Pellets were dissolved in 70% formic acid (FA), gentle shaking at room temperature for 1 h, and then centrifuged at 100,000 × g for 1 h. The supernatants were collected (SDS insoluble/FA soluble). ELISA was performed to quantify SDS soluble and SDS insoluble/FA soluble Aβ using the Aβ40 or Aβ42 ELISA kits from Covance (Emeryville, CA) [24]. The FA extraction solution was neutralized and diluted 20 times with a neutralizing buffer containing 1 M Tris, 0.5 M Na2HPO4, and 0.05% NaN3 before analysis by ELISA. For the measurement of PAI-1 protein in the brain, tissues were homogenized in Tris–HCl buffer containing 0.1% Triton-X 100, pH 8.5. After ultracentrifugation at 100,000 × g for 1 h, the supernatants were used for the measurement of total PAI-1 protein levels using an ELISA kit as we have described before [26]. The results were calculated based on the standard curves and expressed as per protein concentrations.
Immunohistochemical staining of Aβ deposits and low density lipoprotein related protein-1 (LRP-1) expression in mouse brain
Immunohistochemical staining of Aβ deposits in the brain was conducted using monoclonal anti-human Aβ antibody 6E10 (Covance, Emeryville, CA) as previously described [24]. The amyloid load in the cerebral cortex and hippocampus was quantified by determining the percentage of the total section positively labeled by Aβ antibody using Axiovision automatic measurement software (Zeiss, Germany). For detection of the expression of LRP-1 protein in brain microvessels anti mouse LRP-1 light chain monoclonal antibody (5A6, Calbiochem) was used. The number of positive vessels was counted in five fields within cerebral cortex per mouse and the results were expressed as percentage of total number of blood vessels in the same area of the brain [25].
Western analysis
Mouse brain tissues were homogenized in a tissue extraction buffer containing 2% SDS and different protease inhibitors as described above. Western analyses of full-length AβPP and alpha/beta C-terminal fragments (α-/ β-CTFs) were performed using 4–20% gradient gel (Invitrogen). Full-length AβPP proteins were detected with 6E10 antibody (Covance, Emeryville, CA) while α- and β-CTFs were determined using rabbit polyclonal antibody CT695 against the C terminus of AβPP (Invitrogen). LRP-1 protein was detected using anti mouse LRP-1 light chain monoclonal antibody (5A6) while the protein of NADPH oxidase 2 (Nox2), an important producer of reactive oxygen species (ROS), was detected with a polyclonal antibody (Santa Cruz, SC5827). Semi-quantification of the bands was performed by densitometric scanning using Image J software and normalized by β-actin. For detection of 4-hydroxy-2-nonenal (4-HNE) modified proteins, the brain tissues were homogenized and westerns run using non-reducing buffers. 4-HNE modified proteins were revealed by anti-4-HNE antibody (alpha Diagnostic, HNE-11 S).
Zymographic analysis of the activities of tissue type and urokinase type plasminogen activators
The activities of tissue type and urokinase type plasminogen activators (tPA and uPA, respectively) in mouse brain tissue were determined by zymographic analysis according to our previously described protocol [24, 26]. Briefly, equal amounts of proteins were loaded onto 12% SDS polyacrylamide gel containing 1% nonfat dry milk and 5 μg/ml plasminogen. After electrophoresis, the enzyme reactions were initiated by incubating the gel in 0.1 M glycine-NaOH (pH 8.3) at 37°C for 16 h. The lytic bands (tPA and uPA activity) were revealed by Coomassie blue staining and quantified using Image J system. Gels without plasminogen were also run simultaneously to ensure that the lytic bands are due to plasminogen activators. The tPA and uPA bands were identified based on both molecular weight and the loss of lytic activity upon inclusion of the plasmin inhibitor aprotinin (2 μg/ml).
Measurement of plasmin activity in brain tissue
Plasmin activities were measured using a specific chromogenic substrate Tosyl-glycyl-prolyl-lysine-4-nitranilide-acetate (Chromzyme PL, from Roche Applied Sciences) as described before [24]. The reaction mixture contained 33 mM Tris (pH 8.2), 6.4 mM NaCl, and 0.5 mM Chromozym PL. The time-dependent production of 4-nitraniline was followed by monitoring the absorbance at 405 nm. Plasmin activity was calculated using the 4-nitraniline extinction coefficient (ε405 nm = 1 × 104 M−1 cm−1) and expressed as unites per protein concentration.
Measurement of glutathione content in brain tissue
Brain tissues were sonicated in 5% sulfosalicylic acid and centrifuged. GSH concentrations in the supernatant were measured by a redox cycle assay as described before [27]. The protein content was measured by a BCA kit and GSH concentrations were calculated based on standard curve run together with the samples. The results were expressed as nmol/mg protein.
Statistical analysis
Data were expressed as mean ± SEM and evaluated by one-way ANOVA. Statistical significance was determined by Fisher LSD test wherein p < 0.05 were considered statistically significant.
