

Abstract
In the last decade, islet inflammation has emerged as a contributor to the loss of functional β cell mass in both type 1 (T1D) and type 2 diabetes (T2D). Evidence supports that over-nutrition and insulin resistance result in the production of proinflammatory mediators by β cells. In addition to compromising β cell function and survival, cytokines may recruit macrophages into islets, thus augmenting inflammation. Limited, but intriguing, data implies a role of adaptive immune response in islet dysfunction in T2D. Clinical trials validated anti-inflammatory therapies in T2D, while immune therapy for T1D remains challenging. Further research is required to improve our understanding of islet inflammatory pathways, and to identify more effective therapeutic targets for T1D and T2D.
Keywords: β cell, adipocyte, cytokine, immune cells, obesity, clinical trials
Islet inflammation: an emerging and unifying target for diabetes treatment
The current epidemic of T2D is closely associated with increases in obesity [1]. Excessive energy balance results in insulin resistance that is compensated for by increasing insulin secretion. However, insufficient compensation results in T2D, which is characterized by the reduction in islet mass and function. In recent years, overwhelming evidence defines insulin resistance as a state of chronic inflammation involving both innate and adaptive immune responses [1]. Although the presence of islet inflammation is acknowledged for autoimmune destruction of β cells in T1D, new data implicates overlapping pathogenesis between T1D and T2D. Epidemiologic studies suggest that obesity modifies the risk of T1D development [2, 3]. Importantly, small but seminal human studies have also provided evidences that anti-inflammatory therapy can improve glycemia and β cell function in T2D [4, 5]. Here, we focus on recent discoveries (past five years) to discuss the contribution of inflammatory pathways to islet dysfunction in T2D, and to provide updates on the pathogenesis of T1D.
What triggers inflammation in islets under insulin resistance?
Ample evidence from rodent and human studies indicates that in obesity, adipose tissue (AT) inflammation is a major source of pro-inflammatory mediators, and a primary response to excessive caloric intake. AT contributes to inflammation in obesity by means of increased mass, modified adipocyte phenotype, and increased infiltration of immune cells, which affects islet function through humoral and neuronal pathways [1, 6, 7]. In addition, it is noteworthy that pancreatic islets are under similar stress as adipocytes in T2D. The chronic inflammatory state of T2D is reflected in the elevation of circulatory cytokines that potentially affect islets as well as adipocytes [6, 8]. Both islets and adipocytes are exposed to excess glucose and lipids, especially free fatty acids (FFA). Over-nutrition forces adipose tissue to remodel and accommodate enlarged adipocytes, which results in endoplasmic reticulum (ER) stress, hypoxia, and mechanical stresses [9–11]. Under insulin resistance, insulin production increases to meet the high demand, resulting in the expansion of islet mass [12]. Recent findings revealed that obesity is associated with the activation of inflammatory pathway in the hypothalamus, which may alter functions of AT and islets through neuronal regulation [13]. Considering the multiple stressors potentially shared by AT and islets, it is plausible that islets exist also in a chronic inflammatory state, in T2D.
Adipose tissue dysfunction in obesity: a contributor to β cell inflammation in T2D?
The relationship between the pancreatic islet and AT was thought to be unidirectional, by placing insulin secretion as the major determinant of adipocyte glucose uptake and triglyceride storage. However, several recent studies suggest that insulin resistance in AT significantly contributes to β cell failure, through altered secretion of humoral factors from adipocytes and signals from the adipocyte sensory nerve (Figure. 1) [6, 7]. Of particular interest are adipocytokines that are uniquely produced by adipocytes, such as leptin, adiponectin, omentin, resistin, and visfatin, which may contribute to β cell dysfunction during insulin resistance (Box 1). Circulating cytokines may also connect AT inflammation to β cell dysfunction. Overnight exposure of mouse islets to tumor necrosis factor-alpha (TNFα), Interleukin beta (IL-1β), plus Interferon-gamma (IFNγ), at levels comparable to those seen in human obesity, disrupts the regulation of intracellular calcium [8]. Although glucose stimulated insulin secretion (GSIS) was maintained in this study, circulating cytokines might contribute to islet dysfunction after a prolonged period of exposure and when combined with other stresses [8]. TNFα, a cytokine implicated in insulin resistance, reportedly increased islet amyloid polypeptide (IAPP, amylin) expression in β cells with no concurrent expression of proinsulin. This may lead to amyloid production and β cell death [14]. Recent findings showed that the enzyme dipeptidyl peptidase-4 (DPPIV) is secreted by human adipocytes, and therefore may reduce the half-life of DPPIV substrate glucagon-like peptide-1 (GLP-1) with important implications on the insulinotropic effects of this gut peptide on the β cells [15]. Although it is not clear if obesity is associated with increased levels of DPPIV, inhibition of the latter by sitagliptin in a rodent model of obesity and insulin resistance reduced inflammatory cytokine production both in islets and in AT, and improved glucose-stimulated insulin secretion (GSIS) in islets in vitro [16]. Collectively, dysfunctional AT in obesity produces cytokines and peptides that affect islet health and potentially contribute to islet inflammation in T2D.
Figure 1. Dysfunctional adipose tissue in obesity drives β cell inflammation and T2D.
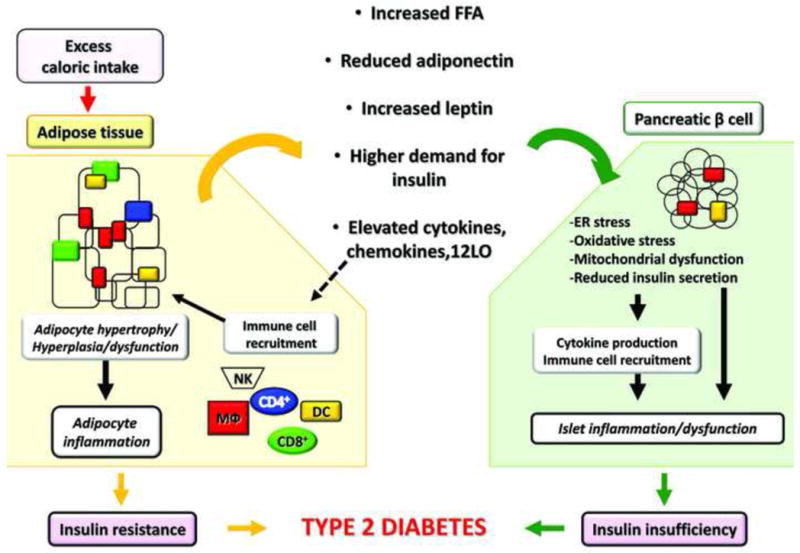
Excess caloric intake progressively induces weight gain and fat accretion leading to obesity. Adipose tissue (AT) is responsible for storage of ~90% of the FFA load following feeding, and therefore, substantial adipocyte remodeling involving primarily hypertrophy and some degree of hyperplasia occurs to accommodate the increasing demand for triglyceride storage. Hypertrophic adipocytes have significantly reduced response to insulin and become progressively more lipolytic liberating excessive FFA. Gene expression of inflammatory proteins and peptides increases resulting in elevated production of cytokines, chemokines, and other inflammatory mediators, such as 12LO. The latter, in turn, contributes to immune cell recruitment and activation within AT, including T cells (CD4+ and CD8+), MΦ, natural killer cells (NK), and dendritic cells (DC). Once in AT, immune cells are an additional source of pro-inflammatory mediators. The array of the adipokines secreted by the adipocytes also changes with increased production of leptin and reduced adiponectin. All of these circulating factors act in an endocrine manner and induce dysfunction in the pancreatic β cells. Elevated FFAs induce lipotoxicity, oxidative stress, mitochondrial dysfunction, and ER stress in the β cells; elevated leptin, coupled with leptin resistance in β cells, may contribute to reduced insulin secretion. Insulin secretion is further reduced by inflammatory mediators originating from either AT or locally produced by the islets and by the infiltrating immune cells. Adiponectin has beneficial effects via AdipoR1 and 2 receptors, which reduce oxidative stress and ER stress in the β cell. Reduced adiponectin expression in obesity minimizes its beneficial effects. Higher demand for insulin due to insulin resistance negatively affects β cells through ER stress and islet expansion. The recruitment of MΦ is also thought to amplify inflammation and islet dysfunction in obesity. It is yet to be determined whether other immune cells are involved in islet inflammation in T2D. The overall inflammatory responses in β cells in obesity contribute to reduced functional β cell mass leading to T2D, when combined with systemic insulin resistance.
