

Abstract
Bax and Bak, two pro-apoptotic Bcl-2 family proteins, have been implicated in acute kidney injury following renal ischemia/reperfusion; however, definitive evidence for a role of these genes in the disease process is lacking. Here we first examined two Bax-deficient mouse models and found that only conditional Bax-deletion specifically from proximal tubules could ameliorate ischemic acute kidney injury. Global (whole mouse) knockout of Bax enhanced neutrophil infiltration without significant effect on kidney injury. In contrast, global knockout of Bak protected mice from ischemic acute kidney injury with improved renal function. Interestingly, in these models, Bax or Bak knockout attenuated renal tubular cell apoptosis without significantly affecting necrotic tubular damage. Cytochrome c release in ischemic acute kidney injury was also suppressed in conditional Bax or global Bak-knockout mice. In addition, Bak deficiency prevented mitochondrial fragmentation in ischemic acute kidney injury. Thus, our gene-knockout studies support a critical role of Bax and Bak in tubular cell apoptosis in ischemic acute kidney. Furthermore, necrosis and apoptosis have distinguishable regulatory functions.
Keywords: renal ischemia, mitochondria, apoptosis, necrosis, proximal tubule
INTRODUCTION
Apoptosis is an important cell death mechanism that functions in various physiological and pathological conditions. In mammalian cells, there are two major apoptotic pathways that are respectively centered around death-receptors and mitochondria. The mitochondrial pathway of apoptosis is characterized by mitochondrial outer membrane permeabilization (MOMP) and consequent release of apoptogenic factors such as cytochrome c. Bcl-2 family proteins are important regulators of MOMP during apoptosis [1, 2]. In this regard, Bax and Bak, two pro-apoptotic proteins with multiple Bcl-2 homology (BH) domains, are the key gate-keeper molecules of MOMP. The anti-apoptotic Bcl-2 proteins, such as Bcl-2 and Bcl-XL, can antagonize Bax and Bak to prevent MOMP. On the contrary, BH3-domain only proteins like Bid, Bim and Puma can activate Bax and Bak to form oligomers in mitochondrial outer membrane resulting in MOMP [1, 2]. Recent research indicates that Bak has a unique role in the regulation of mitochondrial dynamics leading to mitochondrial fragmentation. Importantly, fragmented mitochondria are sensitized to Bax insertion and oligomerization in mitochondrial outer membrane. Thus, Bak and Bax provide the “two-hits” on mitochondria resulting in MOMP and consequent release of apoptogenic factors. Bax/Bak-mediated mitochondrial pathway of apoptosis has been suggested to play important roles in a variety of diseases including acute kidney injury (AKI) [3–12].
Acute kidney injury (AKI) is a kidney disease characterized by rapid loss of kidney function and wide-spread renal tubular cell death including both necrosis and apoptosis [13]. Severe AKI is associated with high mortality and currently there are no effective therapies except supportive care. Clinically, renal ischemia-reperfusion is one of the major causes of AKI [14]. In in vitro models of ischemic AKI, i.e. hypoxia and ATP-depletion in renal proximal tubular cells, Bax and Bak have been shown to play critical roles in apoptosis [10, 15–19]. In vivo, Bax/Bak are induced and activated in ischemic kidney tissues and human kidney allografts after transplantation [6, 20]. Despite these studies, convincing evidence for a pathogenic role of Bax and Bak in ischemic AKI has not been demonstrated by using gene knockout models.
In the current study, we have examined ischemic AKI in several Bax or Bak knockout models. We show that germline Bak -knockout or conditional Bax-knockout from proximal tubules could ameliorate ischemic AKI. Interestingly, in these models tubular apoptosis, but not necrosis, is mainly attenuated. Notably, mitochondrial fragmentation is blocked in Bak-knockout kidney tissues, verifying a role of Bak in the regulation of mitochondrial dynamics in pathological conditions in vivo.
RESULTS
Protection of mice from ischemic AKI by germline knockout of Bak, but not Bax
To investigate the role of Bax and Bak in ischemic AKI, we first started with Bax and Bak germline knockout mice. The depletion of Bax and Bak was confirmed by Western blot of whole kidney lysates (Fig. 1 C & F). Under control conditions, neither Bax knockout [Bax(−/−)] nor Bak knockout [Bak(−/−)] mice showed obvious kidney defects. These mice also had normal kidney function with 30–50 mg/dL blood urea nitrogen (BUN) and 0.3–0.5 mg/dL serum creatinine as wild-type animals [Bax(+/+) or Bak(+/+)] (Fig. 1A, B, D, E). When the mice were challenged with 30 minutes of bilateral kidney ischemia followed by 48 hours of reperfusion, the Bax(−/−) mice showed similar severe renal injury as Bax(+/+) mice, with a huge induction of BUN [ 219.8±60.8 mg/dL (n=5) for Bax(+/+) vs 238.7±39.7 mg/dL (n=6) for Bax (−/−)] and serum creatinine level [2.47±0.86 mg/dL (n=5) for Bax(+/+) vs 2.86±0.39 mg/dL (n=6) for Bax(−/−)] (Fig. 1A, B). In contrast, Bak(−/−) mice were significantly protected from ischemic AKI, showing only mild kidney function loss (BUN of 119.7±28.2 mg/dL, serum creatinine of 0.91±0.28 mg/dL, n=5) compared to Bak(+/+) mice (BUN of 229.8±30.9 mg/dL, serum creatinine of 2.32±0.35 mg/dL, n=6) (Fig. 1D, E).
Fig. 1. Germline knockout of Bak, but not Bax, protects kidney function during ischemia/reperfusion injury.