RESULTS
TBHQ diet caused no obvious toxicities in Aβ PP/PS1 mice
Two and a half month old male AβPP/PS1 mice were fed with a diet containing 1% TBHQ for 6 weeks. The body weight and food intake were monitored during TBHQ feeding. No significant changes in body weight or food intake were observed in TBHQ fed mice as compared with control diet fed mice (data not shown). There was also no significant difference in the mortalities between TBHQ diet and control diet fed mice (data not shown). The results indicate that TBHQ, under the current feeding conditions, causes no significant toxicity.
Effects of TBHQ diet on brain Aβ load in AβPP/PS1 mice
Aβ load in the brain was analyzed by immunohistochemical staining and ELISA techniques. Immunohistochemical staining showed that TBHQ feeding significantly reduced the numbers of senile plaque deposits in cerebral cortex/hippocampal region in AβPP/PS1 mice as compared to control diet fed mice (Fig. 1A, B). ELISA data further showed that TBHQ feeding significantly reduced the levels of both SDS soluble and insoluble Aβ42 but had no significant effect on Aβ40 levels (Fig. 1C, D). No Aβ accumulation was detected in AβPP/PS1 non-transgenic mice with or without TBHQ treatment (data not shown).
Fig. 1.
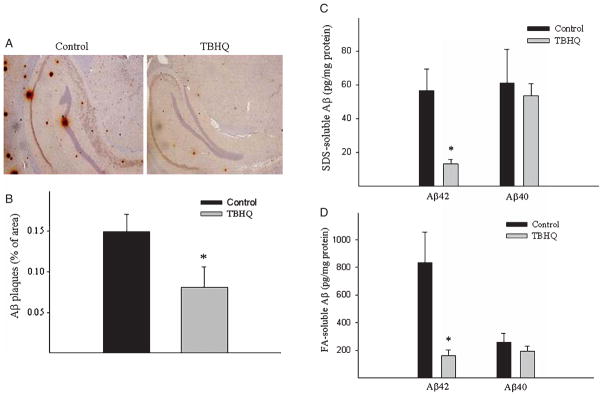
Effect of TBHQ diet on Aβ load in the brain of AβPP/PS1 mice. Two and a half month old male AβPP/PS1 mice were fed with TBHQ or control diet for 6 weeks. Aβ deposits in the brain were revealed by immunohistochemical staining using antibody specific to human Aβ (6E10) (A) and quantified by the histomorphometry system and expressed as percentage of total area of brain (B). The amounts of Aβ40 and Aβ 42, both soluble and insoluble, in the brain were determined by ELISA and calculated based on the total protein content (C&D). *Significantly different from the corresponding control diet fed mice (p < 0.05, n = 5–7).
Effects of TBHQ diet on the expression of full length Aβ PP, α-CTF, and β-CTF in the brain of AβPP/PS1 mice
To explore the mechanism whereby TBHQ reduced brain Aβ load in AβPP/PS1 mice, we measured the levels of full-length AβPP (FL-AβPP), α-C terminal fragments (α-CTF) and β-C terminal fragments (β-CTF) in the brain by Western blot. As shown in Fig. 2, there were no significant differences in the amounts of FL-AβPP (~100 kDa), α-CTF, or β-CTF between control diet and TBHQ diet fed mice. These results suggest that TBHQ diet reduces Aβ load in the brain of AβPP/PS1 mice not by altering AβPP expression/processing or inhibiting Aβ production but by stimulating Aβ degradation and/or efflux from the brain.
Fig. 2.

Effects of TBHQ diet on the amounts of full-length AβPP, α-CTF, and β-CTF in the brain of AβPP/PS1 mice. Full-length AβPP (FL-AβPP) and α/β-CTF in the brain of AβPP/PS1 mice were determined by western analyses using 4–20% gradient SDS-PAGE and anti-human Aβ antibody (6E10) or C terminus of human AβPP (CT695) (top panel). The intensities of the bands were semi-quantified by densitometric scanning and normalized by β-actin (bottom panel). *Significantly different from control diet fed mice (p < 0.05, n = 3–5).
Effects of TBHQ diet on the expression of plasminogen activator inhibitor 1 and the activities of plasminogen activators as well as plasmin in the brain of AβPP/PS1 mice
Plasminogen activator inhibitor 1 (PAI-1) is a primary inhibitor of tissue type and urokinase type plasminogen activator (tPA and uPA), which convert plasminogen into plasmin, a serine protease that plays a critical role in Aβ degradation [28–32]. Our previous studies have shown that PAI-1 expression is increased in the brain of AD patients as well as in animal models of AD; knockout of the PAI-1 gene, on the other hand, reduced brain Aβ burden in AβPP/PS1 transgenic mice [24], suggesting a critical role of PAI-1 in brain Aβ accumulation. To further elucidate the mechanism whereby TBHQ reduced brain Aβ load in AβPP/PS1 mice, we compared PAI-1 protein levels as well as tPA, uPA, and plasmin activities between control diet and TBHQ diet fed mice. ELISA results showed that TBHQ diet significantly reduced PAI-1 protein levels in the brain of AβPP/PS1 mice (Fig. 3A). Associated with inhibition of PAI-1 expression, TBHQ diet increased the activities of tPA and uPA as well as plasmin (Fig. 3B, C and D).
