

Abstract
Schizophrenia is a common mental illness with a large genetic component. Three genome-wide association studies have implicated the major histocompatibility complex gene region on chromosome 6p21.3-22.1 in schizophrenia. In addition, nicotine, which is commonly abused in schizophrenia, affects the expression of central nervous system immune genes. Messenger RNA levels for genes in the 6p21.3-22.1 region were measured in human postmortem hippocampus of 89 subjects. The effects of schizophrenia diagnosis, smoking and systemic inflammatory illness were compared. Cell-specific expression patterns for the class I major histocompatibility complex gene HLA-A were explored utilizing in situ hybridization. Expression of five genes was altered in schizophrenic subjects. Messenger RNA levels for the class I major histocompatibility complex antigen HLA-B were increased in schizophrenic nonsmokers, while levels for smokers were indistinguishable from those of controls. β2 microglobulin, HLA-A and Notch4 were all expressed in a pattern where inflammatory illness was associated with increased expression in controls but not in subjects with schizophrenia. Schizophrenia was also associated with increased expression of Butyrophilin 2A2. HLA-A was expressed in glutamatergic and GABAergic neurons in the dentate gyrus, hilus, and the stratum pyramidale of the CA1-CA4 regions of the hippocampus, but not in astrocytes. In conclusion, the expression of genes from the major histocompatibility complex region of chromosome 6 with likely roles in synaptic development is altered in schizophrenia. There were also significant interactions between schizophrenia diagnosis and both inflammatory illness and smoking.
Genome-wide association studies (GWAS) have demonstrated that the major histocompatibility complex (MHC) gene region on chromosome 6p21.3-22.1 is strongly associated with schizophrenia (Gejman et al., 2011; Purcell et al., 2009; Shi et al., 2009; Stefansson et al., 2009). The MHC region is a gene-rich area with large blocks of genes in high linkage disequilibrium. It is difficult to delineate which genes are responsible for the association with linkage analysis alone. However, information about their pathological affects may be gained by looking at differences in the expression of these genes in schizophrenia. This study investigates the expression of MHC region genes in the human postmortem hippocampus in subjects with schizophrenia and normal controls.
We selected MHC genes with potential brain-specific functions that are also located near SNPs with significant association to schizophrenia in GWAS studies, with the rationale that these genes are likely to exhibit expression changes in schizophrenia. The most studied of these are the class I major histocompatibility complex antigens (MHCI) (Shatz, 2009). In the central nervous system (CNS), MHCI is required for the formation and revision of dendrites during development, as well as for synaptic plasticity in the adult brain (Boulanger, 2009; Corriveau et al., 1998; Huh et al., 2000; Shatz, 2002). MHCI is involved in dendritic pruning, a process of synaptic revision where redundant synaptic connections are eliminated and useful ones are strengthened. Over-expression of MHCI may induce excessive pruning. Observations of decreased prefrontal and temporal brain volume (Pantelis et al., 2005; Shenton et al., 2001) and decreased dendritic spine density (Kolluri et al., 2005; Rosoklija et al., 2007) in schizophrenia have led to renewed interest in over-pruning as a developmental mechanism in this disorder. We investigated four MHCI genes (called human leukocyte antigens, HLA, in humans) including HLA-A, -B, -C and –G, as well as two genes involved in MHCI synthesis and assembly (TAP1 and TAPBP) (Fellerhoff and Wank, 2009). We also measured expression of β2 microglobulin (B2M). B2M is not located on chromosome 6 (it is on chromosome 15q21.1-22.2); however, it is a co-subunit of the MHCI protein, and is required for stable cell surface expression of almost all MHCI molecules.
Class II major histocompatibility proteins (MHCII) may also play an important role in regulating synapse formation and maintenance. These proteins are expressed on microglia and their expression increases when microglia are activated (Gehrmann et al., 1995). Microglia are a part of the innate immune system in the brain. They also play a role in synaptic plasticity by altering the microenvironment of the synapse via cytokine secretion. Activation is accompanied by an increase in secretion of tumor necrosis factor α (TNFα), a cytokine that mediates activity-dependent synaptic scaling (Albensi and Mattson, 2000; Stellwagen and Malenka, 2006). TNFα inhibits long-term potentiation by combined activation of TNF receptor 1 and metabotropic glutamate receptor 5. Microglia also may eliminate dendritic spines by phagocytosis (Blank and Prinz, 2012). Schizophrenia patients have increased numbers of activated microglia and fewer dendritic spines (Radewicz et al., 2000; Rosoklija et al., 2007). We therefore measured expression for three MHCII genes (HLA-DRA, -DQA1, -DRB5) and the TNF gene.
Two other MHC region genes are of potential interest. The Notch4 gene is within 7KB of a SNP with genome-wide significance for association to schizophrenia in two GWAS (Purcell et al., 2009; Stefansson et al., 2009). Other work suggests a significant decrease in expression in the Notch pathway in schizophrenia (Brennand et al., 2011). Butyrophilin 2A2 is an immune cell-surface protein. Messenger RNA levels of this gene in the brain are higher than in all other organs (Smith et al., 2010), however, the gene is little studied and its function in the brain is unknown.
MHC genes in the CNS are regulated by inflammatory factors, including cytokines (Neumann et al., 1997). This fact is relevant in studies of postmortem brain, where many subjects have died in the presence of infection or other types of systemic inflammation such as autoimmune disease. If not included in the analysis, inflammatory illness could act as a confounding factor that may inflate estimates of expression levels for these immune genes. More importantly, many studies have demonstrated an association between immune activation and increased risk for schizophrenia (Brown and Derkits, 2009; Brown et al., 2004; Fellerhoff et al., 2006; Khandaker et al., 2012; Patterson, 2007). Interactions between schizophrenia and inflammatory illness may significantly affect the expression of these genes. Therefore the presence of systemic inflammatory illness was included in our analysis as a factor in MHC gene expression.
Smoking can also act as a confounding factor in studying gene regulation in schizophrenia. MHC expression in schizophrenics could be altered by a high prevalence of smoking, which suppresses immune function (de Leon and Diaz, 2005; Olincy et al., 1997). More than 80% of schizophrenic patients smoke compared to approximately 25% of the general population (de Leon and Diaz, 2005; Leonard et al., 2001). Nicotine and acetylcholine suppress the immune response (Gallowitsch-Puerta and Tracey, 2005). Subjects who smoke may be suppressing the expression of immune genes with brain-specific functions. We therefore included smoking status as a potential covariate in determining MHC gene expression.