Box 1. Adipocytokines and β-cell dysfunction.
Leptin is a key regulator of glucose homeostasis via central and peripheral actions. In obesity, leptin production increases due to adipose mass expansion, however peripheral and central leptin action is attenuated due to leptin resistance [80]. Human and rodent β cells express leptin receptors and leptin resistance in the β cell may lead to β cell dysfunction in T2D associated with obesity [6, 81]. However, the actions of leptin on β cells are complex. In humans, leptin appears to have an anti-proliferative effect on β cells through the PI3K pathway, and inhibits phosphatase with tensin homolog (PTEN) leading to increased activation of the KATP channels and inhibition of GSIS [82]. Also, in human islets, leptin exerted a U-shape response on insulin secretion, with lower concentrations inhibiting insulin release and higher concentrations stimulating it [6]. Therefore, it is possible that in the early stages of insulin resistance, an increase in circulating leptin without a yet compromised signaling mechanism may be responsible for the compensatory hyperinsulinemia. However, in severe obesity characterized by impaired leptin signaling, the latter may reduce insulin secretion, contributing to hyperglycemia and progression to T2D. Adiponectin, another adipocyte derived cytokine, improves insulin sensitivity and vascular function, and its secretion by hypertrophic adipocytes is reduced in obesity. In vivo mouse studies indicate that many of the adiponectin actions may be mediated via generation of sphingosine 1-phosphate [83]. Adiponectin receptors are expressed in primary and clonal β cells and signal through AMPK, PPARγ, PPARα, and p38MAPK [84]. Adiponectin has multiple roles in β cell function and survival. The increase of adiponectin in diabetic Goto-Kakizaki (GK) rats following gastric bypass was associated with a reduction of β cell apoptosis [85] as was in the clonal β cell lines BRIN, BD11, and MIN-1 in response to in vitro PA treatment [86]. In MIN6 cells, adiponectin caused a significant increase in insulin content and secretion through PPARγ in addition to a proliferative effect independent of PPARγ [87]. Therefore, the combined anti-apoptotic and proliferative effects of adiponectin may aid to preserve β cell mass and function, and reduction of adiponectin in obesity and T2D may negate these beneficial effects.
Does “Lipotoxicity” cause islet inflammation?
The increase in serum FFA is a risk factor for the future development of T2D and, is commonly seen in T2D [17]. In addition, fat infiltration of the pancreas and intracellular accumulation of lipids in β cells seen in obesity may increase the local concentration of FFA in islets [17]. The adverse consequences from excessive lipid accumulation, coined “lipotoxicity”, might connect obesity and islet demise in T2D [17]. In isolated islets, the negative effects of FFAs on insulin secretion are most prominent with long chain saturated fatty acids, such as palmitic acid (PA) [17]. PA has been shown to cause ER stress, oxidative stress, ceramide production, and jun N-terminal kinase (JNK) activation, which are all known to provoke inflammatory responses (Figure. 2) [17]. PA induced IL-1β, TNFα, chemokine (C-C motif) ligand 2 (CCL2), IL-6, Chemokine (C-X-C motif) ligand 1 (CXCL1), and IL-8 production, and activated nuclear factor-kappaB (NF-κB) in human islets [18]. The infusion of ethyl-palmitate in mice induced CCL2 and CXCL10 production through the toll like receptor 4 (TLR4)-Myd88 pathway in islets, which supports recruitment of pro-inflammatory M1-type macrophages (MΦ) [19]. However, extrapolation of studies performed with PA to human pathophysiology requires caution. The exposure of cells to pure PA does not necessarily mirror the hyperlipidemia generated in vivo from the mixture of FFA including unsaturated fatty acids such as oleic acid (OA) [20]. It is well known that adverse effects of PA can be substantially lessened when PA is given with OA. Also, it is not clear whether lipid infusion impairs insulin secretion in human studies [17]. While the adverse effects of FFA on islets in an experimental setting are well established, their true contribution to islet demise, in human T2D, merits further clarification.
Figure 2. Major inflammatory mediators and signaling pathways involved in β-cell demise.
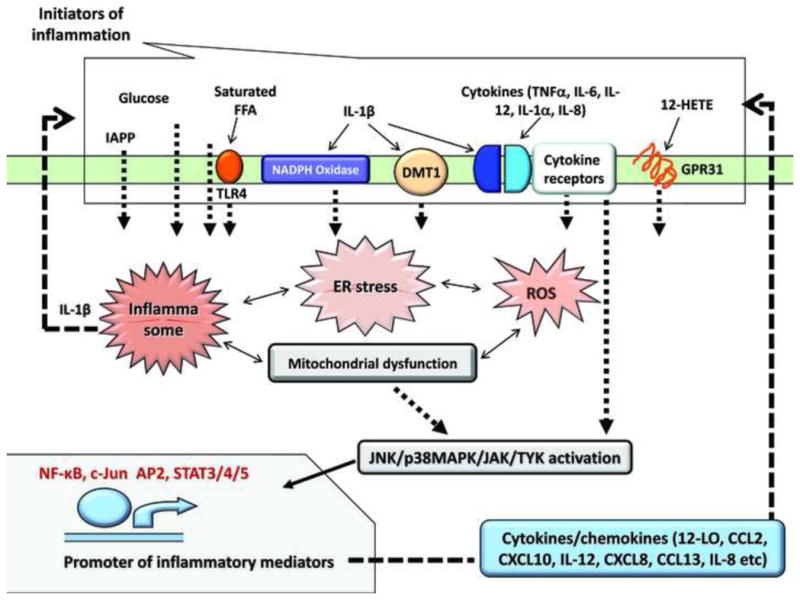
The major triggers of inflammation in islets are shared with the ones that induce inflammation in AT, liver and muscle: excess saturated FFA, lipid mediators such as lipoxygenase products (12-HETE), increased glucose, and pro-inflammatory cytokines and chemokines. IAPP is so far acknowledged as an islet specific inflammatory mediator, and is produced and secreted solely by the β cell. Inflammatory mediators activate cellular stress: saturated FFA and glucose increase ER stress (via CHOP/IRE α1). IL-1β increases the expression of NOX1/4 activating NADPH oxidase and DMT1, which increases iron transport; both lead to an increase in ROS, and together with IAPP, contribute to increased oxidative stress. 12(S)-HETE, produced by 12LO activates the G-protein coupled receptor GPR31, and induces downstream inflammatory signals. Cytokines produced locally or from circulation also activate inflammatory pathways via specific cytokine receptors in β cells. In response to cytokine receptors JAK/TYK, p38MAPK and JNK are activated; also JNK is phosphorylated in response to TLR4 activation or IAPP intracellular aggregation; p38MAPK is activated downstream of GPR31; ROS activate JNK, p38MAPK or NF-κB; activation of MyD88/TRIF downstream of TLR4 also leads to NF-κB activation; other transcription factors activated downstream of kinases are c-Jun, AP2 (downstream of JNK) and STAT3/4/5/(downstream of p38MAPK and JAK/Tyk). A positive feedback loop is operational in the β cell in which inflammatory mediators extrinsic to β cells converge in activating transcription of various cytokines, chemokines, and other inflammatory mediators that in turn act in an autocrine manner to exacerbate the inflammatory response. Also, intracellularly, there is extensive cross talk between signaling molecules involved in oxidative stress, ER stress, mitochondrial dysfunction, and inflammatory responses.
Does oxidative stress induce islet inflammation?