Fig. 3.

Effects of TBHQ diet on PAI-1 protein expression and the activities of tPA, uPA, and plasmin in the brain of AβPP/PS1 mice. (A) PAI-1 protein levels in the brain tissue were determined by ELISA. (B) The activities of tPA and uPA in brain tissue were determined by zymography. (C) the intensities of the bands semi-quantified by densitometric scanning using Image J software. (D) Plasmin activity was determined using a specific chromogenic substrate as described in the Materials and Methods section. *Significantly different from control mice (p < 0.05, n = 5–6).
Effects of TBHQ diet on the expression of low density lipoprotein receptor related protein 1 in the blood-brain barrier and the levels of Aβ in the plasma
LRP-1, a multi-function endocytic receptor expressed on endothelium of the blood-brain barrier (BBB), plays a major role in transporting Aβ out of the brain (efflux) [33–36]. It has been reported that LRP-1 expression in the brain is decreased during aging and in AD, associated with decreased Aβ efflux from and increased Aβ accumulation in the brain [25, 37]. Therefore, we examined whether TBHQ diet reduced brain Aβ load by increasing the expression of LRP-1 at the BBB. Immunohistochemical staining results showed that feeding mice with TBHQ diet significantly increased the abundance of LRP-1 protein at the BBB in AβPP/PS1 mice (Fig. 4A, B). Western analyses further confirmed that LRP-1 protein levels were increased in the brain of TBHQ fed AβPP/PS1 mice as compared with control diet fed AβPP/PS1 mice (Fig. 4C, D).
Fig. 4.

Effects of TBHQ diet on the expression of LRP-1 at the BBB in AβPP/PS1 mice. A) Representative immunostaining pictures of LRP-1 protein in the brains of control-diet and TBHQ-diet fed mice. B) Quantification of LRP-1 positive blood vessels in cerebral cortex/hippocampus, expressed as percentage of total number of blood vessels in the same area of the brain. C) Representative western blotting picture of LRP-1 protein in cerebral cortex and hippocampus of AβPP/PS1 mice. D) Semi-quantitative data of western blotting. *Significantly different from control diet fed AβPP/PS1 transgenic mice (p < 0.05, n = 5).
To further reveal whether increased LRP-1 expression was accompanied by an increase in Aβ efflux from the brain, we measured Aβ40 and Aβ42 levels in the plasma of AβPP/PS1 mice fed with control or TBHQ diet. The results showed that TBHQ diet increased both Aβ40 and Aβ42 levels in the plasma of AβPP/PS1 mice (Fig. 5).
Fig. 5.

Effect of TBHQ on plasma Aβ42 and Aβ40 levels in AβPP/PS1 mice. Aβ40 and Aβ42 levels in the plasma were determined by ELISA. *Significantly different from control diet fed mice (p < 0.05, n = 5).
Effects of TBHQ on glutathione content, NADPH oxidase2 expression, and lipid peroxidation level in the brain of AβPP/PS1 mice
Although it has been reported that TBHQ increases brain antioxidant capacity including the concentration of GSH, the most abundant intracellular free thiol and an important antioxidant, whether TBHQ can alleviate oxidative stress in AD is unknown. NADPH oxidases (Noxs) are important producers of ROS for both phagocytic and non-phagocytic cells while Nox2 is the prototype of NADPH oxidase family. To elucidate whether TBHQ diet can ameliorate oxidative stress levels in AD, we assessed the effects of TBHQ diet on the levels of Nox2 protein, GSH, and lipid peroxidation (4-HNE) in the brain of AβPP/PS1 mice. The results showed that TBHQ diet inhibited Nox2 protein expression (Fig. 6A,B), increased GSH level (Fig. 6C), and suppressed lipid peroxidation (Fig. 6D) in the brain of AβPP/PS1 mice.
Fig. 6.

Effects of TBHQ diet on brain oxidative stress and antioxidant levels in AβPP/PS1 mice. A) Representative western blotting picture of Nox2 protein in the brain of AβPP/PS1 transgenic mice. B) Semi-quantitative data of western blotting. *Significantly different from control diet fed AβPP/PS1 transgenic mice (p < 0.05, n = 5–6). C) GSH content in the brain. *Significantly different from control diet fed mice (p < 0.05, n = 5–6); D) Representative western blotting picture of 4-HNE modified proteins in the brain of AβPP/PS1 mice. β-actin was used to show equal protein loading between samples.