In recent studies, both MHCI and MHCII have been shown to be involved in regulation of synaptic plasticity (Albensi and Mattson, 2000; Shatz, 2009). Plasticity is required for successful learning and memory. In the mature adult brain, these processes are most notably active in the hippocampus, a key brain region for memory formation. The hippocampus undergoes functional losses parallel to those found in frontal cortex in schizophrenia, including volume loss as well as alterations in glutamatergic, GABAergic and neuregulin signaling (Heckers and Konradi, 2002; Law et al., 2007; Mexal et al., 2005). Patients with schizophrenia are impaired in multiple cognitive tasks that are mediated though the hippocampus including sensory gating (Freedman et al., 1996), as well as both spatial and verbal declarative memory (Stone and Hsi, 2011; Stone and Seidman, 2008). In schizophrenia, this cognitive dysfunction is both chronic and disabling. For these reasons we chose to look for differences in MHC gene expression in the hippocampus.
We measured gene expression using quantitative real-time PCR (QRT-PCR) in postmortem hippocampus in 89 subjects. We evaluated changes in gene expression as a function of schizophrenia diagnosis, smoking status, and the presence of systemic inflammatory illness at the time of death. MHCI and MHCII promote processes in synaptic development that decrease dendritic spine density and synapse number (Blank and Prinz, 2012; Shatz, 2002); schizophrenia is associated with both decreased dendritic spine density and reduced synaptic function (Meador-Woodruff and Healy, 2000; Rosoklija et al., 2007). Our hypothesis is that MHCI and MHCII expression is increased in schizophrenia, while Notch4 expression may be decreased. The presence of inflammatory illness would increase expression of these genes, while smoking may suppress it.
Prior to the current study, cell specific expression patterns of MHCI genes have not been explored in the human brain. Since virtually nothing is known about how these genes are expressed in the human brain, and animal models do not fully explain gene function in human brain, it is important to understand which cells express MHC genes in the human brain as a beginning to understanding how these genes are associated with neuropsychiatric disease. We therefore initiated studies to define expression patterns in the human hippocampus. The MHCI gene HLA-A was the most highly expressed MHCI gene in our hippocampal tissue samples and therefore a good candidate for exploring expression patterns. We utilized in situ hybridization and immunofluorescent double labeling to localize hippocampal expression of HLA-A to specific cell types.
METHODS
Subjects
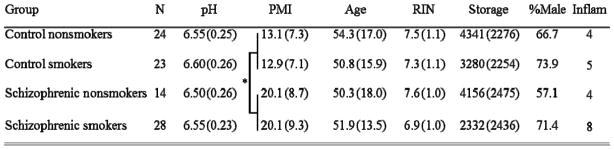
The Schizophrenia Research Center Brain Bank at the University of Colorado Medical Campus provided all of the postmortem brain tissue, which was donated through the Colorado Anatomical Gift Act. The Brain Bank contains brain tissue from subjects with a diagnosis of schizophrenia as well as from unaffected individuals. Control subjects were evaluated by the same procedures as schizophrenic subjects as follows: Pre- and postmortem parameters were recorded for each subject from an extensive review of hospital, autopsy, and neuropathology reports, as well as structured interviews with physicians and family, as described previously (Mexal et al., 2006; Mexal et al., 2005). Based on this information, all subjects were evaluated by two independent psychiatrists and A DSM-IV diagnosis of was confirmed in the affected subjects. Controls were determined not to have any neuropsychiatric disorder by the same procedures. Utilizing these data, group differences in variables that may affect gene expression were evaluated by analysis of variance (ANOVA), to insure that no groups differed on any of the key variables. These variables included brain pH, subject age, tissue storage time, gender, RNA quality as indicated by RNA integrity number (RIN), and postmortem interval (PMI) (Groups are summarized in Table 1; for a detailed table of all subjects, see Table S1). Tissue samples from ninety eight subjects were initially evaluated for the study. Seven samples were excluded due to poor tissue quality (RIN ≤ 5 and/or brain pH < 6). Two additional subjects were excluded due to the use of immunosuppressant drugs (chemotherapy agents and corticosteroids). The remaining 89 samples were included in the analysis. After excluding these nine samples, there were no significant differences among groups for RIN, brain pH, storage time, subject age, or gender distribution. However, schizophrenic subjects had a significantly longer PMI compared to controls.
Table 1.
The Study Cohort.
|
Mean (Standard Deviation). RIN: RNA integrity number; PMI: postmortem interval in hours; Storage: brain storage time in days; age: subject age at death. Inflam: number in the group with an inflammatory condition. There are no significant differences among the groups in RIN, pH, storage time, age, or sex ratio.
Schizophrenic subjects had longer postmortem intervals compared to controls (p < 0.001).
In postmortem brain studies, PMI is defined as the time elapsed between the time of death and the time the brain is removed from the skull. In many brain banks, affected individuals have longer PMIs compared to controls because they are relatively rare, therefore extra time and effort is invested to locate the next of kin and seek consent for tissue donation. Such is the case at the Schizophrenia Research Center Brain Bank at the University of Colorado. Although the groups were matched for all other variables, the schizophrenic subjects had a significantly longer PMI compared to controls (p< 0.001). PMI has been shown to have a limited effect on mRNA and protein assays in postmortem brain. No relationship between PMI and total amounts of RNA has been found for PMIs between 0 and 48 hours (Harrison et al., 1995). The subjects in this study all had PMIs less than 48 hours (range: 2–45 hours) therefore a longer PMI in the affected subjects is unlikely to have an effect on our results. However, to avoid any possible confounding effect we evaluated the effect of PMI on mRNA levels for each gene in the study. If PMI was found to have a significant effect on expression levels for any gene, PMI was included as a covariate in the analyses for that gene. In our samples PMI was only associated with mRNA levels for the TAPBP gene.
Subjects were classified by diagnosis (schizophrenia vs. control). In this brain collection, no information about the level of smoking or severity of infection/inflammation was available for the subjects in the study; therefore smoking status was classified categorically (smoking vs. nonsmoking). Information about inflammatory illness was also classified categorically as the presence or absence of a major infection (e.g. sepsis, pneumonia, encephalitis) or an inflammatory disease (Stevens-Johnson syndrome, rheumatoid arthritis, scleroderma) at the time of death. Diagnoses and causes of death are in Table S1.