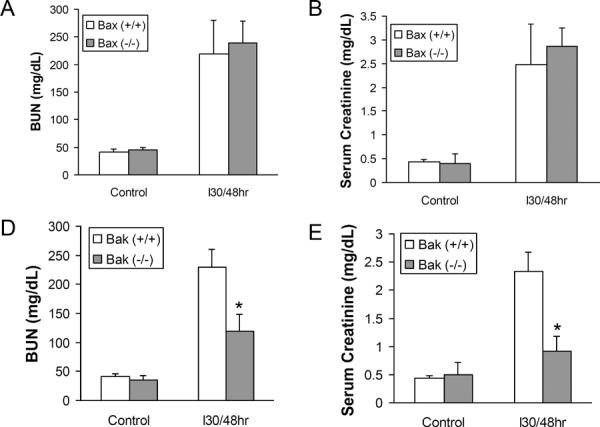
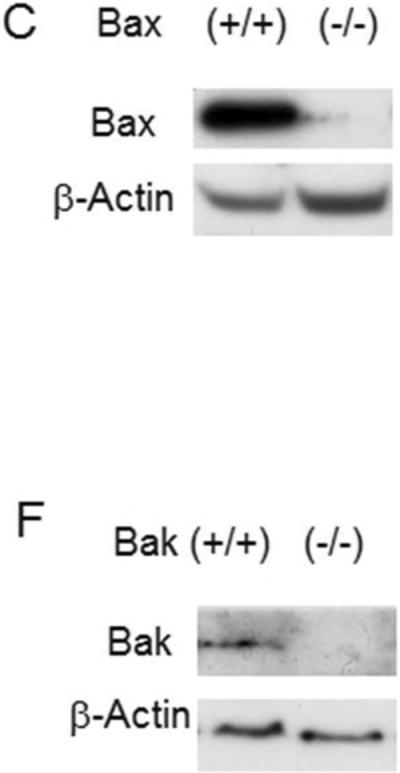
Wild-type, germline Bax knockout, and germline Bak knockout mice were subjected to sham operation or 30 minutes of bilateral renal ischemia followed by 48 hours reperfusion. Blood samples were collected to measure blood urea nitrogen (BUN) and serum creatinine to indicate kidney function. (A) BUN of wild-type and Bax knockout mice. (B) Serum creatinine of wild-type and Bax knockout mice. (C) Immunoblot analysis confirming Bax expression in kidney lysate of wild type mice, but not in Bax knockout tissues. (D) BUN of wild-type and Bak knockout mice. (E) Serum creatinine of wild-type and Bak knockout mice. *, significantly different from treated Bak(+/+) group (P<0.05). (F) Immunoblot analysis confirming Bak expression in kidney lysate of wild type mice, but not in Bak knockout tissue lysate.
We further examined kidney tissues by H & E staining. Regardless of their genotypes, all the mice without renal ischemia/reperfusion showed normal, healthy kidney histology (Fig. 2A, B: Control). After renal ischemia/reperfusion, both Bax(+/+) and Bax(−/−) mice had notable necrotic tubular damage, featured by severe tubular lysis mainly in the outer stripe of the outer medulla, with some in the kidney cortex (Fig. 2A). Surprisingly, despite of functional protection (Fig. 1) Bak knockout mice did not show obvious improvement of tubular necrosis, shown by H & E staining compared to Bak (+/+) group (Fig. 2B). The tubular damage score for the Bak (+/+) and Bak (−/−) tissues after ischemic AKI were 1.83±0.39 and 1.75±0.45 (n=12), respectively, and there was no statistical difference (Fig. 2C).
Fig. 2. Germline knockout of Bax or Bak does not affect tubular necrotic damage during renal ischemia/reperfusion.
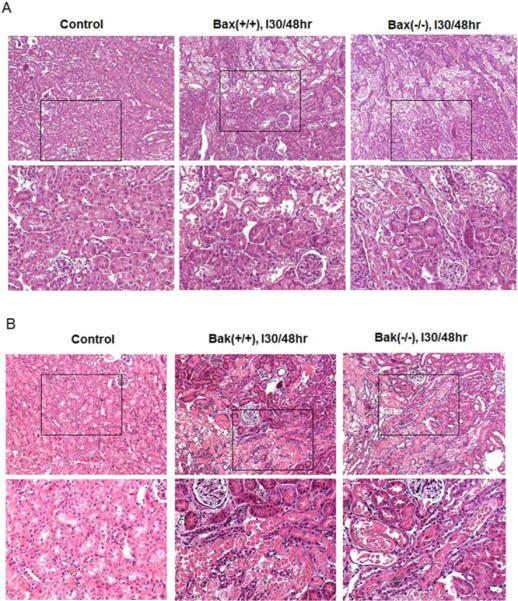
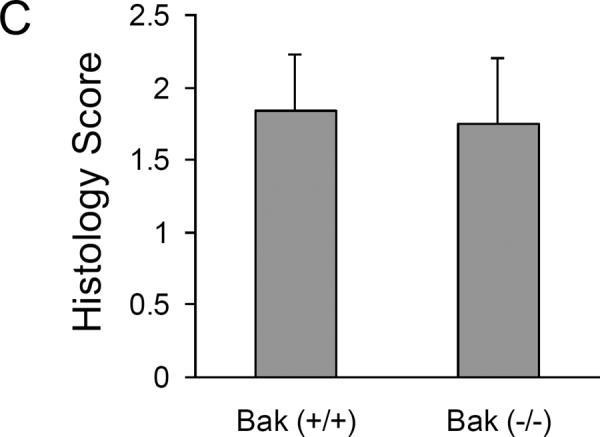
Kidney tissues were collected for H & E staining after sham operation or 30 minutes bilateral renal ischemia and 48 hours reperfusion in mice of the indicated genotypes. (A) Representative H & E images of kidney cortical tissues from wild type and germline Bax knockout mice. The lower panels are the magnified images of the boxed areas in the upper panels. (B) Representative H & E images of kidney cortical tissues from wild type and germline Bak knockout mice. The lower panels are the magnified images of the boxed areas in the upper panels. (C) Histology score of tubular necrotic damage in Bak (+/+) and Bak (−/−) mice after ischemia/reperfusion.
Generation of proximal tubular Bax knockout mouse model and another germline Bak knockout model
The lack of effect of Bax knockout on ischemic AKI in mice (Fig. 1) was surprising, because Bax plays a critical role in cell injury and death in in vitro models of ischemic AKI kidney in cultured kidney tubular cells [10, 15–19]. One explanation was that global Bax knockout affected not only the sensitivity of kidney tubules to ischemic injury but also other participating mechanisms. To test this possibility, we generated a conditional knockout model in which Bax was specifically deleted from kidney proximal tubules (Fig. 3A). For this purpose, male mice with floxed Bax gene and germline Bak deletion were obtained from Korsmeyer lab [21]and bred with females carrying Cre gene driven by the PEPCK promoter (PEPCK-CRE) [22]. After two generations, we obtained offsprings of a variety of Bax and Bak genotypes, of which 3 major categories were selected for our study: 1) Bax knockout specifically in proximal tubular cells [PT-Bax(−/−)]; 2) germline Bak knockout [Bak(−/−)]; and 3) wild type (WT). In addition to genotyping, Bax depletion in proximal tubules in PT-Bax(−/−) mice was confirmed by Western blot analysis of lysates from renal cortical tissues where the majority of cells are proximal tubular cells (Fig. 3B, Supplementary Fig. 1B, C). Immunohistochemical staining of Bax also showed significantly lower Bax expression in renal cortex and outer stripe of outer medulla in PT-Bax(−/−) mice than inner medulla (Supplementary Fig. 1A). As a control, Bak expression was not changed significantly in PT-Bax(−/−) tissues (Supplementary Fig. 1D). The trace signal of Bax in PT-Bax(−/−) cortex was likely due to the existence of other types of renal cells. Similarly, Bak deletion in Bak(−/−) mice was verified(Fig. 3B).