DISCUSSION
AD is a devastating neurodegenerative disease that affects about 27 million people worldwide [38]. Despite extensive study, the etiology of AD is still unclear and there is no effective treatment for this devastating disease. In this study, we showed that feeding 2.5 month old male AβPP/PS1 mice with a diet containing a synthetic phenolic antioxidant TBHQ, which is widely used in food and cosmetic products as a preservative, for 6 weeks dramatically reduced their brain Aβ load. Further studies suggest that TBHQ reduces brain Aβ level probably by 1) suppressing PAI-1 expression/activity and thereby increasing the activities of tPA/uPA and plasmin and Aβ degradation; and 2) increasing LRP-1 expression at the BBB and thereby Aβ efflux from the brain. We also showed that TBHQ increased GSH content and suppressed Nox2 expression as well as lipid peroxidation in the brain of AβPP/PS1 mice. Therefore, it is speculated that TBHQ reduces brain oxidative stress levels in AβPP/PS1 mice, which leads to suppression of PAI-1 but induction of LRP-1 expression and consequently stimulation of Aβ degradation and efflux from the brain (Fig. 7).
Fig. 7.

The hypothetic mechanisms by which TBHQ reduces brain Aβ load in AD. TBHQ increases antioxidant capacity and reduces oxidative stress levels in the brain, which leads to suppression of the expression of PAI-1, a redox regulated protein, and induction of LRP-1 and thereby increased Aβ degradation/efflux from the brain. Solid lines represent evidence demonstrated directly from the present study.
Although it has been well documented that Aβ production is increased in familial AD due to mutations in the AβPP or PS1 and PS2 genes, the mechanism underlying Aβ accumulation in the brain of sporadic AD is unknown. The levels of Aβ in the brain represent a dynamic equilibrium state as a result of their biosynthesis, degradation, influx, and efflux. Increasing evidence suggests that decrease in the clearance capacity, including degradation and efflux, may contribute importantly to Aβ accumulation during aging and in AD. Plasminogen activator inhibitor 1 (PAI-1) is a primary inhibitor of tissue type and urokinase type of plasminogen activators (tPA and uPA, respectively), which convert plasminogen into plasmin, a serine protease that plays a critical role in Aβ degradation [28–32, 39–41]. It was reported that amyloid deposition in the brain was increased in PAI-1 transgenic mice [42, 43]. Previous studies from this and other laboratories showed, on the other hand, that PAI-1 expression was increased in the brain of AD patients and in AβPP or AβPP/PS1 transgenic mice, well established animal models of AD [24, 31, 44, 45]. Knockout of the PAI-1 gene or inhibition of PAI-1 activity with PAI-1 specific inhibitors reduced brain Aβ burden and reversed cognitive deficits in these AD model mice [24, 25]. These data strongly suggest that increased PAI-1 expression contributes importantly to the development of amyloidosis in AD. In this study, we further showed that TBHQ administration significantly reduced brain Aβ load in AβPP/PS1 mice, which was associated with an inhibition of PAI-1 expression and increases in the activities of tPA, uPA, and plasmin (Fig. 3). No significant effects of TBHQ on the expression of full length AβPP or α-/ β-CTFs (Fig. 2). These data further support the notion that PAI-1 plays a pivotal role in AD pathology. These results also suggest that TBHQ reduces brain Aβ load not by inhibiting AβPP/Aβ synthesis but, at least in part, by inhibiting PAI-1 expression and thereby stimulating Aβ degradation.
The mechanism underlying the inhibition of PAI-1 expression in the brain of AβPP/PS1 mice by TBHQ is unknown at the moment. It has been well documented that oxidative stress is increased with age and in AD. It has also been demonstrated that increased oxidative stress contributes importantly to the pathogenesis of AD, including brain Aβ accumulation, although the underlying mechanism remains largely undefined. PAI-1 is an early response protein and can be induced by different cytokines/chemokines/gro wth factors including transforming growth factor beta (TGF-β), tumor necrosis factor alpha (TNFα), and angiotensin II. Interestingly, studies from this and other laboratories have shown that PAI-1 expression is redox regulated and that ROS mediate the induction of PAI-1 by several stimuli in different types of cells [46–52]. The proximal promoter region of the PAI-1 gene contains the binding sites for the activator protein 1 (AP-1), specificity protein 1 (SP-1), and NF-kB, three redox sensitive transcription factors. Our previous studies showed that treatment of fibroblasts with GSH, the most abundance intracellular free thiol and an important antioxidant, or GSH ester blocked the binding of AP-1 and SP-1 to the promoter of the PAI-1 gene and selectively inhibited TGF-β1-induced PAI-1 mRNA expression [26, 46, 53]. Our previous studies further showed that, in addition to GSH, NADPH oxidase inhibitor diphenyleneiodonium and superoxide dismutase/catalase mimetic MnTBaP also dramatically reduced TGF-α-induced PAI-1 protein expression. Therefore, it was hypothesized that the redox status of cell environment regulates PAI-1 expression through modulating the binding of the transcription factors AP-1 and SP-1 to the promoter of the PAI-1 gene. Importantly, previous studies from this lab have shown that GSH concentration decreases with age in several organs including the brain in wild type mice and rats as well as in the plasma of AD patients [54–58]. These data suggest that decrease in GSH level or increase in oxidative stress may underlie the increased PAI-1 expression during aging and in AD. TBHQ can directly scavenge free radicals and also induce the expression of the many enzymes involved in antioxidant defense including glutamate cysteine ligase (GCL), the rate-limiting enzyme in de novo GSH synthesis. In this study, we showed that TBHQ diet increased the concentration of GSH, suppressed the expression of NADPH oxidase 2 (Nox2), and reduced lipid peroxidation level in the brain of AβPP/PS1 mice. These results further confirm antioxidant property of TBHQ. These results also suggest that feeding mice with TBHQ diet suppresses PAI-1 gene expression probably by reducing oxidative stress level in the brain.