Brain Tissue
Human brain was collected at autopsy from local autopsy services following family consent. At autopsy, the brain was weighed, examined for gross pathology, and divided sagittally. One hemisphere was preserved for neuropathological analysis at the macroscopic and microscopic level. Microscopic evaluations included Bielchowsky silver stain on multiple cerebral areas to rule out abnormal neuropathology, such as plaques and tangles associated with Alzheimer’s and other conditions. Patients with neuritic findings or ambiguous neuropathology reports were excluded. The other hemisphere was sliced coronally into 1 cm slices. Hippocampal sections of approximately 1g were dissected from these sections, frozen on dry ice, and stored at −80°C. Hippocampal tissue in each sample for this study was taken from the anterior-most section of the hippocampus that included CA4, CA3, CA2, CA1, dentate gyrus, and subiculum.
For in situ hybridization, 25 hippocampal slices from each of four randomly chosen brains were used to visualize HLA-A expression patterns and localize them to specific cell types (see Table S2).
RNA Isolation and Evaluation
RNA was isolated from human hippocampus with Trizol (Life Technologies, Gaithersburg, MD) following the manufacturer’s protocol, and purified with an RNeasy mini kit (Qiagen, Valencia, CA). RNA integrity numbers (RIN) were derived from 300ng of total RNA using an Agilent 2100 Bioanalyzer and an Agilent 6000 Nano microfluidic chip kit (Agilent Technologies, Santa Clara, CA). The most reliable QRT-PCR gene expression results are obtained with RIN > 5 and amplicon length < 200 bp (Fleige and Pfaffl, 2006; Fleige et al., 2006). Specimens in this study all had RIN scores > 5.1 (mean RIN score = 7.3 ± 1.1) and all amplicons assayed were <200 bp.
Quantitative RT-PCR
cDNA was synthesized from total RNA using the Superscript First Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). Gene expression was evaluated utilizing Taqman® gene expression assays (Life Technologies, Carlsbad, California; for a full list of assays, amplicon lengths and target sequences, see Table S3. PCR was performed in a 7900HT thermal cycler (Life Technologies, Carlsbad, California). Cycling conditions were: 50°C for 2 min, 95°C for 15 min, and 40 cycles of 94°C for 15 s, 58°C for 30 s, and 72°C for 30 s. All assays were done in triplicate. The coefficient of variation for triplicates was <1%. Selection of a gene for normalization of mRNA levels was done by comparison of a panel of housekeeping genes (HIST1H2AG, HIST1H2BJ, GAPDH, B2M and POLR2A) using NormFinder (Andersen et al., 2004). The gene GAPDH was chosen by the NormFinder algorithm. It showed the highest stability and smallest intergroup variation. In order to eliminate between-run variability between the test and normalization genes, GAPDH was run as an endogenous internal control for each sample by utilizing duplex Taqman assays. Messenger RNA levels were then normalized using GAPDH. Duplex assays were screened prior to use and no significant differences in PCR efficiency or threshold cycle were found in duplex vs. singleplex reactions for GAPDH or any of the test genes. For each experiment, a sample without the addition of reverse transcriptase enzyme or without the addition of cDNA was included to assess amplification from genomic DNA and nonspecific product formation, respectively.
In Situ Hybridization Tissue Preparation
Frozen blocks of hippocampal tissue were embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA). Coronal tissue sections 12 μm thick were cut at −18 °C from the embedded blocks on a microtome cryostat (Bright Instrument Company, Huntingdon, England) and thaw-mounted on 3-aminopropyltriethoxysilane coated slides. The mounted sections were quick frozen on dry ice and stored at -−80 °C until use.
Probe Synthesis
A full length HLA-A clone (I.M.A.G.E. Clone I.D.: 3510139) was purchased from American Type Culture Collection (Manassas, VA). A 360 bp fragment containing bp 959–1319 was amplified with Taq polymerase. This region corresponds to exons 5–8, crossing intron/exon borders. These exons correspond to the transmembrane portion of the HLA-A protein. BLAST searches were done to confirm sequence specificity. Probe primers and sequence are in Figure S1.
The fragment was cloned into the vector pDrive and used to transform EZ competent cells with a Qiagen PCR Cloning Kit (Qiagen, Valencia, CA). Plasmid DNA was isolated with a Qiagen Plasmid Mini Kit. A sample of plasmid DNA was digested with EcoRI and run on a 1% agarose gel with ethidium bromide, revealing a single insert band of approximately 360 bp. The clone was sequenced using a 3100 Avant Genetic Analyzer (Applied Biosystems, Foster City, CA). Digoxigenin (DIG) labeled sense and antisense RNA probes were synthesized with a DIG RNA labeling kit (Roche Applied Science, Indianapolis, IN). Labeling efficiency was determined by northern dot blot.
In Situ Hybridization Procedure
Tissue sections were thawed and dried at room temperature for 30 minutes. Sections were rinsed in phosphate-buffered saline (PBS) then fixed with 4% paraformaldehyde in PBS, pH 7.2, for 20 minutes at room temperature. Then they were rinsed in dimethylpyrocarbonate treated water. In situ hybridization was performed with an ISHyb in situ hybridization kit (BioChain, Hayward, CA). For each experiment a DIG labeled sense mRNA probe control was also included as a specificity control.
The slides were rinsed with room temperature PBS, pH 7.4. Nonspecific binding was blocked by incubating in PBS containing 0.3% Triton X-100 (PBS-T) plus 5% normal mouse serum for one hour at room temperature. The probe was visualized with a DyLight-549 conjugated mouse anti-DIG antibody (Jackson ImmunoResearch, West Grove, PA), diluted 1:200 with PBS-T. Nuclei were labeled in 10nm 4′,6-diamidino-2-phenylindole (DAPI nuclear stain). After all labeling procedures were complete, lipofuscin induced autofluorescence was blocked by incubating for 10 minutes at room temperature in 0.1% Sudan Black B dissolved in 70% ethanol as previously described (Newton et al., 2002; Schnell et al., 1999). The tissue was allowed to dry at room temperature, covered with Prolong anti-fade solution (Invitrogen, Carlsbad, CA), coverslipped, allowed to cure at room temperature in the dark for 24 hours, and stored at −20 °C. Slides were viewed under a Nikon Microphot FXA fluorescence microscope and imaged using a Spot RT Slider digital camera (Diagnostic Instruments, Sterling Heights, MI).