Fig. 3. Establishment of proximal tubule-specific Bax-knockout (PT-Bax-KO) mouse model.
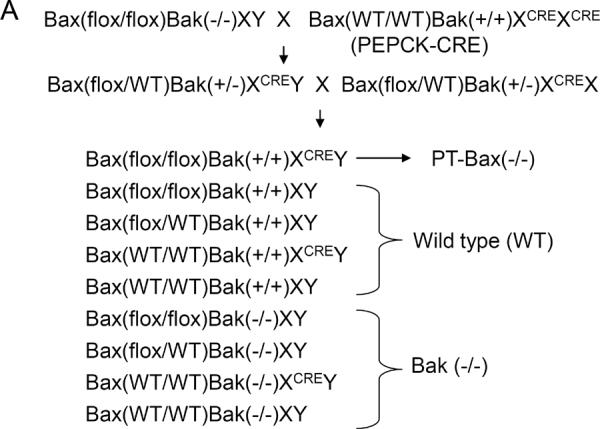
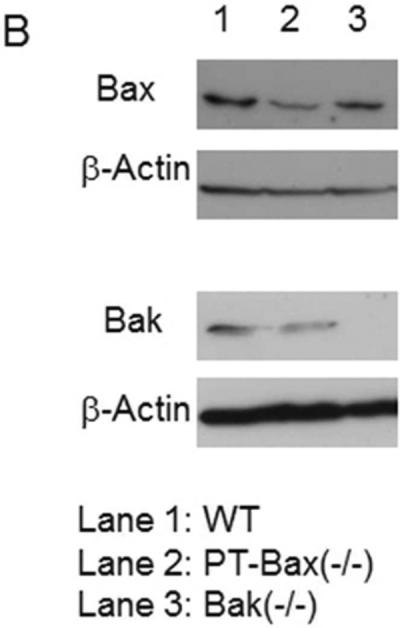
(A) Schematics showing the establishment of the PT-Bax(−/−) mouse model by crossing Bax-floxed Bak-knockout mice with Bax/Bak-proficient PEPCK-Cre mice. Please note, the schematics only lists the three major genotypes that were compared in this study: PT-Bax-KO, wild type, and Bak-KO. (B) Immunoblot analysis of renal cortical lysate confirming Bax deletion in PT-Bax(−/−) mice and Bak deletion in Bak(−/−) mice.
Suppression of ischemic AKI, but not tubular necrosis, in PT-Bax(−/−) mice and Bak(−/−) mice
Under control conditions, the mice of different Bax and Bak genotypes showed normal renal function (BUN level in the range of 20–40 mg/dL and serum creatinine level in the range of 0.3–0.5 mg/dL) (Fig. 4A, B). After 30 minutes of bilateral renal ischemia and 48 hours reperfusion, wild-type mice (WT, n=8) had severe renal injury with BUN increased to 249.1±85.0 mg/dL and serum creatinine increased to 1.82±0.64 mg/dL. In contrast, both PT-Bax(−/−) and Bak(−/−) mice showed only mild kidney injury after the same treatment of renal ischemia/reperfusion. As shown in Fig. 4, the BUN level of PT-Bax(−/−) group (n=8) was 119.4±48.5 mg/dL and serum creatinine was 0.80±0.34 mg/dL. Similarly, Bak(−/−) mice (n=8) only showed a smaller increase in BUN and serum creatinine after renal ischemia-reperfusion than wild-type mice.
Fig. 4. Kidney function during ischemia/reperfusion injury is protected in PT-Bax-KO and germline Bak-KO mice.
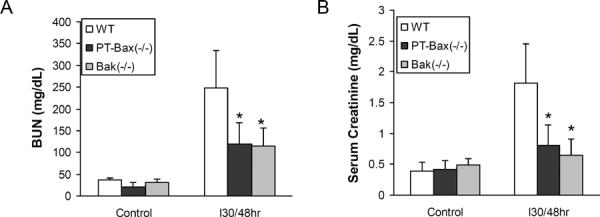
Blood samples were collected after 30 minutes of bilateral renal ischemia and 48 hours of reperfusion or sham operation to measure BUN and creatinine. (A) BUN. (B) Serum creatinine. *, statistically significant difference compared to WT group (P<0.05).
Although both PT-Bax(−/−) and Bak(−/−) mice were protected from renal ischemia-reperfusion-induced renal functional loss or failure, these mice showed similar necrotic tubular damage as WT tissues (Fig. 5). As shown in Fig. 5A, the sham-operated control kidneys had normal tubular morphology. After renal ischemia/reperfusion, there was severe tubular necrosis in all genotypes of mice with the majority of the tubules lysed in the outer stripe of the outer medulla and some tubules lysed in the cortex. The histology scores did not show significant differences in tubular necrosis in PT-Bax(−/−), Bak(−/−), or WT mice (Fig. 5B).
Fig. 5. Tubular necrotic damage during renal ischemia/reperfusion is not suppressed in PT-Bax-KO and germline Bak-KO mice.

Kidney tissues with or without 30 minutes of ischemia and 48 hours of reperfusion were collected and processed for H & E staining. (A) Representative H & E staining of cortical tissues. The lower panels are magnified images from the boxed areas in the upper panels. (B) Histology score of necrotic tubular damage in kidney cortical tissues collected after renal ischemia/reperfusion in the mice of indicated genotypes.