LRP1 is a multi-function endocytic receptor expressed on endothelium of the BBB, which plays a major role in transporting Aβ out of the brain [33–36]. It has been reported that the expression of LRP-1 at the BBB is decreased in AD, a pre-AD stage referred as amnestic mild cognitive impairment, and in well established animal model of AD, associated with a decreased Aβ efflux and an increased accumulation of Aβ in the brain [25, 37, 59–61]. LRP-1 expression/activity in the brain is also decreased with increasing age [25, 62]. Furthermore, it has been reported that LRP-1 antisense selectively decreased LRP-1 expression, reduced BBB clearance of Aβ42, increased brain Aβ42 level, and impaired learning function in mice [63]. Together, these results suggest that decrease in the expression of LRP-1 during aging and in AD contributes importantly to the pathophysiology of AD. In this study, we showed that TBHQ feeding significantly increased the expression of LRP-1 protein at the BBB, which was accompanied by elevation of plasma Aβ40 and Aβ42 levels. These data suggest that TBHQ reduces brain Aβ load in AβPP/PS1 mice probably also through increasing LRP-1 expression and thereby Aβ efflux from the brain.
The mechanism underlying the decrease in the expression of LRP-1 in the brain during aging and in AD is unknown. Although there is no published data showing that LRP-1 gene expression is redox regulated, a recent study showed that LRP-1 protein in the hippocampus from AD patients was oxidatively modified (4-HNE modification) [64]. The authors proposed that Aβ induced oxidative modification of its transporter LRP1, leading to decreased Aβ efflux and increased Aβ deposition in the brain [64]. Whether increased oxidative stress is responsible for the decrease of LRP-1 expression during aging and in AD and whether TBHQ induces LRP-1 expression by reducing oxidative stress level in the brain of AβPP/PS1 mice is unknown and warrant further investigation. Understanding the mechanism regulating the expression of LRP-1, a major Aβ transporter from the brain to the blood, may lead to discovery of novel therapeutic agents for this devastating disease.
Overall, the present study demonstrated for the first time that TBHQ administration reduced brain Aβ level in AβPP/PS1 mice. The results also suggest that THBQ does so by suppressing oxidative stress and thereby the expression/activity of PAI-1 and by stimulating the expression of LRP-1, which leads to increased degradation/clearance of Aβ from the brain. Together, the results suggest that TBHQ may have therapeutic potential for AD.
Acknowledgments
The work was supported by grants from National Institute of Aging (NIA, AG016029) and National Heart, Lung, and Blood Institute (HL088141) to Rui-Ming Liu (RML); an Award from University of Alabama at Birmingham School of Public Health to RML; grants from NIA (AG031846), Alzheimer’s Association (IIRG-09-131791), and American Health Assistance Foundation (A2010328) to Ling Li.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=882).