Double Fluorescent Labeling
HLA-A mRNA expression was visualized by in situ hybridization combined with immunofluorescent counterstaining to localize cell type specific markers. A primary antibody against the neurotransmitter glutamate was used to label glutamatergic cells; an antibody for GFAP was used to label astrocytes. An antibody against both the 65 and 67 kDa isoforms of glutamate decarboxylase (GAD65/GAD67) was used to label GABAergic interneurons. For all experiments, a deletion control without primary antibody was also performed as a specificity control.
Nonspecific binding was blocked by incubating in PBS-T plus 5% normal donkey serum. Slides were rinsed with PBS, placed in a humidity box and covered with 100–200ul of PBS-T plus a primary antibody (rabbit anti-glutamate (1:500), anti-GFAP (1:500), (Millipore, Billerica, MA) or anti-GAD65/GAD67 (1:200) (Abcam, Cambridge, MA). A deletion control without primary antibody was included. The humidity box was sealed and incubated overnight at 4°C.
Slides were rinsed in room temperature PBS, placed in a humidity box with 100–200ul FITC conjugated donkey anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA) diluted 1:100 in PBS-T, and incubated at 37°C for 3 hours, minimizing light exposure. Slides were rinsed four times for 10 minutes in 37°C PBS.
Statistical Analysis
Ninety eight brain tissue samples were initially evaluated for the study. Seven samples were excluded due to poor tissue quality (RIN ≤ 5 and/or brain pH < 6). Two additional subjects were excluded due to the use of immunosuppressant drugs (chemotherapy agents and corticosteroids). The remaining 89 samples were included in the analysis.
Subjects were classified by diagnosis (schizophrenia vs. control), smoking status (smoking vs. nonsmoking) and presence of inflammatory disease. Information about the level of smoking was not available for the subjects. Individuals with a major infection at the time of their death according to chart review (e.g. sepsis, pneumonia, encephalitis) or who had an inflammatory disease (Stevens-Johnson syndrome, rheumatoid arthritis, scleroderma) were given a group designation (inflammatory condition vs. no such condition). This inflammatory factor was used in the analysis as a potential variable in determining gene expression.
To determine whether the proportion of males to females was different across the experimental groups, a chi-squared test was performed. Group means were compared for PMI, brain pH, subject age, storage time, and RIN score using an ANOVA.
For each subject, mean normalized expression values for the gene of interest were calculated using GAPDH mRNA levels. Expression values were log2 transformed to approximate statistical test assumptions. To investigate the effects of schizophrenia, smoking and inflammation on gene expression, a multivariate linear regression was used that included main and two-way interaction terms for the three categorical variables of diagnosis, smoking status, and the presence of systemic inflammation at the time of death. Since PMI had a significant effect on expression levels for TAPBP, this factor was also included as a covariate in the model for that gene. Backwards variable selection was used to eliminate non-significant effects and identify a reduced model for each gene. The subject groupings for each gene were therefore determined by these variables and interactions. These groupings are presented in the results graphs for each gene (Figures 2–7). Main effects were constrained to remain in the model if an interaction was selected. Least-squares means estimates were calculated for parameters with significant effects and used to illustrate effect sizes. These estimates were adjusted for all other parameters in the model. Hypotheses about significant differences between groups were tested using linear contrasts performed with the groups of interest. To adjust for multiple testing, false discovery rate controlling (FDR) adjustments (Benjamini and Hochberg, 1995) were globally performed. All p-values presented are therefore empirical p-values generated by FDR adjustment.
Figure 2. Interactive effects of schizophrenia and inflammatory illness on HLA-A levels.

Least squares means of log transformed expression relative to GAPDH. Means are adjusted for all other effects in the model. A. Controls with inflammatory illness had significantly increased HLA-A expression compared to those with no inflammatory illness [F(1,87)= 12.77; p = 0.002; fold Δ = 1.6]. This difference in expression did not occur in the schizophrenic subjects. B. Smokers had significantly lower HLA-A expression [F(1,87) = 5.26; p = 0.038; fold Δ = 1.2]. Bars are one S.E.M.
Figure 7. Interactive effects of Smoking and inflammatory illness on HLA-DRA and TAP1 expression.

Least squares means of log transformed expression relative to GAPDH. Means are adjusted for all other effects in the model. A. Effects on HLA-DRA expression. Nonsmokers with an inflammatory illness had significantly increased HLA-DRA expression compared to those with no inflammatory illness (t = 3.67; p = 0.002; fold Δ = 3.3). This difference in expression did not occur in the smokers. B. Effects on TAP1 expression. Nonsmokers with an inflammatory illness had significantly increased TAP1 expression compared to those with no inflammatory illness [F(1,87) = 14.47; p = 0.002; fold Δ = 2.0]. This difference in expression did not occur in the smokers. Bars are one S.E.M.
The HLA-C gene was expressed in a binary, “on-off” fashion. Binary logistic regression was used to analyze expression of this gene, with main and interaction terms for diagnosis, smoking status, and the presence of systemic inflammation at the time of death. All analyses were performed using SAS 9.3 statistical software (SAS, Cary, NC).
RESULTS
Parameters That Affect Gene Expression Profile
We compared gene expression by quantitative real-time PCR (QRT-PCR) in human postmortem hippocampal tissue from 89 subjects. Tissue samples included 24 nonsmoking controls (four in this group had an inflammatory condition at the time of death), 23 control smokers (five had an inflammatory condition), 14 nonsmoking subjects with schizophrenia (four had an inflammatory condition) and 28 schizophrenic smokers (eight had an inflammatory condition). For summarized subject data see Table 1; detailed information for each brain can be found in Table S1. Messenger RNA for a total of thirteen genes was initially measured (see Table 2 for a list of these genes and pertinent references). Nine of these genes were expressed at high enough levels in the hippocampus for further analysis. Univariate effects for each variable were explored before including covariates and interactions. A table of these effects can be found in Table S4 in the supplementary material.
Table 2.