Pancreatic islets are vulnerable to oxidative stress due to low antioxidant defense. The elevated glucose and FFA levels, commonly seen in T2D, are known to cause oxidative stress [17, 21]. Microarray analysis of human islets obtained by laser microdissection showed differential regulation of oxidative stress genes in T2D donors, compared with controls, implicating oxidative stress in islet dysfunction [22]. Oxidative stress provokes inflammatory responses through activation of JNK, NF-κB, and p38MAPK (Figure 2) [23]. Recently, divalent metal transporter 1 (DMT1) was proposed as an additional pathway by which IL-1β promotes oxidative stress in islets [24]. DMT1 is an iron transporter that increases cellular iron uptake and iron-catalyzed formation of reactive oxygen species (ROS). IL-1β treatment up-regulates DMT1 in islets, while the deletion of DMT1 protectsβ cells from IL-1β-induced apoptosis. Moreover DMT1 deficient mice are protected against low dose streptozotocin induced diabetes (T1D model) and high fat diet induced diabetes (T2D model) [24]. Phagocyte-like NADPH oxidase (NOX) and 12 lipoxygenase (12LO) are additional potential mediators of oxidative stress in islets upon cytokine exposure [25].
Evidence supporting ER stress as an inducer of islet inflammation
The role of ER stress in β cell dysfunction in T2D (Box 2) is well recognized. Also, ER stress provokes cellular inflammatory pathways, which are most likely operational in β cells (Box 2) [26, 27]. In human islets, chemically-induced ER stress by cyclopiazonic acid increased cytokine expression and NF-κB activation [18]. Recently, thioredoxin interacting protein (TXNIP) has been shown to increase rapidly in islets during ER stress provoked by thapsigargin (depletes calcium store in ER). TXNIP up-regulation resulted in IL-1β and IL-6 production through the initiation of the inflammasome [28, 29]. TXNIP is also implicated in the induction of oxidative stress through its interaction with thioredoxin, a critical redox protein in cells. TXNIP expression is regulated by glucose in human islets and plays a role in glucose-induced β cell death [30]. Therefore, TXNIP may be a key transducer of glucotoxicity, oxidative stress, and ER stress, feeding into various inflammatory pathways in islets (Figure 2).
Box 2. ER stress and inflammatory responses.
Studies of human pancreatic sections imply that nuclear CCAAT/-enhancer-binding protein homologous protein (CHOP) and several ER stress proteins play a crucial role in T2D (reviewed in [18, 27]). Islet β cells are highly specialized in the production of insulin, which constitutes 30 to 50% of total protein. ER stress plays major role in islet dysfunction in humans harboring rare mutations in the insulin gene (mutant Ins-gene-induced diabetes of youth, MIDY). Also mutations in and ER stress related genes, result in clinical syndromes namely the Wolfran syndrome (with mutations in WFS1, a protein that regulates Ca2+ store in ER) and the Wolcott-Ralison syndrome (with mutations in the pancreatic eukaryotic translation initiation factor 2-alpha kinase 3 (EIF2AK3) and infancy-onset diabetes). In T2D, β cells are forced to increase insulin secretion under insulin resistance and hyperglycemia, which may create ER stress in the absence of rare mutations. Also, insulin misfolding may be more prevalent than previously considered. The CDK5 regulatory subunit associated protein 1-like 1 (CDKAL1) variant is one of T2D susceptibility loci identified from genome wide association studies (GWAS) and has been shown to increase insulin misfolding [88]. CDKAL1 is a methylthiotransferase critical for accurate translation of AAA and AAG codons to lysine, and misreading of lysine in proinsulin results in ER stress due to mutated insulin [88]. In addition to protein translation load, nutritional overload of glucose and fatty acid causes ER stress reflecting the important role of ER as a nutritional sensor [17]. Once ER stress is provoked, several pathways connect ER stress to inflammation [26]. Three ER-localized proteins, inositol-requiring 1α (IRE1α), double-stranded RNA-dependent protein kinase (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6), are well established sensors of ER stress and initiate signaling cascades in response to ER stress. PERK and IRE1α activate NF-κB, resulting in proinflammatory cytokine and chemokine production. NF-κB is bound with the NF-κB inhibitor IκB and sequestrated in cytoplasm at baseline. PERK suppresses the translation of IκB through eIF2α, while IRE1α promotes the degradation of IκB through the activation of its kinase, IKK. IRE1α also activates JNK, another signaling pathway with established role in the proinflammatory responses. Since cytokines also provoke ER stress in β cells, ER stress may play a critical role in the pathogenesis of T1D as well [27].
Is IAPP a unique mediator of islet inflammation?
In T2D, one can see accumulation of amyloid in islets. There continues to be a debate as to whether IAPP is responsible for the decline in functional β cell mass, or simply a marker of β cell demise [31]. The active role of IAPP was previously proposed based on the observation that mice overexpressing human IAPP show activation of the JNK pathway and β cell apoptosis [32]. Recently, studies showed that IAPP aggregates may actively contribute to islet dysfunction in T2D by provoking an inflammatory response [31, 33]. Aggregates of IAPP activated NLRP3-containing inflammasomes in bone marrow-derived dendritic cells resulting in IL-1β production in a MyD88 dependent manner [31, 33]. Furthermore, β cells directly respond to IAPP aggregates by producing the chemokine CCL2, that attracts MΦ [31]. Islets expressing human IAPP transplanted into diabetic mice rapidly develop amyloid deposition in islets along with MΦ accumulation. Moreover, an IL-1β antagonist can reduce amyloid deposition, MΦ recruitment, and hyperglycemia [31]. It still remains to be determined whether IAPP serves as a unique antigen for an adaptive immune response in T2D.
Does the gut contribute to islet inflammation?
There exists a heightened appreciation of the connection between the gut and the development of T1D and T2D. Gut microbes manifest unique changes in obesity that are associated with the increase in circulatory lipopolysaccharide (LPS) [34]. LPS mediates reduction in insulin gene expression and insulin secretion in human, rat, and mouse islets via activation of inflammatory pathway involving TLR4 and NF-κB. Thus, LPS may serve as one contributing factor for islet dysfunction in T2D [35]. A recent review suggested that gut permeability in the NOD model of TID, is increased in the presence of pathogenic bacteria, and this leads to the acceleration of insulitis [36]. Further evidence suggests that colonization of the gut by specific bacterial species alters the development of autoimmunity in NOD mice and can modify the cytokine and chemokine profile[36]. However, evidence that gut microbes contribute to islet demise is circumstantial and lacks validation in humans. It is possible that advances in this rapidly moving area may provide useful information in the understanding of islet inflammation.
The amplification of inflammatory responses in islets
Several pathways exist that result in inflammatory responses in islets. Once inflammatory mediators are produced in islets, they can cause ER stress, oxidative stress, mitochondrial dysfunction, apoptosis, and other adverse reactions that amplify the downward spiral of cellular dysfunction (Figure. 2). The interactive nature of these pathways are exemplified in the study showing that mild ER stress, not sufficient to cause apoptosis, increases the sensitivity of islet cells to IL-1β along with up-regulation of inflammatory mediators CCL2, CXCL1, iNOS and Fas [37]. The amplification of inflammatory responses likely plays a role in the loss of β cells in both T1D and T2D, and thus potentially serves as a unifying therapeutic target.
Who are the key players in islet inflammation?