References
- 1.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A[beta] peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 2.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJF, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against [beta]-Amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 3.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukumoto H, Takahashi H, Tarui N, Matsui J, Tomita T, Hirode M, Sagayama M, Maeda R, Kawamoto M, Hirai K, Terauchi J, Sakura Y, Kakihana M, Kato K, Iwatsubo T, Miyamoto M. A noncompetitive BACE1 inhibitor TAK-070 ameliorates Aβ pathology and behavioral deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2010;30:11157–11166. doi: 10.1523/JNEUROSCI.2884-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kimura R, Devi L, Ohno M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J Neurochem. 2010;113:248–261. doi: 10.1111/j.1471-4159.2010.06608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu AL, Dong ZH, Sun MJ. Protective effect of N-acetyl-L-cysteine on amyloid beta-peptide-induced learning and memory deficits in mice. Brain Res. 2006;1109:201–206. doi: 10.1016/j.brainres.2006.06.042. [DOI] [PubMed] [Google Scholar]
- 7.Tucker S, Ahl M, Bush A, Westaway D, Huang X, Rogers JT. Pilot study of the reducing effect on amyloidosis in vivo by three FDA pre-approved drugs via the Alzheimer’s APP 5′ untranslated region. Curr Alzheimer Res. 2005;2:249–254. doi: 10.2174/1567205053585855. [DOI] [PubMed] [Google Scholar]
- 8.Quinn JF, Bussiere JR, Hammond RS, Montine TJ, Henson E, Jones RE, Stackman RW., Jr Chronic dietary alpha-lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice. Neurobiol Aging. 2007;28:213–225. doi: 10.1016/j.neurobiolaging.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Hager K, Kenklies M, McAfoose J, Engel J, Munch G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease–a 48 months follow-up analysis. J Neural Transm Suppl. 2007:189–193. doi: 10.1007/978-3-211-73574-9_24. [DOI] [PubMed] [Google Scholar]
- 10.Alamed J, Chaiyasit W, McClements DJ, Decker EA. Relationships between free radical scavenging and antioxidant activity in foods. J Agric Food Chem. 2009;57:2969–2976. doi: 10.1021/jf803436c. [DOI] [PubMed] [Google Scholar]
- 11.Liu RM, Shi MM, Giulivi C, Forman HJ. Quinones increase gamma-glutamyl transpeptidase expression by multiple mechanisms in rat lung epithelial cells. Am J Physiol. 1998;274:L330–L336. doi: 10.1152/ajplung.1998.274.3.L330. [DOI] [PubMed] [Google Scholar]
- 12.Liu RM, Hu H, Robison TW, Forman HJ. Differential enhancement of gamma-glutamyl transpeptidase and gamma-glutamylcysteine synthetase by tert-butylhydroquinone in rat lung epithelial L2 cells. Am J Respir Cell Mol Biol. 1996;14:186–191. doi: 10.1165/ajrcmb.14.2.8630269. [DOI] [PubMed] [Google Scholar]
- 13.Liu RM, Hu H, Robison TW, Forman HJ. Increased gamma-glutamylcysteine synthetase and gamma-glutamyl transpeptidase activities enhance resistance of rat lung epithelial L2 cells to quinone toxicity. Am J Respir Cell Mol Biol. 1996;14:192–197. doi: 10.1165/ajrcmb.14.2.8630270. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J. Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol Sci. 2005;83:313–328. doi: 10.1093/toxsci/kfi027. [DOI] [PubMed] [Google Scholar]
- 15.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duvoix A, Schnekenburger M, Delhalle S, Blasius R, Borde-Chiché P, Morceau F, Dicato M, Diederich M. Expression of glutathione S-transferase P1-1 in leukemic cells is regulated by inducible AP-1 binding. Cancer Lett. 2004;216:207–219. doi: 10.1016/j.canlet.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Prince M, Li Y, Childers A, Itoh K, Yamamoto M, Kleiner HE. Comparison of citrus coumarins on carcinogen-detoxifying enzymes in Nrf2 knockout mice. Toxicol Lett. 2009;185:180–186. doi: 10.1016/j.toxlet.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdel-Wahab MH. Potential neuroprotective effect of t-butylhydroquinone against neurotoxicity-induced by 1-methyl-4-(2′-methylphenyl)-1,2,3,6-tetrahydropyridine (2′-methyl-MPTP) in mice. J Biochem Mol Toxicol. 2005;19:32–41. doi: 10.1002/jbt.20053. [DOI] [PubMed] [Google Scholar]
- 20.Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P, Murphy TH. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem. 2005;280:22925–22936. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- 21.Shah ZA, Li RC, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of reactive oxygen species in modulation of Nrf2 following ischemic reperfusion injury. Neuroscience. 2007;147:53–59. doi: 10.1016/j.neuroscience.2007.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu XY, Li CY, Bu H, Li Z, Li B, Sun MM, Guo YS, Zhang L, Ren WB, Fan ZL, Wu DX, Wu SY. The neuroprotective potential of phase II enzyme inducer on motor neuron survival in traumatic spinal cord injury in vitro. Cell Mol Neurobiol. 2008;28:769–779. doi: 10.1007/s10571-007-9219-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siebert A, Desai V, Chandrasekaran K, Fiskum G, Jafri MS. Nrf2 activators provide neuroprotection against 6-hydroxydopamine toxicity in rat organotypic nigrostriatal cocultures. J Neurosci Res. 2009;87:1659–1669. doi: 10.1002/jnr.21975. [DOI] [PubMed] [Google Scholar]