Genes evaluated in this study.
| Gene symbol* | Official Gene Name | Location | References |
|---|---|---|---|
| HLA-A | major histocompatibility complex, class I, A | 6p21.3 - MHCI region | Purcell et al. (2009)- alleles associated with rs3130375, a SNP significant at 3.66 × 10−7 |
| HLA-B | major histocompatibility complex, class I, B | Mexal et al. (2005) - HLA-A expression increased in post-mortem hippocampus of schizophrenic nonsmokers | |
| HLA-C | major histocompatibility complex, class I, C | Shatz et al. (2002)- MHCI essential for axono-dendritic remodeling | |
| HLA-G | major histocompatibility complex, class I, G | ||
|
| |||
| B2M | beta-2 microglobulin (co-subunit of MHCI) | 15q21-q22.2 | Mexal et al. (2005)- expression increased in post-mortem hippocampus of schizophrenic nonsmokers |
|
| |||
| TAP1 | transporter 1, ATP-binding cassette, sub-family B (MHCI synthesis) | 6p21.3 - MHCII region | Fellerhoff et al. (2009) - A1/B1 genotype associated with schizophrenia with odds ratio 14.1 |
|
| |||
| TAPBP | TAP binding protein; AKA tapasin, (MHCI chaperone protein) | 6p21.3 - extended MHCII region |
Purcell et al. (2009)- SNP within 20KB associated at 4.49 × 10−5 Fellerhoff et al. (2009)- 01/01 genotype associated with schizophrenia. |
|
| |||
| HLA-DRA | major histocompatibility complex, class II, DR alpha | 6p21.3 - MHCII region |
Purcell et al. (2009) - DRB and DQA alleles associated with rs3130375, a SNP significant at 3.66 × 10−7; DRA within 20KB of a SNP associated at 8.34 × 10−6 Shi et al. (2009) HLA-DQA1- upstream and intronic SNPs significant at 6.88 × 10−8 and 8.87 × 10−8 respectively |
| HLA-DRB5 | major histocompatibility complex, class II, DR beta 5 | ||
| HLA-DQA1 | major histocompatibility complex, class II, DQ alpha 1 | ||
|
| |||
| NOTCH4 | notch 4 | 6p21.3 - MHCII region |
Purcell et al. (2009), Stefansson et al. (2009) - genome wide association of a SNP 7KB from the gene Brennand et al. (2011)- significant decrease in expression in the NOTCH pathway in schizophrenia |
|
| |||
| TNF | tumor necrosis factor alpha | 6p21.3 |
Albensi and Mattson (2000) - Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Stellwagen and Malenka (2006) - Synaptic scaling mediated by glial TNF-alpha |
|
| |||
| BTN2A2 | butyrophilin, subfamily 2, member A2 | 6p22.1 |
Purcell et al. (2009) SNP within 20KB associated at 1.18 × 10−5 Smith et al. (2010) - Brain has the highest mRNA levels of BTN2A2 compared to all other organs |
Gene symbol = official gene symbol designated by the HUGO Gene Nomenclature Committee (HGNC). Official gene name = official HGNC full length name of the gene. References = previous studies that have demonstrated genetic association or differential expression in schizophrenia.
Human MHCI and MHCII genes were initally called human leukocyte antigens (HLA) prior to understanding the full scope of their function. This designation has been retained in the official gene symbols, but not in the HGNC full length names of the genes.
RNA integrity and mRNA levels for the Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene were not significantly different among the experimental groups. Gender, age, storage time, and brain pH were not different. Subjects with schizophrenia had a longer mean post mortem interval (PMI) [mean PMI = 20.1 ± 9.0 (SD) hours] compared to controls (mean = 13.0 ± 7.1 hours; p < 0.001). However, PMI only affected mRNA levels of the TAPBP gene, with increasing PMI associated with decreased transcript.
Relative Expression Levels of MHCI Genes
HLA-A was the most highly expressed MHCI gene assayed. In controls it was expressed in essentially a 1:1 ratio with β2-microglobulin (see Figure 1). HLA-B and HLA-C were expressed 3.5 and 5-fold less respectively compared to HLA-A. There was no detectable expression of HLA-G in any of the subjects.
Figure 1. Relative expression levels of B2M and three HLA genes in control subjects without an inflammatory condition.

Mean expression compared to GAPDH, expressed as a percentage. Bars are one S.E.M. HLA-G does not have a bar because there was no detectable expression of this gene.
Hippocampal MHC Gene Expression in Subjects with Schizophrenia
To investigate the effects of schizophrenia, smoking and inflammatory illness on gene expression, multivariate linear regression modeling was used that included main and interaction terms for diagnosis, smoking status, the presence of a systemic inflammatory illness at the time of death. A reduced model containing all significant effects was produced for each gene analyzed. Global adjustment for multiple testing was performed utilizing false discovery rate (FDR) methods, therefore all p-values given are empirical p-values generated by FDR adjustment (adj p).
For HLA-A, mRNA levels differed across schizophrenia diagnosis and the presence of inflammatory disease at the time of death (adj p = 0.045 for the interaction). Controls with an inflammatory illness had significantly increased HLA-A expression compared to controls with none (adj p = 0.002; fold Δ = 1.6; see Figure 2A). This difference in expression did not occur in the schizophrenic subjects. Controls with an inflammatory illness also had significantly higher HLA-A expression compared to schizophrenics with such illness (adj p = 0.023; fold Δ= 1.5). Smoking also had an effect on HLA-A levels. Smokers had decreased expression in all subjects (adj p= 0.038, fold Δ= 1.2; Figure 2B) after adjusting for the schizophrenia and inflammatory illness groups.
For the gene HLA-B, mRNA levels differed across groups defined by diagnosis and smoking status (adj p = 0.017 for the interaction). Figure 3A illustrates the interaction between these two variables. Schizophrenic nonsmokers had significantly increased HLA-B expression compared control nonsmokers (adj p = 0.005; fold Δ = 2.3), and compared to schizophrenic smokers (adj p < 0.001; fold Δ = 2.7; Figure 3A). There was also an interaction between the effects of smoking and inflammatory illness (adj p = 0.022 for the interaction). Nonsmokers with an inflammatory illness at the time of death had significantly increased HLA-B expression compared to those with no inflammatory illness (adj p = 0.001; fold Δ = 2.4). This difference in expression did not occur in the smokers (Figure 3B).
Figure 3. Interactive effects of schizophrenia, smoking and inflammatory illness on HLA-B expression.

Least squares means of log transformed expression relative to GAPDH. Means are adjusted for all other effects in the model. A. Schizophrenic nonsmokers had significantly increased HLA-B expression compared Control nonsmokers [F (1,87)= 10.69; p = 0.005; fold Δ = 2.3], and compared to schizophrenic smokers [F(1,87) = 22.99; p < 0.001; fold Δ = 2.7]. B. Nonsmokers with an inflammatory illness had significantly increased HLA-B expression compared to those with no inflammatory illness [F(1,87) = 18.15; p = 0.001; fold Δ = 2.4]. This difference in expression did not occur in the smokers. Bars are one S.E.M.