Less renal tubular apoptosis in PT-Bax (−/−) mice and Bak(−/−) mice after ischemic AKI
In addition to necrosis, apoptosis is another major mechanism of tubular cell death in ischemic AKI [23–25]. We performed TUNEL assay to examine renal apoptosis, a relatively reliable method for detecting apoptosis in kidney tissues [26]. All the kidney tissues with 30 minutes of ischemia followed by 48 hours of reperfusion showed positive TUNEL staining, while very few TUNEL-positive cells were identified in the sham-operated renal tissue (Fig. 6A). Compared to their WT littermates, PT-Bax(−/−) and Bak(−/−) mice had much less renal apoptosis (Fig. 6A) with statistically significant differences [60±21 cells/mm2 for WT, 13±11 cells/mm2 for PT-Bax(−/−) and 19±11cells/mm2 for Bak(−/−)] (Fig. 6B).
Fig. 6. Renal tubular apoptosis during ischemia/reperfusion is attenuated in PT-Bax-KO and germline Bak-KO mice.
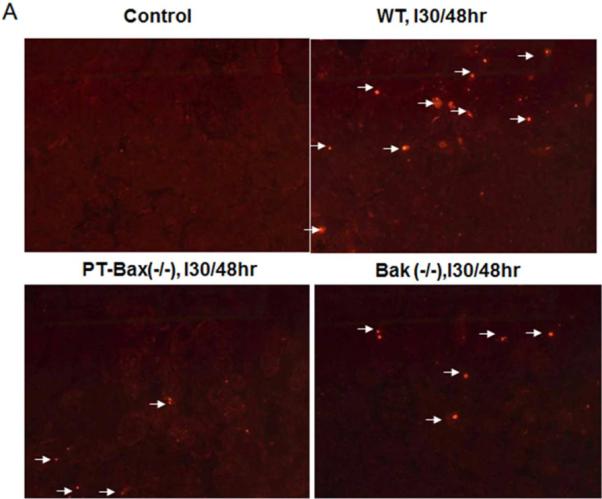
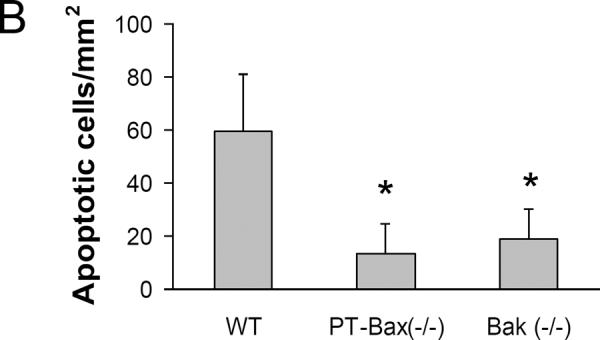
Mice of indicated genotypes were subjected to sham control treatment or 30 minutes of renal ischemia with 48 hours of reperfusion to collect kidney tissues for TUNEL assay. (A) Representative TUNEL images. Arrows point to TUNEL-positive cells. (B) Quantification of apoptotic cells. *, statistically significant difference compared to WT group.
The Bax/Bak- mediated apoptotic pathway is featured by cytochrome c release from mitochondria. In the tubular cells of normal control kidneys, cytochrome c was contained in mitochondria, which showed a pattern of granulated staining (Fig. 7A upper panels). After ischemic injury, some tubular cells showed evenly distributed cytochrome c signals in the cytosol and the signal was strengthened due to the shrinkage of apoptotic cells (Fig. 7A, lower panels). The cells with cytochrome c release were identified in kidneys with 30 minutes of bilateral renal ischemia and 12 hours of reperfusion. Notably, the numbers of cell with cytochrome c release were significantly decreased in PT-Bax(−/−) kidneys (13.4±18.5 cells/mm2) and Bak(−/−) kidneys (20.0±18.5 cells/mm2), compared to WT kidneys (47.4±36.6 cells/mm2) (Fig. 7B).
Fig. 7. Cytochrome c release during ischemia/reperfusion is suppressed in PT-Bax-KO and germline Bak-KO mice.
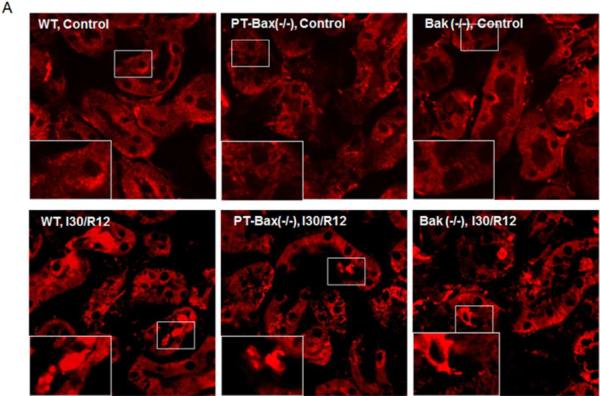

Kidney tissues were collected after 30 minutes of renal ischemia and 12 hours of reperfusion for immunofluorescence analysis of cytochrome c. (A) Representative images showing cytochrome c released after 12 hours reperfusion. Insert images are magnified from boxed areas. (B) Counting of cells with cytochrome c release. Fifteen to nineteen images were randomly taken for each group and the number of cells with cytochrome c release was counted. *, statistically significant difference compared to WT group (P<0.05).
Neutrophil infiltration increase in germline Bax knockout mice after ischemic AKI
It is intriguing that proximal tubule-specific Bax knockout mice, but not germline Bax knockout mice, were protected from ischemic AKI (Figs. 1–7). Global Bax knockout in mice was reported to induce hyperplasia in immune cells [27]. We hypothesized that that renal ischemia-reperfusion may induce a heightened inflammatory response in global Bax knockout mice, which neutralizes the tubular protective effect of Bax deletion in these animals. To test this possibility, we examined neutrophil and macrophage infiltration in kidneys after ischemic AKI in germline Bax knockout mice and PT-Bax (−/−) mice and their wild type littermates. As shown in Fig. 8, ischemic AKI induced marked neutrophil infiltrations in kidney tissues in all genotype mice (Fig. 8). Notably, neutrophil infiltration was significantly higher in germline Bax knockout tissues than that of wild type littermate kidneys (Fig. 8A), while neutrophil infiltration in PT-Bax(−/−) mice was comparable to their wild type (Fig. 8B). Macrophage infiltration was also significantly induced after ischemic AKI, but there were no significant differences between Bax knockout mice and their wild type littermates (data not shown).
Fig. 8. Significantly more neutrophils infiltrate into kidney in germline Bax knockout mice after ischemia/reperfusion injury.