- 24.Liu RM, van Groen T, Katre A, Cao D, Kadisha I, Ballinger C, Wang L, Carroll SL, Li L. Knockout of plasminogen activator inhibitor 1 gene reduces amyloid beta peptide burden in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2011;32:1079–1089. doi: 10.1016/j.neurobiolaging.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vayalil PK, Olman M, Murphy-Ullrich JE, Postlethwait EM, Liu RM. Glutathione restores collagen degradation in TGF-beta-treated fibroblasts by blocking plasminogen activator inhibitor-1 expression and activating plasminogen. Am J Physiol Lung Cell Mol Physiol. 2005;289:L937–L945. doi: 10.1152/ajplung.00150.2005. [DOI] [PubMed] [Google Scholar]
- 27.Beutler E. Effect of flavin compounds on glutathione reductase activity: In vivo and in vitro studies. J Clin Invest. 1969;48:1957–1966. doi: 10.1172/JCI106162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Nostrand WE, Porter M. Plasmin cleavage of the amyloid beta-protein: Alteration of secondary structure and stimulation of tissue plasminogen activator activity. Biochemistry. 1999;38:11570–11576. doi: 10.1021/bi990610f. [DOI] [PubMed] [Google Scholar]
- 29.Periz G, Fortini ME. Proteolysis in Alzheimer’s disease. Can plasmin tip the balance? EMBO Rep. 2000;1:477–478. doi: 10.1093/embo-reports/kvd124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer’s disease brains. EMBO Rep. 2000;1:530–535. doi: 10.1093/embo-reports/kvd107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J Neurosci. 2003;23:8867–8871. doi: 10.1523/JNEUROSCI.23-26-08867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Exley C, Korchazhkina OV. Plasmin cleaves Abeta42 in vitro and prevents its aggregation into beta-pleated sheet structures. Neuroreport. 2001;12:2967–2970. doi: 10.1097/00001756-200109170-00042. [DOI] [PubMed] [Google Scholar]
- 33.Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2009;8:16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zlokovic BV. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics. 2008;5:409–414. doi: 10.1016/j.nurt.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27:909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bates KA, Verdile G, Li QX, Ames D, Hudson P, Masters CL, Martins RN. Clearance mechanisms of Alzheimer’s amyloid-beta peptide: Implications for therapeutic design and diagnostic tests. Mol Psychiatry. 2009;14:469–486. doi: 10.1038/mp.2008.96. [DOI] [PubMed] [Google Scholar]
- 37.Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH. Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aderinwale OG, Ernst HW, Mousa SA. Current therapies and new strategies for the management of Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2010;25:414–424. doi: 10.1177/1533317510372372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashi T, Price D, Walker D, Scheff S, McGillis JP, Rydel RE, Estus S. The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci. 2000;20:3937–3946. doi: 10.1523/JNEUROSCI.20-11-03937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melchor JP, Pawlak R, Chen Z, Strickland S. The possible role of tissue-type plasminogen activator (tPA) and tPA blockers in the pathogenesis and treatment of Alzheimer’s disease. J Mol Neurosci. 2003;20:287–289. doi: 10.1385/JMN:20:3:287. [DOI] [PubMed] [Google Scholar]
- 41.Turner AJ, Nalivaeva NN. New insights into the roles of metalloproteinases in neurodegeneration and neuroprotection. Int Rev Neurobiol. 2007;82:113–135. doi: 10.1016/S0074-7742(07)82006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eren M, Gleaves LA, Atkinson JB, King LE, Declerck PJ, Vaughan DE. Reactive site-dependent phenotypic alterations in plasminogen activator inhibitor-1 transgenic mice. J Thromb Haemost. 2007;5:1500–1508. doi: 10.1111/j.1538-7836.2007.02587.x. [DOI] [PubMed] [Google Scholar]
- 43.Vaughan DE, De Taeye BM, Eren M. PAI-1 antagonists: Predictable indications and unconventional applications. Curr Drug Targets. 2007;8:962–970. doi: 10.2174/138945007781662364. [DOI] [PubMed] [Google Scholar]