HLA-C was expressed in a binary pattern. Forty-two out of the 89 subjects expressed no detectable HLA-C, while the remainder displayed moderate expression (threshold cycle ~ 28). Schizophrenic subjects tended to be more likely to be non-expressers, however this was not statistically significant after adjustment for multiple testing (χ2= 3.94, adj p = 0.063). Among subjects who expressed HLA-C, there was no difference in expression between controls and schizophrenics.
B2M mRNA levels differed across groups defined by schizophrenia diagnosis and the presence of inflammatory illness (adj p = 0.011 for the interaction). Controls with an inflammatory illness at the time of death had significantly increased B2M expression compared to those with no inflammatory illness (adj p < 0.001; fold Δ = 1.8; see Figure 4A). This difference in expression did not occur in the schizophrenic subjects. A similar interaction was found between smoking and inflammatory illness (adj p = 0.032 for the interaction). Nonsmokers with inflammatory illness at the time of death had significantly increased B2M expression compared to those with no inflammatory illness (adj p = 0.001; fold Δ = 1.8). This difference in expression did not occur in the smokers (Figure 4B)
Figure 4. Interactive effects of schizophrenia, smoking and inflammatory illness on B2M expression.

Least squares means of log transformed expression relative to GAPDH. Means are adjusted for all other effects in the model. A. Controls with an inflammatory illness had significantly increased B2M expression compared to those with no inflammatory illness [F(1,87) = 24. 21; p < 0.001; fold Δ = 1.8]. This difference in expression did not occur in the schizophrenic subjects. B. Nonsmokers with an inflammatory illness had significantly increased B2M expression compared to those with no inflammatory illness [F(1,87) = 18.15; p = 0.001; fold Δ = 1.8). This difference in expression did not occur in the smokers. Bars are one S.E.M.
Notch4 expression differed across groups defined by schizophrenia and inflammatory illness in a pattern similar to those found in B2M and HLA-A (adj p = 0.022 for the interaction; see figure 5). Controls with an inflammatory illness had significantly increased Notch4 expression compared to those with no inflammatory illness (adj p = 0.004; fold Δ = 1.8, Figure 5A). This difference in expression did not occur in the schizophrenic subjects. Smoking decreased expression of this gene in all subjects (adj p= 0.031, fold Δ = 1.3, Figure 5B) after adjusting for the schizophrenia and inflammatory illness groups.
Figure 5. Interactive effects of schizophrenia and smoking on Notch4 expression.

Least squares means of log transformed expression relative to GAPDH. Means are adjusted for all other effects in the model. A. Controls with inflammation had significantly increased Notch4 expression compared to those with no inflammation [F(1,87) = 11.1; p = 0.004; fold Δ = 1.8]. This difference in expression did not occur in the schizophrenic subjects. B. Smokers had significantly lower expression than nonsmokers [F(1,87) = 5.85; p = 0.031; fold Δ = 1.3]. Bars are one S.E.M.
For the butyrophilin 2A2 gene, Expression was increased in schizophrenia (adj p= 0.023, fold Δ = 1.4 ; Figure 6A). Expression was decreased in smokers, but this was only marginally significant after correction for multiple testing (adj p= 0.057, fold Δ = 1.3; Figure 6B).
Figure 6. Effects of schizophrenia and smoking BTN2A2 expression.

A. Subjects with schizophrenia had significantly higher levels of BTN2A2 mRNA [F(1,87) = 6.56; p = 0.023; fold Δ = 1.4]. B. Smokers had lower BTN2A2 levels [F(1,87) = 4.28; p = 0.057; fold Δ = 1.3]. Bars are 1 S.E.M.
Genes with No Expression Changes in Schizophrenia
HLA-DRA levels differed only across the combined effects of smoking and inflammatory disease at the time of death (adj p = 0.023 for the interaction). Nonsmokers with an inflammatory illness had significantly increased HLA-DRA expression compared to those with no inflammatory illness (adj p = 0.002; fold Δ = 3.3; see Figure 7A). This increase in expression did not occur in the smokers.
Similarly, TAP1 levels differed only across the combined effects of smoking and inflammatory illness at the time of death (adj p = 0.045 for the interaction). Nonsmokers with an inflammatory illness had significantly increased TAP1 expression compared to those with no inflammatory illness (adj p = 0.002; fold Δ = 2.0; see Figure 7B). This difference in expression did not occur in the smokers for this gene.
Expression of the TAPBP gene was associated only with PMI: increased PMI was associated with decreased transcript (β = −0.01; adj p = 0.023).
Genes with Poor Expression in the Hippocampus
For four genes, mRNA levels detected by the Taqman assay were too low for accurate analysis (mean threshold cycle >34). These included HLA-G, HLA-DQA1, HLA-DRB5 and TNF.
In Situ Hybridization
The in situ procedure was performed with sense riboprobes as a negative control. The antisense probe labeled cells bright red (Figure S2-A in the supplemental material). The sense probe produced a much weaker signal (Figure S2, B).
HLA-A mRNA was very well expressed, allowing visualization of the architecture of the whole hippocampus. Labeled HLA-A mRNA was visible in cells of the granule cell layer of the dentate gyrus, the hilus, and the stratum pyramidale of the CA1, CA2, CA3, and CA4 regions of the hippocampus (Figure 8).
Figure 8. HLA-A mRNA in the Human Hippocampus.

This slice was taken from a control nonsmoking subject (subject SL283, see Table S1) but the qualitative pattern in all subjects tested was the same. DG = dentate gyrus; gc = granule cells; CA1-4 = cornu ammonis areas 1–4. SI = subiculum. This image is a composite of 85 photomicrographs taken at 4X magnification. Bar = 1mm.
To localize HLA-A expression to specific cell types, in situ hybridization was combined with immunofluorescent labeling for cell-type specific proteins. For astroglia, fluorescein isothiocyanate (FITC, green) immunofluorescent labeling for glial fibrillary acidic protein (GFAP) was combined with in situ hybridization for HLA-A mRNA (DyLight 549, red). Cells that labeled for HLA-A mRNA were not observed to contain filaments labeled for GFAP and cells that labeled for GFAP did not label for HLA-A (Figure 9, A-C). The two labels appeared to be mutually exclusive.
Figure 9. Double Labeling for HLA-A and Cell Specific Markers in the Hippocampus.