Mice were subjected to sham operation of 30 minutes of renal ischemia followed by 48 hours of reperfusion. Kidney tissues were stained for counting of neutrophils. (A) Neutrophil infiltration counting in germline Bax knockout mice and the wild type littermates. *, statistically significant difference compared to Bax(+/+) group (P<0.05). (B) Neutrophil infiltration counting in PT-Bax-KO mice and the wild type littermates.
Inhibition of mitochondrial fragmentation by Bak knockout
Recent research has established a critical role of mitochondrial fragmentation in mitochondrial outer membrane permeabilization and consequent release of cytochrome c. Using gene knockout cells, we further suggested Bak as a key regulator of mitochondrial fragmentation during cell injury and stress [17–19]. Despite these observations, in vivo evidence for the role of Bak in mitochondrial fragmentation under apoptotic conditions was lacking. Therefore, we compared mitochondrial fragmentation in Bak(−/−) and Bak(+/+) mice (Fig. 9). Under control conditions, most of the kidney tubular cells had long and filamentous mitochondria and only a small number of tubular cells showed fragmented mitochondria in both Bak(+/+) (8.7% cells) and Bak(−/−) (14.0% cells) kidneys. The number of cells with mitochondrial fragmentation increased significantly to 41.4% in Bak(+/+) kidneys after 30 minutes ischemia and 15 minutes reperfusion. However, in Bak(−/−) mice, the induction of mitochondrial fragmentation by ischemia/reperfusion in kidney tissues was blocked and only 15.9% of cells showed fragmented mitochondria.
Fig. 9. Inhibition of mitochondrial fragmentation during ischemia/reperfusion in germline Bak-KO mice.
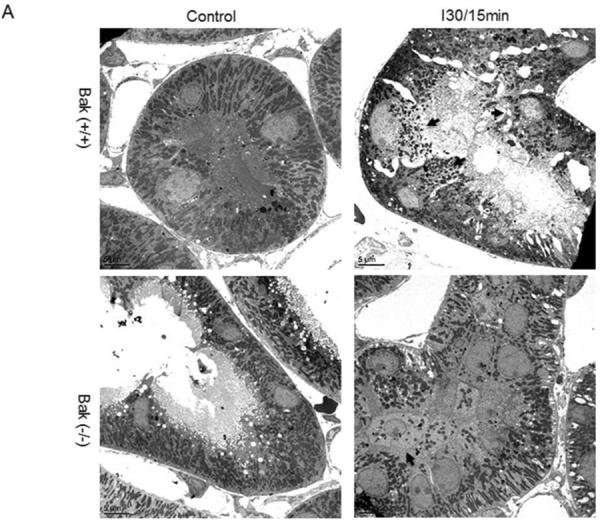
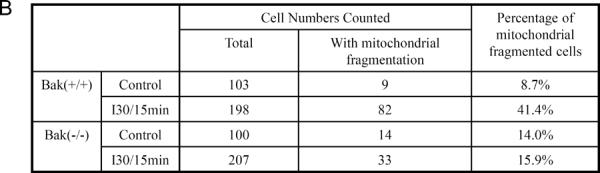
Kidney tissues were collected after 30 minutes of ischemia and 15 minutes of reperfusion and then processed for electron microscopy. (A) Representative images showing the morphology of mitochondria. Arrows point to the cells with fragmented mitochondria. (B) Quantification of the cells with mitochondrial fragmentation. The cells with filamentous and fragmented mitochondria were counted to determine the percentage of cells with mitochondrial fragmentation.
DISCUSSION
Bax and Bak have been proposed to function in ischemic AKI for a long time. However, definitive in vivo evidence for the pathological roles of Bax and Bak in this disease condition has been lacking, except for some initial studies showing Bax and Bak activation in ischemically injured kidney tissues [6, 16, 20]. In this study, we dissected the roles of Bax and Bak during ischemic AKI using several gene knockout mouse models. Three major conclusions can be drawn from these knockout experiments: 1) while global knockout of Bax does not protect against ischemic AKI, conditional knockout of Bax from proximal tubules is renoprotective; 2) global knockout of Bak protect against ischemic AKI and one of the underlying mechanisms may involve the maintenance of mitochondrial dynamics and integrity; 3) Bax and Bak knockout blocks kidney tubular apoptosis during ischemic AKI without affecting tubular necrosis, supporting the existence of distinct mechanisms for these two forms of cell death.
The initial experiments with the germline Bax knockout mouse model were surprising because global deletion of Bax did not show any renoprotective effects and as a matter of fact, after ischemic injury both BUN and serum creatinine levels in these mice were marginally higher than wild type animals (Fig. 1). This result was not consistent with the role of Bax in ischemic AKI as suggested by earlier studies. In addition, the observation was in contrast to our previous work demonstrating kidney protection by germline Bax knockout in the AKI model of cisplatin nephrotoxicity [28]. To address this puzzling, negative effect, we established a conditional knockout model in which Bax was specifically deleted from kidney proximal tubules. In these PT-Bax knockout mice, we demonstrated that ischemic AKI was markedly ameliorated (Figs. 4–7). Based on these results, we conclude that Bax in proximal tubular cells indeed plays an important role in tubular cell injury and death and ischemic AKI. Then, why global deletion of Bax is not effective? One plausible explanation is that, global deletion of Bax from all tissues and cells affects not only kidney proximal tubules but also other injury-related factors. As a multifactorial process, ischemic AKI involves a complex pathogenesis [29, 30]. In addition to intrinsic kidney tubular damage, other injury mechanisms such as inflammation and vascular dysfunction may also be affected by the deletion of Bax. In this regard, mice with global Bax knockout were reported to have hyperplasia in the immune system including thymocytes and B cells [27]. Considering the involvement of immune cells and associated inflammation in ischemic AKI [31–34], we speculate that renal ischemia-reperfusion may induce a heightened inflammatory response in global Bax knockout mice, which neutralizes the tubular protective effect of Bax deletion in these animals. On the contrary, in the conditional Bax knockout model, Bax is only ablated from kidney proximal tubules resulting in protection, while other pathogenic mechanisms are not altered. In line with this possibility, significantly higher neutrophil infiltration is induced during ischemic AKI in global Bax knockout mice that wild type or PT-Bax knockout mice (Fig. 8). The fact that global Bax knockout protected against cisplatin-induced AKI [28], but not ischemic AKI (present study, Fig.1), suggests that some pathogenic mechanisms in these two AKI models are distinguishable.