Current knowledge of T1D pathogenesis
Epidemiologic studies have shown that the prevalence of T1D is increasing, and that obesity is a risk factor for T1D [2, 3]. Thus, autoimmune processes responsible for T1D may interact with risk factors associated with T2D. Some argue that aggressive insulitis and relentless β cell loss seen in T1D are distinct from the slow decline of β cell mass seen in T2D, which is further discussed in Box 3. However, there is still debate over which might occur first, unchecked β cell apoptosis leading to an autoimmune response, or the autoimmune response leading to an increase in β cell apoptosis. There is evidence that, in the absence of inflammation, the level of β cell apoptosis does not contribute to diabetes initiation [38]. Others have shown that β cell apoptosis occurs as a result of IFNγ and TNFα mediated activation of STAT1 signaling and subsequent Bim activation, a BH3-only activator protein crucial to the apoptosis cascade in β cells [39]. While it is clear that autoimmunity is necessary for disease progression, the initiation of insulitis in T1D may be affected by factors also involved in T2D (Box 3). For the initiation of islet inflammation, cells must first infiltrate the peri-islet capsule that innately protects islets [40], although autoimmune infiltration is able to overcome this protection. It has been shown that heparan sulfate protects pancreatic islets from ROS, but heparanase from mononuclear cells can negate this protection [41]. TLRs are generally responsible for innate immune recognition of viral and bacterial infections, thereby promoting further immune responses. Several papers have shown a role for TLR2 [42], TLR4 [43], and TLR9 [44] in the development of T1D, and there is evidence of dysregulated TLR signaling in pre-diabetic, autoantibody positive patients, when compared to normal healthy subjects [45]. Subsequent to TLR signaling, it is clear that chemokines and cytokines can contribute to β cell decline. Normal human islets treated with IL-1β, IFNγ, and TNFα show significant up regulation in the expression of CCL5, CCL8, CCL22, CXCL9, CXCL10, and CX3CL1, as assayed by microarray analysis, qRT-PCR analysis, and immunofluorescent staining [46]. Studies of chemokine expression in T1D patients revealed a pronounced expression of CXCL10 in diabetics versus normal controls [46]; however, this increase may not translate to elevated serum levels [45]. The role of IL-17 in the development of T1D remains unclear. Recent studies suggest that IL-17 is not necessary for T1D development [47, 48], but that CD8+, IL-17-producing T cells can potentiate T1D pathogenesis [49]. Th1 cytokines, such as IL-12, are implicated in autoimmunity and may play a role in T1D and well as T2D [50]. It is clear that T1D development and progression are determined by multiple factors, including β cell inflammation.
Box 3. T1D and T2D: Commonalities and differences.
There is no doubt that the autoimmune destruction of β cells is a central pathology of T1D. However, as the obesity epidemic has seen the simultaneous increase in both T1D and T2D, several key observations suggest the need to reassess the distinction between T1D and T2D. Systemic review of nine clinical studies supports the association of obesity and subsequent T1D development [3]. Both auto-antibodies and islet reactive T cells are detected in blood of some individuals with T2D [89]. As discussed above, accumulating evidence supports the involvement of innate immunity in T2D, likely triggered by AT inflammation, lipotoxicity, ER stress, oxidative stress, and other pathways activated by obesity and insulin resistance. Furthermore, IL-12 is shown to have a role in the pathogenesis of both TID and T2D [50]. It is plausible that these insults on islets associated with obesity, may exacerbate insulitis and the autoimmune process in T1D. A recent study reported that NOD mice show signs of ER stress and NF-κB activation at a pre-diabetic stage along with insulitis, indicating a close relationship between inflammatory pathways and other cell distress pathways [90]. Anti-inflammatory therapies including IL-1β, DMT1, and 12LO inhibitors have shown efficacy in animal models of T1D and T2D, supporting that anti-inflammatory therapy may stand as a unifying target for diabetes [24, 52, 72, 91, 92]. The pioneering work by Brooks-Worrell, et al. further present the possibility that adaptive immunity may be involved in large population of diabetes including clinically labeled T2D, and that islet reactive T cells in peripheral blood may serve as a new biomarker for immunomodulatory therapy for both T1D and T2D [89, 93]. However, one needs to note that there remain differences between the two forms of diabetes including typical clinical presentation, rate of β cell destruction, profiles of autoantibodies, and histology of islets [18, 93, 94]. Indeed, neither T2D risk loci identified by GWAS, nor body weight led to T1D progression, at least in the first degree relatives of T1D [95]. Therefore, efforts should continue to critically evaluate common features and differences between T1D and T2D. These results should provide a deeper insight into the complex pathogenesis and new ways to halt β cell demise in both forms of diabetes.
Humoral factors implicated in the islet inflammation in T2D
The microarray study of laser-captured microdissected islets from human T2D pancreata showed statistically significant increases in IL-8, CCL2, and CCL13, while showing a trend for increased expression for IL-1β, IL-6, and CXCL1 [18]. IL-12 and CXCL10 are other proinflammatory cytokines increased in islets isolated from T2D donors compared with non-diabetic donors [50, 51]. One early instigator of inflammation in the islets is 12LO, the enzyme that converts arachidonic acid to 12-HPETE, which is subsequently converted to the highly pro-inflammatory 12(S)-HETE by glutathione peroxidase. 12(S)-HETE can influence downstream production of pro-inflammatory cytokines, including IL-12 and IFN-γ [52]. As 12LO is produced in both islets and MΦ, its role is becoming appreciated in the pathogenesis of both T1 and T2D [52, 53].
What are the possible consequences of cytokine production?
Cytokines produced in islets impair islet functions through oxidative stress, ER stress, and downstream activation of NF-κB, JNK, p38MAPK, p53, STAT4, intrinsic mitochondrial death pathway, and others (Figure 2) [18, 50, 54–56]. Secreted frizzled-related protein 4 (SFRP4) is released from islets upon exposure to IL-1β, and is up-regulated in human islets from T2D donors [57]. Interestingly, SFRP4 impairs insulin secretion and may be one key contributor to islet dysfunction in T2D [57]. MicroRNAs are also potential mediators of islet dysfunction. MiR-21, mir-34a, and miR-146a are increased after IL-1β and TNFα treatment, in human islets, and their reduction confers protection against cytokine-induced impairment of GSIS [58]. One of the gene targets transcriptionally regulated via NF- κB activation is inducible nitric oxide synthase (Nos2 or iNOS). A recent study identified that posttranslational hypusination (addition of the amino acid hypusine) of eukaryotic translation initiation factor 5A (eIF5A) regulates Nos2 expression upon cytokine stimulation in insulin secreting cells, and the inhibition of hypusination protects islets from cytokine mediated islet demise ex vivo and in vivo [59]. Furthermore, cytokines can recruit immune cells that are a critical component for the inflammatory process. CCL2, a chemoattractant associated with T2D risk, is increased in human T2D islets, and is induced by PA in islets [18, 19]. However CCL2 may not be sufficient to recruit MΦ at the levels commonly seen in islets. Levels in the order of 100 fold above normal were required to generate insulitis and hyperglycemia in transgenic mice overexpressing CCL2 [60]. To further complicate the matter, CCL2 overexpression protected NOD mice against the development of diabetes by increasing tolerogenic CD11b+CD11c+ dendritic cells [61]. CXCL8 and CXCL10 are other chemokines produced by islet cells that are chemotactic for neutrophils, MΦ, and monocytes [19, 54, 55]. Thus a combination of several chemokines together may be at work during the development of islet inflammation.
Are there immune cells in T2D islets and do they contribute to β cell demise?
The presence and functional involvement of immune cells in islets provides strong evidence for inflammation as a contributing factor to islet dysfunction. Islets harbor residential MΦ and monocyte populations [55]. Histological studies show significant increases of CD68+ MΦ to ~1.5% of islets in T2D, supporting the notion that several innate immune response players are associated with T2D [55]. The increase in MΦis also reported in several animal models of diabetes through histological and flow cytometric analyses [19, 55, 62]. The functional contribution of MΦ was tested by depleting MΦ systemically using clodronate liposomes in diabetic KKAy mice on high fat diet, and in leptin receptor-deficient db/db mice [19]. The treatment reduced M1-type MΦ in islets, restored the expression levels of insulin and pancreatic and duodenal homeobox 1 (Pdx1), and improved an index of insulin secretion [19]. Further studies are required to address whether the positive effects on islets are due to removal of islet MΦ, or also depend on MΦ in other tissues, including AT. Dendritic cells and lymphocytes residing in islets are believed to play critical roles in adaptive immune responses that result in autoimmune destruction of β cells in T1D. At least in histological studies, neither T cells nor B cells are prominent in islets of human T2D or animal models of T2D [55, 62]. However, peripheral blood T cells from 42% of clinically defined T2D patients react against islet extracts, despite of the lack of islet antibodies in these patients, suggesting that adaptive immunity may be involved at a significant frequency in clinically defined T2D (Box 3) [63].