(A–C) Double Labeling for HLA-A and GFAP in a group of cells in the CA3 region. HLA-A mRNA was detected by in situ hybridization and labeling with a DyLight 549 conjugated antibody (red). GFAP was detected by FITC labeled antibodies (green). Nuclei were labeled with DAPI nuclear stain (blue).
A. Fluorescence microscopy image showing HLA-A positive cells.
B. Fluorescence microscopy showing intermediate filaments labeled for GFAP.
C. Merged image with all three wavelengths. HLA-A labeled cells do not appear to contain filaments labeled for GFAP.
(D–F) Double Labeling for HLA-A and Glutamate in a group of cells is in the CA3 region. HLA-A mRNA was detected by in situ hybridization and labeling with a DyLight 549 conjugated antibody (red). Glutamate was detected with a FITC labeled antibody (green). Nuclei were labeled with DAPI nuclear stain (blue).
D. Fluorescence microscopy image showing HLA-A positive cells.
E. Fluorescence microscopy showing glutamate labeled cells.
F. Merged image with all three wavelengths.
(G–I) Double labeling for HLA-A and GAD65/GAD67 in the dentate gyrus polymorphic layer. HLA-A mRNA was detected by in situ hybridization and labeling with a DyLight 549 conjugated antibody (red). GAD65 and GAD67 were detected by a FITC labeled antibody (green). Nuclei were labeled with DAPI nuclear stain (blue).
G. Fluorescence microscopy image showing HLA-A positive cells.
H. Fluorescence microscopy showing GAD65/GAD67 labeled cells.
I. Merged image with all three wavelengths. Bar = 25 microns.
When cells were dually labeled for HLA-A mRNA and glutamate, the two fluorescent tags followed a similar distribution in the cell bodies and processes, producing a yellow color (Figure 9, D–F).
A FITC labeled antibody against both the 65 and 67 kDa isoforms of glutamate decarboxylase (GAD65/GAD67) was used to label GABAergic interneurons. All observed cells that were positive for GAD also labeled for HLA-A mRNA. The HLA-A label (red) was primarily visible in the cell body, but also in cell processes at low intensity (Figure 9G). GAD labeling (green) was present in cell bodies but was more intense in cell processes (Figure 9H). This staining pattern produced bi-colored cells with yellow/orange cell bodies and green processes (Figure 9I). These labeling patterns were identical in all four subjects chosen for in situ hybridization, regardless of diagnosis, smoking status, or the presence of systemic inflammation at the time of death.
DISCUSSION
QRT-PCR was used to measure changes in immune gene expression in the post-mortem hippocampus and to evaluate the effects of schizophrenia diagnosis, smoking, and the presence of systemic inflammatory illness at the time of death. In situ hybridization was used to locate and characterize cells expressing HLA-A, the most highly expressed MHC gene.
HLA-A mRNA was found in the granular cells of the dentate gyrus, in the hilus, and in the stratum pyramidale of the CA1-CA4 regions of the hippocampus. HLA-A mRNA was observed in cells that express glutamate and GAD65/67, but not those expressing GFAP. This suggests that HLA-A is expressed in glutamatergic neurons and GABAergic interneurons but not in astrocytes. This is the first study to look at HLA-A expression in the human brain and to localize it to specific cell types. Future studies might further define expression in specific neuronal subtypes (for example parvalbumin expressing cells) and look at the expression of HLA-B and –C.
We identified two genes that were over-expressed in schizophrenia after adjustment for all other variables: HLA-B and BTN2A2. The butyrophilins are a little-studied group of MHC-associated membrane proteins. In T-cells, binding of BTN2A2 receptors decreases secretion of interferon gamma and interleukin-2 (Smith et al., 2010). The role of butyrophilins in the brain has not been studied. In immune function they transduce signals from cell to cell that inhibit activation of transcription factors such as NF-κB. NF-κB signaling is critically involved in synapse formation and spine maturation. NF-κB blockade in the forebrain is associated with reduced levels of mature spines (Schmeisser et al., 2012), a finding that has been observed in schizophrenia (Rosoklija et al., 2007). This is consistent with previous studies that have found abnormal immune function in schizophrenia.
HLA-B is a member of the MHCI gene family. Its expression was increased in schizophrenia, but only in the nonsmokers. Levels for smokers were indistinguishable from those of controls. This pattern could indicate that HLA-B is over-expressed in schizophrenia, but smoking returns expression levels to within normal limits. In addition, over the entire group (including both schizophrenics and controls), inflammation significantly increased HLA-B expression in nonsmoking subjects, but not in smokers. This suggests that inflammatory illness may further increase the high levels of an HLA-B found in the nonsmoking schizophrenic subjects.
HLA-A, β2 microglobulin, and Notch4 expression displayed a similar expression pattern with respect to schizophrenia. Expression of these genes was increased significantly in control subjects who had systemic inflammatory illness at the time of their death, but this difference in expression was not seen in the schizophrenic subjects. This pattern could indicate that up-regulation of these genes normally occurs with such inflammation, but in schizophrenia, this up-regulation fails to occur.
Taken together these expression data suggest a scenario of aberrant MHCI expression in schizophrenia which is exacerbated during infection and other systemic inflammation. B2M is required for stable expression of almost all MHCIs; its expression may represent the maximum quantity of MHCI proteins on the cell surface. Both the HLA-A and HLA-B proteins require dimerization with β2 microglobulin for stable expression. HLA-B expression was increased 2.3 fold in nonsmoking schizophrenic subjects. These data suggest that in these subjects, increased quantities of HLA-B are competing for dimerization with a limited quantity of β2 microglobulin. This may cause a shift from HLA-A to HLA-B expression on the cell surface. Additionally, in the presence of inflammation, HLA-A failed to up-regulate in schizophrenic subjects (smokers and nonsmokers included). This could cause a further shift toward HLA-B expression in schizophrenia during inflammatory illness. The significance of these shifts is unclear because the roles of different MHCI genes in the human brain have not been determined. In animal models, different MHCI genes are expressed in unique subsets of neurons throughout the CNS (Boulanger, 2009). The different expression patterns seen in HLA-A and HLA-B may reflect heterogeneous roles in neuronal function. Individuals with this aberrant expression pattern may be more susceptible to destruction of synapses during infectious illness. Further, infection or inflammation during important periods in brain development may increase risk for schizophrenia. Several different lines of study have previously demonstrated a link between prenatal or early childhood exposure to infection and schizophrenia (Brown and Derkits, 2009; Khandaker et al., 2012; Patterson, 2007).