Another interesting observation from this study is that, while both conditional knockout of Bax and global deletion of Bak markedly suppressed tubular cell apoptosis in ischemic AKI, necrotic tubular damage was not affected in either of the models. Functionally, the mice from these models were significantly protected from ischemic AKI (Fig. 4). These results support for a role of tubular apoptosis in the pathogenesis of ischemic AKI. It is however noteworthy that the renoprotection in these knockout models was incomplete, suggesting the existence of pathogenic mechanisms other than tubular apoptosis. Such non-apoptotic mechanism apparently includes tubular necrosis. The observation that only tubular apoptosis, and not necrosis, was attenuated in the knockout model is particularly interesting, because in most of previous studies, tubular apoptosis and necrosis were ameliorated simultaneously by renoprotective approaches [17, 18, 35–39]. The correlated changes in necrosis and apoptosis in these previous studies may be caused by the close relationship between necrosis and apoptosis during AKI. For example, necrosis and apoptosis are often induced in the same injured kidney tissues. While their induction may depend on the severity of the injury, necrosis can be secondary to apoptosis; in other words, during prolonged injury, apoptotic cells may breakdown their plasma membrane at late stage showing a necrotic morphology. On the other hand, necrosis and apoptosis also have their own distinct features and regulatory mechanisms. In this aspect, an earlier study by Kelly, Dagher and colleagues showed that Guanosine supplementation reduced apoptosis and protected kidneys against ischemic AKI without affecting necrotic tissue damage[40]. Our current results further support the notion that in spite of their connections, necrosis and apoptosis have their own specific regulatory mechanisms that can be used to dissect and distinguish them from each other under disease conditions. Bax was recently suggested to regulate necrosis by promoting mitochondria fusion [41]. However, in our current study necrotic tubular damage was not affected by either global or proximal tubule specific Bax knockout. Consistently, Bax did not have a critical role in the regulation of mitochondrial dynamics during cell injury [19]. The cause of the discrepancy between the studies is not clear, but it may be related to the differences in the experimental models examined.
Finally, the present study demonstrates the first in vivo evidence for the regulation of mitochondrial dynamics by Bak (Fig. 9). Mitochondria were traditionally described in a static status with mixed morphologies of “threads” and “beads”. However, the research in recent years has revealed a highly dynamic nature of mitochondria, which constantly undergo fission and fusion[42]. Importantly, mitochondrial dynamics changes in response to the cellular condition. In normal, unstressed cells, mitochondrial fission and fusion are maintained to preserve a filamentous mitochondrial morphology. When cells are stressed or injured, mitochondrial dynamics is rapidly switched to fission resulting in mitochondrial fragmentation, which contributes to mitochondrial outer membrane permeabilization and release of apoptotic factors. In previous studies, we and others found that Bak has a unique role in promoting mitochondrial fragmentation by interacting with mitochondrial fusion proteins called mitofusins [19, 43]. Moreover, the fragmented mitochondria are sensitized to Bax insert, leading to enhanced mitochondrial outer membrane permeabiliztion and apoptosis [17]. In the present study, we showed that mitochondrial fragmentation during ischemic AKI was blocked in Bak-knockout mouse kidney, demonstrating clear-cut in vivo evidence for a role of Bak in mitochondrial fragmentation in the cells under pathological conditions (Fig 9). Notably, the blockade of mitochondrial fragmentation in Bak(−/−) tissues was accompanied by the suppression of mitochondrial leakage of cytochrome c and lower tubular apoptosis(Figs. 7 and 9), further supporting the pathogenic role of mitochondrial fragmentation.
In conclusion, this study has demonstrated the roles of Bax and Bak in tubular cell apoptosis during ischemic AKI by using several gene knockout mouse models. This study has also revealed that necrosis and apoptosis have distinguishable regulatory niches. In addition, using gene knockout models this study has demonstrated the first in vivo evidence for a role of Bak in mitochondrial fragmentation under pathological conditions.
METHODS
Animals
The animal experiments were conducted in accordance with the animal use protocols approved by the Institutional Animal Care and User Committees of Charlie Norwood VA Medical Center and Georgia Health Sciences University. All animals (mice) were maintained in Charlie Norwood VA Medical Center animal facility with a 12/12-hour light/dark cycle and free access to food and water.
Bax germline knockout mice, Bak germline knockout mice (C57BL/6 background) and C57BL/6 mice were originally purchased from Jackson Laboratory (Bar Harbor, ME, USA). Heterozygous males and females [Bax (+/−)] were mated to produce wild-type [Bax(+/+) and Bax homozygous knockout mice [Bax (−/−)]. The genotypes of the mice were determined by PCR to identify male littermates for experiments as described previously [28]. Bak knockout mice were maintained as homozygous to compare with wild-type C57BL/6 mice.
The mouse line with floxed Bax gene in germ-line Bak knockout background was originally obtained from the laboratory of Dr. Stanley Korsmeyer at Dana-Farber Cancer Institute (Boston, MA) [21] with a mixed genetic background of 129X1/SvJ and C57BL/6. The PEPCK-CRE mouse line was originally provided by Dr. Volker Haase at Vanderbilt University School of Medicine (Nashville, TN) [22] with a mixed genetic background of Balb/C, 129Sv/J, and C57BL/6. These two mouse lines were crossed as depicted in Results (Fig. 3A) to produce proximal tubule-Bax knockout mice, Bak knockout mice, and wild- type littermates. The genotypes of all the mice were determined by PCR, and male littermates were used for the study.
Ischemic acute kidney injury
To induce ischemic AKI, 8 to10 week-old-male mice were subjected to bilateral renal pedicle clamping as described in our previous studies [35, 39, 44]. Briefly, mice were anesthetized with 50–60 mg/kg pentobarbital by intraperitoneal injection. Renal pedicles were exposed by flank incisions and were clamped for 30 min before release. Kidneys were collected at different reperfusion times as indicated in the article. Sham-operated mice experienced a similar procedure except for the renal pedicle clamping. The body temperature of the mice was maintained at 37 °C by a homoeothermic blanket system (Harvard Apparatus, Holliston, MA) during the whole procedure.
Measurement of renal function
Blood samples were collected when mice were sacrificed. Serum samples were obtained after blood clotting and centrifugation. Renal function was measured with analytical kits from Biotron Diagnostics (Hemet, CA) for blood urea nitrogen (BUN) and from Stanbio Laboratory (Boerne, TX) for serum creatinine, respectively.