Emerging therapeutics that target inflammatory pathways
T1D therapy
As summarized in recent reviews, immune modulatory therapy has been pursued for T1D since cyclosporine reduced the decline of insulin production in T1D in 1980s. However, this approach has yet to offer clinically durable safety and efficacy [64, 65]. Neither preventing B cell responses by using an antibody against the B cell surface protein CD20, Rituximab [66], nor blocking IL-1β responses via Anakinra [64, 67] or Canakinumab showed clear responses. (http://professional.diabetes.org/presentations_details.aspx?session=3988 presented at the 72nd scientific session, American Diabetes Association) have long-lasting benefits on recently diagnosed patients. There is a possibility that a combination of anti-CD3 paired with anti-IL-1β blockade will provide more beneficial results [68]. Another new target in the horizon may be anti-serpinB13 antibodies, as increases in these antibodies lead to decreases in anti-insulin autoantibodies in the NOD model [69]. Furthermore, a deficiency in anti-serpinB13 antibodies correlates with early onset of T1D in humans [69]. As the initiating factors of T1D are not yet clear, we are left to try to modify downstream targets, rather than having a clear means of prevention. Considering the complex pathogenesis of T1D, combination therapy rather than targeting single cytokine or cell type may be needed [64, 65].
IL-1β
IL-1β has emerged recently as a master cytokine in islets that triggers and amplifies inflammatory responses (Figure 2) [54, 55]. Recent studies of human T2D islets using gene co-expression analysis also identified IL-1 binding genes and IL-1 receptor activity as gene clusters with strong association to T2D [57]. Discussed in Box 4, clinical trials have shown 0.5~1% reduction of A1c with increase in the indexes of insulin secretion. In addition to the cost and the size of trials reporting the efficacy, there are several reservations for IL-1β-based therapy. In human islets, PA induces IL-1β, TNFα, CCL2, IL-6, CXCL1, and IL-8 production. IL-1β antagonism was sufficient to abolish the up-regulation of many cytokines upon PA treatment but was unable to prevent apoptosis [18]. Low dose of IL-1β for the limited period was shown to increase β cell proliferation and insulin secretion [54]. Reflecting the role of IL-1β as the upstream regulator of cytokine production in islets, its blockade also reduces IL-6 production in islets. However, IL-6 may support insulin secretion and islet function by augmenting GLP-1 secretion at least when tested in a non-inflammatory condition [70]. Therefore, a more selective treatment may offer better responses.
Box 4. Clinical studies targeting inflammatory pathway in islets.
IL-1β based studies
The landmark study in 2007 showed that IL-1α and IL-1β antagonism by the naturally occurring antagonist IL-1ra (Anakinra) improved glycemic control (0.46% drop in A1c) of uncontrolled T2D (pretreatment A1c 8.7%) in a 13-week placebo-controlled, double-blinded, randomized trial [4]. The improvement in glycemic control is likely due to better insulin secretion, since C-peptide was increased and the proinsulin/insulin ratio was reduced with little changes in insulin sensitivity [54, 55]. IL-1β antibodies (XOMA-052, Canakinamab) have a longer half-life compared with recombinant IL-1ra, and block IL-1β without blocking IL-1α. XOMA-052 administered to T2D (A1c ~9%) reduced A1c by 0.85% with an increase in both C-peptide and insulin sensitivity after 3 month [96]. Diacerin, a medication marketed for osteoarthritis in some countries, reduces IL-1β production. In a randomized double-blinded, placebo-controlled trial, the 2-month treatment of drug naïve T2D with diacerin increased insulin secretion without changes in insulin sensitivity [97]. However, it should be noted that studies published so far include relatively small numbers of subjects suggesting caution in interpretation. Currently the CANakinumab anti-inflammatory Thrombosis Outcomes Study (CANTOS) trial is underway. The study is a multi-national, event-driven, intent-to-treat protocol enrolling 17,200 stable, post-myocardial (MI) subjects with persistent elevation of C-reactive protein (CRP) with primary endpoints of nonfatal MI, nonfatal stroke, and cardiovascular death [98]. Since new onset diabetes will be also monitored, the study may provide valuable information about its efficacy for T2D. Moreover, it is critical to know whether new therapeutics will have cardiovascular benefit for overall mortality and morbidity benefits [98].
Salsalate
TINSAL-T2D is a multi-center randomized clinical trial that tested the efficacy of salsalate for T2D. Stage I results have been published [5]. A stage II trial involving 3.5 gm daily of salsalate in addition to other therapy, up to 26 weeks, is also completed. Preliminary results were presented at the Scientific Meeting of American Diabetes Association in June 2012 (http://www.medscape.com/viewarticle/765478). 286 T2D patients, neither on injectables nor thiazolidinediones, were enrolled in double-blinded, randomized prospective study. There was 0.24% drop in A1c along with 11mg/dl reduction in fasting glucose (both p<0.001). The index of insulin secretion was increased. The hypoglycemic effect of salsalate is potentially underestimated in the study, since the use of other hypoglycemic agents declined in the patients on salsalate. Reduction in neutrophils, but no change in CRP was noted in peripheral blood. A small rise in urinary albumin, LDL, and weight were reported as a potential concern.
Nonacetylated salicylate
This is another class of medications aggressively pursued for its efficacy in T2D treatment [55]. Nonacetylated salicylates, such as salsalate, have very weak activity as cyclooxygenase inhibitors, but they suppress NF-κB action that is not only downstream of IL-1β activation, but also central to cellular inflammatory responses, ER stress, and oxidative stress. This class of medications may also have beneficial metabolic effects through the activation of AMPK [71]. Targeting-INflammation Using SALsalate in Type 2 Diabetes (TINSAL-T2D) is a multi-center randomized trail (Box 4). Preliminary results show that the index of insulin secretion was increased, validating the islet NF-κB pathway as a therapeutic target. Although A1 reduction has been modest, this low cost and orally active compound holds promise, especially if it shows cardioprotective effects, which are being tested in the TINSAL-CVD (cardio vascular disease) trial.
Emerging targeted therapies
12LO as a target
12LO is implicated in insulin resistance and islet dysfunction, as it produces proinflammatory arachidonic acid products, and is up-regulated in islets of both T1D and T2D models [52]. 12LO activity is also increased by hyperglycemia and by proinflammatory cytokines in human islets [52, 53]. Gene deletion and pharmacological suppression of 12LO prevented the development of diabetes in NOD mice (T1D model), Zucker Diabetic Fatty (ZDF) rat (T2D model), and diet induced obese mice (T2D model) [52, 72]. 12LO activation is also implicated in AT inflammation and atherogenesis [52]. Thus, the inhibition of 12LO potentially addresses insulin resistance, β cell dysfunction, and cardiovascular complications simultaneously, making it a promising target for both T1D and T2D. New therapeutic agents are being developed to target these pathways [53].
The role of histone deacetylases
Recently, histone deacetylases (HDAC) I, IIA, IIB, III, and IV have been proposed as a novel target to control inflammatory responses in a wide array of diseases including diabetes [73]. The anti-inflammatory role of HDAC inhibitors is partly explained by NF-κB inhibition, via the acetylation of its p65 subunits. Although its efficacy in human T2D remains to be determined, several lines of evidences support the role of HDAC inhibition in β cell preservation: genetic linkage study revealed that loci in 6q21 associated with both T1D and T2D lie near HDAC2: an HDAC inhibitor was shown to reduce cytokine induced NF-κB activation in β cells and to protect against apoptosis: HDACIIA may aid to expand β cell mass: and an orally active inhibitor against class I and II HDAC, ITF2357, showed efficacy in the prevention of STZ induced diabetes [74]. However, the precise mechanism by which HDACs operate in β cells is complex as might involve multiple potential targets and different specific substrates for each HDAC [75]. Activation of Sirtuin 1 (Sirt1), a NAD+ dependent HDAC class III deacetylase, may play an anti-inflammatory role in islets. Sirt1 expression is reduced in pancreatic islets after cytokine exposure and its overexpression prevents cytokine induced β cell damages through NF-κB [76]. Nicotinamide mononucleotide (NMN), a metabolite that augments sirtuin action, rescues islets from reduced insulin secretion after IL-1 β and TNFα exposure [77]. Further studies that identify targets of each class of HDACs in human islets under inflammatory conditions will aid in therapeutic application of this emerging class of agents.