- 44.Cacquevel M, Launay S, Castel H, Benchenane K, Cheenne S, Buee L, Moons L, Delacourte A, Carmeliet P, Vivien D. Ageing and amyloid-beta peptide deposition contribute to an impaired brain tissue plasminogen activator activity by different mechanisms. Neurobiol Dis. 2007;27:164–173. doi: 10.1016/j.nbd.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 45.Jacobsen JS, Comery TA, Martone RL, Elokdah H, Crandall DL, Oganesian A, Aschmies S, Kirksey Y, Gonzales C, Xu J, Zhou H, Atchison K, Wagner E, Zaleska MM, Das I, Arias RL, Bard J, Riddell D, Gardell SJ, Abou-Gharbia M, Robichaud A, Magolda R, Vlasuk GP, Bjornsson T, Reinhart PH, Pangalos MN. Enhanced clearance of Abeta in brain by sustaining the plasmin proteolysis cascade. Proc Natl Acad Sci U S A. 2008;105:8754–8759. doi: 10.1073/pnas.0710823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vayalil PK, Iles KE, Choi J, Yi A-K, Postlethwait EM, Liu R-M. Glutathione suppresses TGF-beta-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1281–L1292. doi: 10.1152/ajplung.00128.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu RM, Choi J, Wu JH, Gaston Pravia KA, Lewis KM, Brand JD, Mochel NS, Krzywanski DM, Lambeth JD, Hagood JS, Forman HJ, Thannickal VJ, Postlethwait EM. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J Biol Chem. 2010;285:16239–16247. doi: 10.1074/jbc.M110.111732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang Z, Seo JY, Ha H, Lee EA, Kim YS, Han DC, Uh ST, Park CS, Lee HB. Reactive oxygen species mediate TGF-beta1-induced plasminogen activator inhibitor-1 upregulation in mesangial cells. Biochem Biophys Res Commun. 2003;309:961–966. doi: 10.1016/j.bbrc.2003.08.102. [DOI] [PubMed] [Google Scholar]
- 49.Uchida Y, Ohba K, Yoshioka T, Irie K, Muraki T, Maru Y. Cellular carbonyl stress enhances the expression of plasminogen activator inhibitor-1 in rat white adipocytes via reactive oxygen species-dependent pathway. J Biol Chem. 2004;279:4075–4083. doi: 10.1074/jbc.M304222200. [DOI] [PubMed] [Google Scholar]
- 50.Hong HK, Song CY, Kim BC, Lee HS. ERK contributes to the effects of Smad signaling on oxidized LDL-induced PAI-1 expression in human mesangial cells. Transl Res. 2006;148:171–179. doi: 10.1016/j.trsl.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 51.Vulin AI, Stanley FM. Oxidative stress activates the plasminogen activator inhibitor type 1 (PAI-1) promoter through an AP-1 response element and cooperates with insulin for additive effects on PAI-1 transcription. J Biol Chem. 2004;279:25172–25178. doi: 10.1074/jbc.M403184200. [DOI] [PubMed] [Google Scholar]
- 52.Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu R-M, Liu Y, Forman HJ, Olman M, Tarpey MM. Glutathione regulates transforming growth factor-beta-stimulated collagen production in fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2004;286:L121–L128. doi: 10.1152/ajplung.00231.2003. [DOI] [PubMed] [Google Scholar]
- 54.Liu R-M, Choi J. Age-associated decline of gamma-glutamylcysteine synthetase gene expression in rats. Free Radic Biol Med. 2000;28:566–574. doi: 10.1016/s0891-5849(99)00269-5. [DOI] [PubMed] [Google Scholar]
- 55.Liu RM. Down-Regulation of gamma-Glutamylcysteine Synthetase Regulatory Subunit Gene Expression in Rat Brain Tissue During Aging. J Neurosci Res. 2002;68:344–351. doi: 10.1002/jnr.10217. [DOI] [PubMed] [Google Scholar]
- 56.Liu RM, Dickinson DA. Decreased synthetic capacity underlies the age-associated decline in glutathione content in Fisher 344 rats. Antioxid Redox Signal. 2003;5:529–536. doi: 10.1089/152308603770310176. [DOI] [PubMed] [Google Scholar]
- 57.Liu H, Wang H, Shenvi S, Hagen TM, Liu R-M. Glutathione Metabolism during Aging and in Alzheimer Disease. Ann NY Acad Sci. 2004;1019:346–349. doi: 10.1196/annals.1297.059. [DOI] [PubMed] [Google Scholar]
- 58.Liu H, Harrell LE, Shenvi S, Hagen T, Liu RM. Gender differences in glutathione metabolism in Alzheimer’s disease. J Neurosci Res. 2005;79:861–867. doi: 10.1002/jnr.20424. [DOI] [PubMed] [Google Scholar]
- 59.Donahue JE, Flaherty SL, Johanson CE, Duncan JA, 3rd, Silverberg GD, Miller MC, Tavares R, Yang W, Wu Q, Sabo E, Hovanesian V, Stopa EG. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006;112:405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 60.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 61.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J Neurosci Res. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M, Ghiso J, Frangione B, Zlokovic BV. Brain clearance of Alzheimer’s amyloid-beta40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target. 2002;10:359–368. doi: 10.1080/10611860290031831. [DOI] [PubMed] [Google Scholar]
- 63.Jaeger LB, Dohgu S, Hwang MC, Farr SA, Murphy MP, Fleegal-DeMotta MA, Lynch JL, Robinson SM, Niehoff ML, Johnson SN, Kumar VB, Banks WA. Testing the neurovascular hypothesis of Alzheimer’s disease: LRP-1 antisense reduces blood-brain barrier clearance, increases brain levels of amyloid-beta protein, and impairs cognition. J Alzheimers Dis. 2009;17:553–570. doi: 10.3233/JAD-2009-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Owen JB, Sultana R, Aluise CD, Erickson MA, Price TO, Bu G, Banks WA, Butterfield DA. Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: Implications for Abeta accumulation in AD brain. Free Radic Biol Med. 2010;49:1798–1803. doi: 10.1016/j.freeradbiomed.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]