Immunoblot analysis
Kidney cortical lysate samples were subjected to electrophoresis with Mini-gel system from Bio-Rad (Hercules, CA) and then transferred to PVDF membrane. The blot was blocked by 5% milk and then incubated with specific primary antibody. Finally, the blots were exposed to the horseradish peroxidase-conjugated secondary antibody, and antigens on the blots were revealed using an enhanced chemiluminescence kit (Pierce, Rockford, IL)
Histology
Fresh kidney samples were fixed in 4% paraformaldehyde overnight at 4°C, followed by paraffin-embedding. 4-mm thick sections were subjected to hematoxylin-eosin staining. Slides were checked in a blind manner, and representative images were recorded. The lysed tubules as indicated in supplement Fig. 4 were considered as necrotic. The histological tubular necrosis was also scored as follows: 0, no damage; 1, 0–25% damage; 2, 26–50% damage; 3, 50–75% damage; 4, >75% damage.
TUNEL assay
Paraffin-embedded renal sections were rehydrated, permeabilized in 0.1M sodium citrate for 2hrs at 60°C, and incubated with a TUNEL reaction mixture from in situ Cell Death Detection Kit (Roche Applied Science, Indianapolis, IN) for 1hr at 37°C. The slides were mounted with ProLong® Gold Antifade Reagent and examined with fluorescence microscopy. To count TUNEL-positive cells, 10 images were randomly taken in the outer medulla and kidney cortex and the number of apoptotic cells was counted in a blind way.
Immunofluorescence staining
For immunofluorescence of cytochrome c, kidneys were perfused with M-Zamboni fixative (paraformaldehyde/picric acid), followed by M-Zamboni fixation overnight at 4°C. The tissue was balanced in 30% sucrose in PBS at 4°C and then embedded in O.C.T. compound for cryo-section. The cryo-section was immediately kept in PBS for the following staining steps. After blocking in a buffer containing 2% bovine serum albumin, 0.2% non-fat milk, 2% normal goat serum and 0.8% Triton-X100 in PBS, the slides were incubated with specific primary antibody followed by Cy3-labelled secondary antibody. After mounting with ProLong® Gold Antifade Reagent (Invitrogen, Carlsbad, CA), representative images were obtained by confocal microscopy. To count the cells with cytochrome c release, 15 to19 images were randomly taken in the renal cortex and outer medulla and the number of cells with cytochrome c release were counted in a blind way.
Examination of mitochondrial morphologyby electron microscopy
Kidneys were perfused with 10 ml of 10 U/ml heparin and 50 ml fixative (100 mM sodium cacodylate, 2 mM CaCl2, 4 mM MgSO4, 4% paraformaldehyde, and 2.5% glutaraldehyde). After overnight post-fixation in the same fixative at 4°C, a ~1mm3 tissue block containing both cortical and outer medulla part was cut from each kidney. The blocks were processed for standard sample preparation in the Electron Microscopy facility of Georgia Health Sciences University. Pictures with a scale bar were randomly taken from each sample and the length of mitochondria in the cells was measured by NIH ImageJ software. The cells with less than 1% long filamentous mitochondria (>2 μm in length) were considered as cells with mitochondrial fragmentation [18].
Immunohistochemical staining
Paraffin-embedded renal sections were rehydrated and steamed for 1 hr in 10mM sodium citrate with 0.05% Tween 20 for antigen retrieval. The sections were blocked with 3% H2O2 in PBS for 10 min, followed by 1hr exposure to blocking buffer of 2% BSA, 0.2% non-fat milk, 2% normal goat serum, 0.8% Triton X-100 in PBS and overnight incubation with primary antibodies diluted in blocking buffer in 4°C. After Avidin/Biotin blocking kits treatment (Vector Laboratories, Burlingame, CA), the sections were incubated with biotin labeled secondary antibody. The signals were amplified with TSA™ Biotin System (PerkinElmer, Waltham, MA) and developed with Vecstatin Elite ABC kit and ImmPACT DAB Peroxidase Substrate (Vector Laboratories, Burlingame, CA). The slides were counterstained with hematoxylin and mounted after dehydration. The antibodies used were anti-Neutrophil and anti-Macrophage (Abcam, Cambridge, MA). To count neutrophils, 10 images were randomly taken in the outer medulla and kidney cortex and the number of neutrophils was counted in a blind way.
Statistics
Data were expressed as means± SD. Statistical differences between two groups were determined by Student's t-test with Microsoft Excel 2003. P<0.05 was considered to be significantly different.
Supplementary Material
Acknowledgements
We thank Dr. Stanley Korsmeyer and his laboratory at Dana-Farber Cancer Institute (Boston, MA) for kindly providing the mouse line with floxed Bax gene in germ-line Bak knockout background. We also thank Dr. Volker Haase at Vanderbilt University School of Medicine (Nashville, TN) for kindly providing the PEPCK-CRE mouse line.
Sources of Support: Scientist Development Grant from American Heart Association (Qingqing Wei) and grants from the Department of Veterans' Affairs and the National Institutes of Health of U.S.A (Zheng Dong).
Footnotes
DISCLOSURE The authors have nothing to disclose.