Other potential agents
Some of the agents clinically available for T2D and metabolic disease potentially improve islet function through anti-inflammatory properties. The FAT-1 transgenic mouse, that expresses the C. elegans fat-1 gene encoding an n-3 fatty acid desaturase that converts n-6 to n-3 fatty acids (which is absent in mammals) shows augmented production of n-3 polyunsaturated fatty acids, is protected against diabetes development after multiple low dose streptozotocin (STZ) injection and displays lower levels of IL-1β, TNFα, NF-κB and 12-HETE [78]. The thiazolidinediones also known as glitazones, an established treatment for T2D and peroxisome proliferator-activated receptor gamma (PPARγ) agonists, were also shown to have anti-inflammatory properties in islets [79].
Concluding remarks
Inflammatory pathways are likely part of a complex, intermingled chain of events that converges in the reduction of functional β cell mass in both T1D and T2D. Recent advances in the field point to substantial overlap in the pathogenesis of β cell loss between T1D and T2D: insulin resistance and obesity may modify T1D development, while signs of adaptive immunity have been documented in some T2D cases. Chronic inflammation also plays a critical role in insulin resistance and macrovascular disease. Thus, the next frontier in the clinical treatment of diabetes will likely involve a targeted approach to block one or more of the most relevant pathways leading to β cell dysfunction with potential cardiovascular benefits.
Outstanding questions.
Is it possible that similar initiating factors can manifest differentially in TID and T2D based upon different interaction sites (i.e., perhaps in T2D initiation of disease takes place in AT, whereas in T1D disease begins in the islets themselves, leading to altered disease progression)?
Obesity increases the risk of T1D, while substantial populations of clinical T2D patients harbor circulatory T cells against islet antigens. Are T1D and T2D two separate entities of the disease, or do they represent a spectrum of one disease with the highest prominence of autoimmune destruction of β cells being T1D and the lowest end being T2D?
Does islet inflammation affect the maintenance of differentiated status of β cells?
Are neutrophils increased at the initiation of islet inflammation? Do M2 MΦ aid in the regression of islet inflammation? Do dendritic cells in T2D islets present islet antigens and initiate adaptive immune response? What are the subtypes and functional status of immune cells involved in T2D islets?
Does the contribution and reversibility of inflammation vary depending on disease duration, the extent of islet inflammation, or the severity of distal inflammation such as AT inflammation? Do these factors predict responses to anti-inflammatory therapy? Will novel inflammatory targets such as HDACs, 12LO, and IL-12 provide additional benefits?
adipocytokines from different AT depots differentially contribute to islet inflammation and dysfunction? What are the signaling and key molecules involved in the developing of leptin resistance in the β cell? Do pro-inflammatory adipocytokines affect the α, δ, ε, and PP cells? If so, are they relevant to β cell demise? Do obesity associated neuronal changes cause inflammatory changes in islets?
What is the translational value of rodent data with focus on inflammatory pathways for the human pathophysiology?
Acknowledgments
We apologize that we were unable to cite many outstanding works performed by investigators in the field due to space limitations. Funding support for the authors include Juvenile Research Foundation grant (Nadler JL), American Diabetes Association (Morris MA), National Institutes of Health (R01-HL112605 to Nadler JL, R01-DK090490 to Imai Y), Ferguson and Iacocca foundation (Nadler J), and Merck (Dobrian, AD).
Glossary
- Adaptive Immune Response
Immune response in which B and T cells develop specific, long-term, protective immunity against a pathogen. After initial adaptive responses, cells are able to respond more quickly and more effectively
- Adipocytokine
Adipose secreted cytokines in responses to internal and external stimuli. Some such as leptin, adiponectin, omentin, resistin and visfatin, are unique to adipocytes and serve as humoral regulators of energy homeostasis
- Autoimmunity
Any number of aberrant immune responses in which the immune system (both innate and adaptive arms) attacks the body leading to a variety of disease pathologies
- Clonal β cells
Sincethe primary culture of β cells usually does not proliferate, several transformed cells are established that acquire clonal expansion capacity while maintaining nutritionally regulated insulin secretion. These include MIN1, MIN6 and βTC-3 cells (mouse), and INS cells (rat). These cell lines are useful research tools, but tend to have reduced insulin gene expression and GSIS compared with islets
- Dendritic Cells
Innate immune cells that function as professional antigen presenting cells for the cells of the adaptive immune response. They present antigens to and activate B and T cells
- Endoplasmic reticulum (ER) stress
The ER is the cellular machinery engages in protein processing, lipid biogenesis, Ca2+ storage, and nutritional sensing. To keep the integrity of the organelle, increased load for peptide synthesis results in unfolded protein response (UPR), an integral pathway aiming to reduce translational load to ER (adaptive UPR). Excessive stress provokes cell death signals including proinflammatory cytokine production (terminal UPR)
- Glucagon-like peptide -1 (GLP-1)/dipeptidyl peptidase-4 (DPP-IV) inhibitor
GLP-1 is a short-lived gut peptide derived from pro-glucagon and secreted from L-cells upon food ingestion to augment insulin secretion (it acts as an incretin hormone). GLP-1 preserves islet mass in animal models. DPP-IV rapidly degrades GLP-1 and DPP-IV inhibitors enhance endogenously secreted GLP-1 actions by increasing half-life, and currently serve as anti-diabetic agents
- Histone deacetylases (HDAC)
are a class of enzymes that maintains lysine residues of histone in deacetylated form and keeps DNA in a compact transcriptionally inactive form. HDACs targets also include a variety of cellular proteins
- Inflammasome
An oligomeric protein complex comprised of caspase 1, NALP, PYCARD, and sometimes caspase 5, which is responsible for the activation of inflammatory processes. This complex is key to the production of IL-1β
- Innate Immune Response
Non-specific and immediate mechanism by which the body defends itself against infection. Innate responders promote the adaptive immune response
- Islet amyloid polypeptide (IAPP)
also called amylin, is a 37 amino acid peptide co-secreted with insulin from β cells, and the major component of the islet amyloid
- Oxidative stress
Oxidative stress results from the production of reactive oxygen species (ROS), molecules with chemically reactive oxygen beyond cells reduction capacity. Excess ROS have deleterious effects on the cells by causing hyperoxidation of DNA, protein, and lipids
- Toll like receptors (TLR)
Pattern-recognition receptors that recognize molecules released from exogenous pathogens and endogenous cell damages. TLR4 recognizes LPS and saturated fatty acid, and plays an important role in adipose inflammation. Myd88 serves as adaptor proteins for intracellular signaling of TLR that includes NF- κB activation
- Th1 Cytokines
IFN-γ and TGF-β are produced by Th1 T cells in response to IL-12 production by macrophages and dendritic cells, and suppress production of the Th2 cytokine IL-4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Das A, Mukhopadhyay S. The evil axis of obesity, inflammation and type-2 diabetes. Endocrine, metabolic & immune disorders drug targets. 2011;11:23–31. doi: 10.2174/187153011794982086. [DOI] [PubMed] [Google Scholar]
- 2.Ziegler AG, et al. Accelerated progression from islet autoimmunity to diabetes is causing the escalating incidence of type 1 diabetes in young children. Journal of autoimmunity. 2011;37:3–7. doi: 10.1016/j.jaut.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verbeeten KC, et al. Association between childhood obesity and subsequent Type 1 diabetes: a systematic review and meta-analysis. Diabetic medicine: a journal of the British Diabetic Association. 2011;28:10–18. doi: 10.1111/j.1464-5491.2010.03160.x. [DOI] [PubMed] [Google Scholar]
- 4.Larsen CM, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. The New England journal of medicine. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 5.Goldfine AB, et al. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Annals of internal medicine. 2010;152:346–357. doi: 10.1059/0003-4819-152-6-201003160-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunmore SJ, Brown JE. The role of adipokines in beta-cell failure of type 2 diabetes. The Journal of endocrinology. 2013;216:T37–45. doi: 10.1530/JOE-12-0278. [DOI] [PubMed] [Google Scholar]
- 7.Tsui H, et al. ‘Sensing’ the link between type 1 and type 2 diabetes. Diabetes/metabolism research and reviews. 2011;27:913–918. doi: 10.1002/dmrr.1279. [DOI] [PubMed] [Google Scholar]