REFERENCES
- 1.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122:437–441. doi: 10.1242/jcs.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baines CP. The cardiac mitochondrion: nexus of stress. Annu Rev Physiol. 72:61–80. doi: 10.1146/annurev-physiol-021909-135929. [DOI] [PubMed] [Google Scholar]
- 4.Chopra M, Reuben JS, Sharma AC. Acute lung injury:apoptosis and signaling mechanisms. Exp Biol Med (Maywood) 2009;234:361–371. doi: 10.3181/0811-MR-318. [DOI] [PubMed] [Google Scholar]
- 5.Dagher PC. Apoptosis in ischemic renal injury: roles of GTP depletion and p53. Kidney Int. 2004;66:506–509. doi: 10.1111/j.1523-1755.2004.761_7.x. [DOI] [PubMed] [Google Scholar]
- 6.Gobe G, Zhang XJ, Cuttle L, et al. Bcl-2 genes and growth factors in the pathology of ischaemic acute renal failure. Immunol Cell Biol. 1999;77:279–286. doi: 10.1046/j.1440-1711.1999.00826.x. [DOI] [PubMed] [Google Scholar]
- 7.Hagberg H, Mallard C, Rousset CI, et al. Apoptotic mechanisms in the immature brain: involvement of mitochondria. J Child Neurol. 2009;24:1141–1146. doi: 10.1177/0883073809338212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nickells RW, Semaan SJ, Schlamp CL. Involvement of the Bcl2 gene family in the signaling and control of retinal ganglion cell death. Prog Brain Res. 2008;173:423–435. doi: 10.1016/S0079-6123(08)01129-1. [DOI] [PubMed] [Google Scholar]
- 9.Robertson CL, Scafidi S, McKenna MC, et al. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp Neurol. 2009;218:371–380. doi: 10.1016/j.expneurol.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saikumar P, Venkatachalam MA. Role of apoptosis in hypoxic/ischemic damage in the kidney. Semin Nephrol. 2003;23:511–521. doi: 10.1053/s0270-9295(03)00130-x. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Wei Q, Wang CY, et al. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279:19948–19954. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- 12.Wei Q, Dong Z. Regulation and pathological role of bid in ischemic acute kidney injury. Ren Fail. 2007;29:935–940. doi: 10.1080/08860220701641165. [DOI] [PubMed] [Google Scholar]
- 13.Devarajan P. Cellular and molecular derangements in acute tubular necrosis. Curr Opin Pediatr. 2005;17:193–199. doi: 10.1097/01.mop.0000152620.59425.eb. [DOI] [PubMed] [Google Scholar]
- 14.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto K, Tomita N, Yoshimura S, et al. Hypoxia-induced renal epithelial cell death through caspase-dependent pathway: role of Bcl-2, Bcl-xL and Bax in tubular injury. Int J Mol Med. 2004;14:633–640. [PubMed] [Google Scholar]
- 16.Saikumar P, Dong Z, Patel Y, et al. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene. 1998;17:3401–3415. doi: 10.1038/sj.onc.1202590. [DOI] [PubMed] [Google Scholar]
- 17.Brooks C, Cho SG, Wang CY, et al. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol. 300:C447–455. doi: 10.1152/ajpcell.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks C, Wei Q, Cho SG, et al. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks C, Wei Q, Feng L, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castaneda MP, Swiatecka-Urban A, Mitsnefes MM, et al. Activation of mitochondrial apoptotic pathways in human renal allografts after ischemiareperfusion injury. Transplantation. 2003;76:50–54. doi: 10.1097/01.TP.0000069835.95442.9F. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi O, Fisher J, Suh H, et al. Essential role of BAX, BAK in B cell homeostasis and prevention of autoimmune disease. Proc Natl Acad Sci U S A. 2005;102:11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80:29–40. doi: 10.1038/ki.2011.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pabla N, Wei Q, Dong Z. Essentials of Apoptosis: A Guide for Basic and Clinical Research. 2nd editon 2009. Apoptosis in acute kidney injury; pp. 565–579. [Google Scholar]
- 25.Padanilam BJ. Cell death induced by acute renal injury: a perspective on the contributions of apoptosis and necrosis. Am J Physiol Renal Physiol. 2003;284:F608–627. doi: 10.1152/ajprenal.00284.2002. [DOI] [PubMed] [Google Scholar]
- 26.Kelly KJ, Plotkin Z, Vulgamott SL, et al. P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. J Am Soc Nephrol. 2003;14:128–138. doi: 10.1097/01.asn.0000040596.23073.01. [DOI] [PubMed] [Google Scholar]
- 27.Knudson CM, Tung KS, Tourtellotte WG, et al. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 28.Wei Q, Dong G, Franklin J, et al. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007;72:53–62. doi: 10.1038/sj.ki.5002256. [DOI] [PubMed] [Google Scholar]
- 29.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7:189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 31.Portilla D, Okusa MD. T cells and T-cell receptors in acute renal failure. Kidney Int. 2006;69:208–210. doi: 10.1038/sj.ki.5000128. [DOI] [PubMed] [Google Scholar]
- 32.Jang HR, Gandolfo MT, Ko GJ, et al. B cells limit repair after ischemic acute kidney injury. J Am Soc Nephrol. 21:654–665. doi: 10.1681/ASN.2009020182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Renner B, Strassheim D, Amura CR, et al. B cell subsets contribute to renal injury and renal protection after ischemia/reperfusion. J Immunol. 185:4393–4400. doi: 10.4049/jimmunol.0903239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burne-Taney MJ, Ascon DB, Daniels F, et al. B cell deficiency confers protection from renal ischemia reperfusion injury. J Immunol. 2003;171:3210–3215. doi: 10.4049/jimmunol.171.6.3210. [DOI] [PubMed] [Google Scholar]
- 35.Wei Q, Yin XM, Wang MH, et al. Bid deficiency ameliorates ischemic renal failure and delays animal death in C57BL/6 mice. Am J Physiol Renal Physiol. 2006;290:F35–42. doi: 10.1152/ajprenal.00184.2005. [DOI] [PubMed] [Google Scholar]
- 36.Wu H, Ma J, Wang P, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 21:1878–1890. doi: 10.1681/ASN.2009101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujimoto K, Hosotani R, Wada M, et al. Ischemia-reperfusion injury on the pancreas in rats: identification of acinar cell apoptosis. J Surg Res. 1997;71:127–136. doi: 10.1006/jsre.1997.5151. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S, Allen DA, Kieswich JE, et al. Dexamethasone ameliorates renal ischemia-reperfusion injury. J Am Soc Nephrol. 2009;20:2412–2425. doi: 10.1681/ASN.2008080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei Q, Bhatt K, He HZ, et al. Targeted deletion of Dicer from proximal tubules protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 21:756–761. doi: 10.1681/ASN.2009070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly KJ, Plotkin Z, Dagher PC. Guanosine supplementation reduces apoptosis and protects renal function in the setting of ischemic injury. J Clin Invest. 2001;108:1291–1298. doi: 10.1172/JCI13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whelan RS, Konstantinidis K, Wei AC, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waxman AB, Kolliputi N. IL-6 protects against hyperoxia-induced mitochondrial damage via Bcl-2-induced Bak interactions with mitofusins. Am J Respir Cell Mol Biol. 2009;41:385–396. doi: 10.1165/rcmb.2008-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei Q, Wang MH, Dong Z. Differential gender differences in ischemic and nephrotoxic acute renal failure. Am J Nephrol. 2005;25:491–499. doi: 10.1159/000088171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.