- 8.Dula SB, et al. Evidence that low-grade systemic inflammation can induce islet dysfunction as measured by impaired calcium handling. Cell calcium. 2010;48:133–142. doi: 10.1016/j.ceca.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiological reviews. 2013;93:1–21. doi: 10.1152/physrev.00017.2012. [DOI] [PubMed] [Google Scholar]
- 11.Sun K, et al. Adipose tissue remodeling and obesity. The Journal of clinical investigation. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saisho Y, et al. beta-Cell Mass and Turnover in Humans: Effects of obesity and aging. Diabetes care. 2013;36:111–117. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai D, Liu T. Hypothalamic inflammation: a double-edged sword to nutritional diseases. Annals of the New York Academy of Sciences. 2011;1243:E1–39. doi: 10.1111/j.1749-6632.2011.06388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai K, et al. TNF-alpha acutely upregulates amylin expression in murine pancreatic beta cells. Diabetologia. 2011;54:617–626. doi: 10.1007/s00125-010-1972-9. [DOI] [PubMed] [Google Scholar]
- 15.Lamers D, et al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. 2011;60:1917–1925. doi: 10.2337/db10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobrian AD, et al. Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice. American journal of physiology Endocrinology and metabolism. 2011;300:E410–421. doi: 10.1152/ajpendo.00463.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Raalte DH, Diamant M. Glucolipotoxicity and beta cells in type 2 diabetes mellitus: target for durable therapy? Diabetes research and clinical practice. 2011;93(Suppl 1):S37–46. doi: 10.1016/S0168-8227(11)70012-2. [DOI] [PubMed] [Google Scholar]
- 18.Igoillo-Esteve M, et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia. 2010;53:1395–1405. doi: 10.1007/s00125-010-1707-y. [DOI] [PubMed] [Google Scholar]
- 19.Eguchi K, et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell metabolism. 2012;15:518–533. doi: 10.1016/j.cmet.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 20.Brookheart RT, et al. As a matter of fat. Cell metabolism. 2009;10:9–12. doi: 10.1016/j.cmet.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lightfoot YL, et al. Oxidative stress and beta cell dysfunction. Methods Mol Biol. 2012;900:347–362. doi: 10.1007/978-1-60761-720-4_17. [DOI] [PubMed] [Google Scholar]
- 22.Marselli L, et al. Gene expression profiles of Beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PloS one. 2010;5:e11499. doi: 10.1371/journal.pone.0011499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamb RE, Goldstein BJ. Modulating an oxidative-inflammatory cascade: potential new treatment strategy for improving glucose metabolism, insulin resistance, and vascular function. International journal of clinical practice. 2008;62:1087–1095. doi: 10.1111/j.1742-1241.2008.01789.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen JB, et al. Divalent Metal Transporter 1 Regulates Iron-Mediated ROS and Pancreatic beta Cell Fate in Response to Cytokines. Cell metabolism. 2012;16:449–461. doi: 10.1016/j.cmet.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Weaver JR, et al. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Molecular and cellular endocrinology. 2012;358:88–95. doi: 10.1016/j.mce.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 26.Garg AD, et al. ER stress-induced inflammation: does it aid or impede disease progression? Trends in molecular medicine. 2012;18:589–598. doi: 10.1016/j.molmed.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 27.Eizirik DL, et al. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia. 2013;56:234–241. doi: 10.1007/s00125-012-2762-3. [DOI] [PubMed] [Google Scholar]
- 28.Oslowski CM, et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell metabolism. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lerner AG, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell metabolism. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J, et al. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes. 2008;57:938–944. doi: 10.2337/db07-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montane J, et al. Metabolic stress, IAPP and islet amyloid. Diabetes, obesity & metabolism. 2012;14(Suppl 3):68–77. doi: 10.1111/j.1463-1326.2012.01657.x. [DOI] [PubMed] [Google Scholar]
- 32.Subramanian SL, et al. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia. 2012;55:166–174. doi: 10.1007/s00125-011-2338-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masters SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nature immunology. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 35.Amyot J, et al. Lipopolysaccharides impair insulin gene expression in isolated islets of Langerhans via Toll-Like Receptor-4 and NF-kappaB signalling. PloS one. 2012;7:e36200. doi: 10.1371/journal.pone.0036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atkinson MA, Chervonsky A. Does the gut microbiota have a role in type 1 diabetes? Early evidence from humans and animal models of the disease. Diabetologia. 2012;55:2868– 2877. doi: 10.1007/s00125-012-2672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miani M, et al. Mild endoplasmic reticulum stress augments the proinflammatory effect of IL-1beta in pancreatic rat beta-cells via the IRE1alpha/XBP1s pathway. Endocrinology. 2012;153:3017–3028. doi: 10.1210/en.2011-2090. [DOI] [PubMed] [Google Scholar]
- 38.Carrington EM, et al. Reducing or increasing b-cell apoptosis without inflammation does not affect diabetes initiation in neonatal NOD mice. Eur J Immunol. 2011;41:2238–2247. doi: 10.1002/eji.201141476. [DOI] [PubMed] [Google Scholar]
- 39.Barthson J, et al. Cytokines tumor necrosis factor-alpha and interferon-gamma induce pancreatic beta-cell apoptosis through STAT1-mediated Bim protein activation. The Journal of biological chemistry. 2011;286:39632–39643. doi: 10.1074/jbc.M111.253591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irving-Rodgers HF, et al. Molecular composition of the peri-islet basement membrane in NOD mice: a barrier against destructive insulitis. Diabetologia. 2008;51:1680–1688. doi: 10.1007/s00125-008-1085-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ziolkowski AF, et al. Heparan sulfate and heparanase play key roles in mouse beta cell survival and autoimmune diabetes. The Journal of clinical investigation. 2012;122:132–141. doi: 10.1172/JCI46177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim DH, et al. Inhibition of autoimmune diabetes by TLR2 tolerance. J Immunol. 2011;187:5211–5220. doi: 10.4049/jimmunol.1001388. [DOI] [PubMed] [Google Scholar]
- 43.Devaraj S, et al. Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine. 2011;55:441–445. doi: 10.1016/j.cyto.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, et al. TLR9 blockade inhibits activation of diabetogenic CD8+ T cells and delays autoimmune diabetes. J Immunol. 2010;184:5645–5653. doi: 10.4049/jimmunol.0901814. [DOI] [PubMed] [Google Scholar]
- 45.Alkanani AK, et al. Dysregulated toll-like receptor-induced interleukin-1beta and interleukin-6 responses in subjects at risk for the development of type 1 diabetes. Diabetes. 2012;61:2525–2533. doi: 10.2337/db12-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarkar SA, et al. Expression and regulation of chemokines in murine and human type 1 diabetes. Diabetes. 2012;61:436–446. doi: 10.2337/db11-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joseph J, et al. IL-17 silencing does not protect nonobese diabetic mice from autoimmune diabetes. J Immunol. 2012;188:216–221. doi: 10.4049/jimmunol.1101215. [DOI] [PubMed] [Google Scholar]
- 48.Van Belle TL, et al. Development of autoimmune diabetes in the absence of detectable IL-17A in a CD8-driven virally induced model. J Immunol. 2011;187:2915–2922. doi: 10.4049/jimmunol.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saxena A, et al. Tc17 CD8+ T cells potentiate Th1-mediated autoimmune diabetes in a mouse model. J Immunol. 2012;189:3140–3149. doi: 10.4049/jimmunol.1103111. [DOI] [PubMed] [Google Scholar]
- 50.Taylor-Fishwick DA, et al. Production and function of IL-12 in islets and beta cells. Diabetologia. 2013;56:126–135. doi: 10.1007/s00125-012-2732-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulthess FT, et al. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell metabolism. 2009;9:125–139. doi: 10.1016/j.cmet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 52.Dobrian AD, et al. Functional and pathological roles of the 12- and 15-lipoxygenases. Progress in lipid research. 2011;50:115–131. doi: 10.1016/j.plipres.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma K, et al. 12-Lipoxygenase Products Reduce Insulin Secretion and {beta}-Cell Viability in Human Islets. The Journal of clinical endocrinology and metabolism. 2010;95:887–893. doi: 10.1210/jc.2009-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donath MY, et al. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends in endocrinology and metabolism: TEM. 2010;21:261–267. doi: 10.1016/j.tem.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 55.Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nature reviews Immunology. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 56.Grunnet LG, et al. Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes. 2009;58:1807–1815. doi: 10.2337/db08-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahdi T, et al. Secreted frizzled-related protein 4 reduces insulin secretion and is overexpressed in type 2 diabetes. Cell metabolism. 2012;16:625–633. doi: 10.1016/j.cmet.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 58.Roggli E, et al. Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic beta-cells. Diabetes. 2010;59:978–986. doi: 10.2337/db09-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maier B, et al. The unique hypusine modification of eIF5A promotes islet beta cell inflammation and dysfunction in mice. The Journal of clinical investigation. 2010;120:2156–2170. doi: 10.1172/JCI38924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin AP, et al. Increased expression of CCL2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes. 2008;57:3025–3033. doi: 10.2337/db08-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kriegel MA, et al. Pancreatic islet expression of chemokine CCL2 suppresses autoimmune diabetes via tolerogenic CD11c+ CD11b+ dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3457–3462. doi: 10.1073/pnas.1115308109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones HB, et al. Variation in characteristics of islets of Langerhans in insulin-resistant, diabetic and non-diabetic-rat strains. International journal of experimental pathology. 2010;91:288–301. doi: 10.1111/j.1365-2613.2010.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brooks-Worrell BM, et al. Identification of autoantibody-negative autoimmune type 2 diabetic patients. Diabetes care. 2011;34:168–173. doi: 10.2337/dc10-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Staeva TP, et al. Recent lessons learned from prevention and recent-onset type 1 diabetes immunotherapy trials. Diabetes. 2013;62:9–17. doi: 10.2337/db12-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tooley JE, et al. New and future immunomodulatory therapy in type 1 diabetes. Trends in molecular medicine. 2012;18:173–181. doi: 10.1016/j.molmed.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pescovitz MD, et al. Effect of rituximab on human in vivo antibody immune responses. The Journal of allergy and clinical immunology. 2011;128:1295–1302. e1295. doi: 10.1016/j.jaci.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sumpter KM, et al. Preliminary studies related to anti-interleukin-1beta therapy in children with newly diagnosed type 1 diabetes. Pediatric diabetes. 2011;12:656–667. doi: 10.1111/j.1399-5448.2011.00761.x. [DOI] [PubMed] [Google Scholar]
- 68.Ablamunits V, et al. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes. 2012;61:145–154. doi: 10.2337/db11-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Czyzyk J, et al. Enhanced anti-serpin antibody activity inhibits autoimmune inflammation in type 1 diabetes. J Immunol. 2012;188:6319–6327. doi: 10.4049/jimmunol.1200467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ellingsgaard H, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nature medicine. 2011;17:1481–1489. doi: 10.1038/nm.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hawley SA, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tersey SACJ, Rosenberg L, Taylor-Fishwick DA, Mirmira RG, Nadler JL. Amelioration of type 1 diabetes following treatment of non-obese diabetic mice with INGAP and lisofylline. Journal of Diabetes Mellitus. 2012;2:251–257. doi: 10.4236/jdm.2012.22040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Novotny GW, et al. Transcriptional and translational regulation of cytokine signaling in inflammatory beta-cell dysfunction and apoptosis. Archives of biochemistry and biophysics. 2012;528:171–184. doi: 10.1016/j.abb.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 74.Christensen DP, et al. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med. 2011;17:378–390. doi: 10.2119/molmed.2011.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lundh M, et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia. 2012;55:2421–2431. doi: 10.1007/s00125-012-2615-0. [DOI] [PubMed] [Google Scholar]
- 76.Lee JH, et al. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes. 2009;58:344–351. doi: 10.2337/db07-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Caton PW, et al. Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia. 2011;54:3083–3092. doi: 10.1007/s00125-011-2288-0. [DOI] [PubMed] [Google Scholar]
- 78.Bellenger J, et al. High pancreatic n-3 fatty acids prevent STZ-induced diabetes in fat-1 mice: inflammatory pathway inhibition. Diabetes. 2011;60:1090–1099. doi: 10.2337/db10-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kono T, et al. PPAR-gamma activation restores pancreatic islet SERCA2 levels and prevents beta-cell dysfunction under conditions of hyperglycemic and cytokine stress. Mol Endocrinol. 2012;26:257–271. doi: 10.1210/me.2011-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Patel SB, et al. Leptin: linking obesity, the metabolic syndrome, and cardiovascular disease. Curr Hypertens Rep. 2008;10:131–137. doi: 10.1007/s11906-008-0025-y. [DOI] [PubMed] [Google Scholar]
- 81.Morioka T, et al. Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. The Journal of clinical investigation. 2007;117:2860–2868. doi: 10.1172/JCI30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang L, et al. Deletion of Pten in pancreatic ss-cells protects against deficient ss-cell mass and function in mouse models of type 2 diabetes. Diabetes. 2010;59:3117–3126. doi: 10.2337/db09-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Holland WL, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wijesekara N, et al. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. The Journal of biological chemistry. 2010;285:33623–33631. doi: 10.1074/jbc.M109.085084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chai F, et al. Adiponectin downregulates hyperglycemia and reduces pancreatic islet apoptosis after roux-en-y gastric bypass surgery. Obesity surgery. 2011;21:768–773. doi: 10.1007/s11695-011-0357-6. [DOI] [PubMed] [Google Scholar]
- 86.Brown JE, et al. Regulation of beta-cell viability and gene expression by distinct agonist fragments of adiponectin. Peptides. 2010;31:944–949. doi: 10.1016/j.peptides.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 87.Rao JR, et al. Adiponectin increases insulin content and cell proliferation in MIN6 cells via PPARgamma-dependent and PPARgamma-independent mechanisms. Diabetes Obes Metab. 2012;14:983–989. doi: 10.1111/j.1463-1326.2012.01626.x. [DOI] [PubMed] [Google Scholar]
- 88.Wei FY, et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. The Journal of clinical investigation. 2011;121:3598–3608. doi: 10.1172/JCI58056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brooks-Worrell B, Palmer JP. Immunology in the Clinic Review Series; focus on metabolic diseases: development of islet autoimmune disease in type 2 diabetes patients: potential sequelae of chronic inflammation. Clinical and experimental immunology. 2012;167:40–46. doi: 10.1111/j.1365-2249.2011.04501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tersey SA, et al. Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61:818–827. doi: 10.2337/db11-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ehses JA, et al. IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13998–14003. doi: 10.1073/pnas.0810087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thomas HE, et al. IL-1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes. 2004;53:113–121. doi: 10.2337/diabetes.53.1.113. [DOI] [PubMed] [Google Scholar]
- 93.Brooks-Worrell B, et al. Biomarkers and immune-modulating therapies for type 2 diabetes. Trends in immunology. 2012;33:546–553. doi: 10.1016/j.it.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 94.Cnop M, et al. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 95.Winkler C, et al. Lack of association of type 2 diabetes susceptibility genotypes and body weight on the development of islet autoimmunity and type 1 diabetes. PloS one. 2012;7:e35410. doi: 10.1371/journal.pone.0035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cavelti-Weder C, et al. Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes care. 2012;35:1654–1662. doi: 10.2337/dc11-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ramos-Zavala MG, et al. Effect of diacerein on insulin secretion and metabolic control in drug-naive patients with type 2 diabetes: a randomized clinical trial. Diabetes care. 2011;34:1591–1594. doi: 10.2337/dc11-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ridker PM, et al. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) American heart journal. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]