There is now an extensive body of research linking inflammation to other psychiatric disorders, especially major depressive disorder (Krishnadas and Cavanagh, 2012; Miller et al., 2009). Inflammatory molecules appear to disrupt normal neural function and connectedness. Research has yet to uncover what determines why one individual experiences a psychotic disorder while another develops an affective one, given similar psychological or physiological stressors. Individual genetic background likely plays a role. In addition, the timing of the exposure to inflammation may also be an important factor. Earlier exposures, especially prenatal or early childhood ones, may be more disruptive to neurodevelopment, resulting in greater cognitive impairment and psychosis.
In this collection of brain samples, which contains both subjects with schizophrenia and unaffected controls, smoking had a significant effect on expression of most of the genes that were analyzed. Smoking decreased expression of both HLA-A and Notch4. Smoking also appears to interact with inflammation so that it inhibits the up-regulation that occurs during inflammatory illness in nonsmokers. This pattern was found in HLA-B, B2M, TAP1 and HLA-DRA. These patterns are meaningful for future studies of immune genes in schizophrenia because schizophrenia patients tend to smoke more than the general public (de Leon and Diaz, 2005). Measuring expression of these genes without accounting for smoking may produce misleading results. Stimulation of nicotinic receptors might be a mechanism for these effects. The α7nAchR is a likely candidate as a regulator due to its role in the inhibition of cytokine release, which is unique among the nicotinic receptors (Wang et al., 2003). Since there have been recent clinical trials of α7nAchR agonists to improve cognition in schizophrenia (Freedman et al., 2008), these medications may also become a factor in this research.
Previous studies have reported conflicting results for the effect of schizophrenia on HLA-A expression (Kano et al., 2011; Mexal et al., 2005); however, these studies did not account for inflammatory illness in the subjects. This could have spuriously affected the outcomes of these studies. One factor that may act as a confounder in this and other studies is the use of psychotropic medications. All of the schizophrenic subjects in this study were prescribed one or more medications at the time of their death. These included various antipsychotics, mood stabilizers, and other adjunctive medications. Noncompliance is also common in this population and is difficult to document. Several studies have suggested that antipsychotic drugs can regulate the immune response by altering cytokine secretion; however the pattern of changes is complex and conflicts from one study to the next (Cazzullo et al., 2002; Chen et al., 2012; Kim et al., 2001; Zhang et al., 2004).
The MHC gene region on chromosome 6p21.3-22.1 is a large block of over 100 genes which is consistently associated with schizophrenia in GWAS studies. These genes are so tightly linked that is difficult to narrow the association with current techniques. We evaluated nine of these genes for expression changes in schizophrenia. Expression of five was altered in subjects with schizophrenia. These are primarily MHCI genes with putative roles in the modification of dendritic connections and in synaptic plasticity.
Supplementary Material
Highlights.
Class I major histocompatibility genes are differentially expressed in the hippocampus in schizophrenia, with a shift from HLA-A to HLA-B expression.
Acknowledgments
The authors would like to acknowledge Dr. Brandie Wagner all of her advice and consultation on the statistical analysis.
Footnotes
Conflict of interest statement:
All authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Andersen CL, Jensen JL, Ørntoft TF. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Research. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B- Methodological. 1995;57:289–300. [Google Scholar]
- Blank T, Prinz M. Microglia as modulators of cognition and neuropsychiatric disorders. Glia. 2012 doi: 10.1002/glia.22372. (ePub ahead of print) [DOI] [PubMed] [Google Scholar]
- Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64:93–109. doi: 10.1016/j.neuron.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Derkits EJ. Prenatal Infection and Schizophrenia: A Review of Epidemiologic and Translational Studies. American Journal of Psychiatry. 2009;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, Perrin M, Gorman JM, Susser ES. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry. 2004;161:889–895. doi: 10.1176/appi.ajp.161.5.889. [DOI] [PubMed] [Google Scholar]
- Cazzullo CL, Sacchetti E, Galluzzo A, Panariello A, Adorni A, Pegoraro M, Bosis S, Colombo F, Trabattoni D, Zagliani A, Clerici M. Cytokine profiles in schizophrenic patients treated with risperidone: A 3-month follow-up study. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2002;26:33–39. doi: 10.1016/s0278-5846(01)00221-4. [DOI] [PubMed] [Google Scholar]
- Chen ML, Tsai TC, Wang LK, Lin YY, Tsai YM, Lee MC, Tsai FM. Risperidone modulates the cytokine and chemokine release of dendritic cells and induces TNF-α-directed cell apoptosis in neutrophils. International Immunopharmacology. 2012;12:197–204. doi: 10.1016/j.intimp.2011.11.011. [DOI] [PubMed] [Google Scholar]
- Corriveau RA, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/s0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- de Leon J, Diaz FJ. A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophrenia Research. 2005;76:135–157. doi: 10.1016/j.schres.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Fellerhoff B, Laumbacher B, Mueller N, Gu S, Wank R. Associations between Chlamydophila infections, schizophrenia and risk of HLA-A10. Mol Psychiatry. 2006;12:264–272. doi: 10.1038/sj.mp.4001925. [DOI] [PubMed] [Google Scholar]
- Fellerhoff B, Wank R. Transporter associated with antigen processing and the chaperone tapasin: Are non-classical HLA genes keys to the pathogenesis of schizophrenia? Medical Hypotheses. 2009;72:535–538. doi: 10.1016/j.mehy.2008.12.036. [DOI] [PubMed] [Google Scholar]
- Fleige S, Pfaffl MW. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 2006;27:126–139. doi: 10.1016/j.mam.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Fleige S, Walf V, Huch S, Prgomet C, Sehm J, Pfaffl M. Comparison of relative mRNA quantification models and the impact of RNA integrity in quantitative real-time RT-PCR. Biotechnology Letters. 2006;28:1601–1613. doi: 10.1007/s10529-006-9127-2. [DOI] [PubMed] [Google Scholar]
- Freedman MDR, Olincy MDA, Buchanan MDR, Harris PDJ, Gold PDJ, Johnson PDL, Allensworth MAD, Guzman-Bonilla BAA, Clement MSWPDB, Ball RNCMSM, Kutnick MDJ, Pender BSV, Martin MDL, Stevens PDK, Wagner PDB, Zerbe PDG, Soti PDF, Kem PDW. Initial Phase 2 Trial of a Nicotinic Agonist in Schizophrenia. American Journal of Psychiatry. 2008;165:1040–1047. doi: 10.1176/appi.ajp.2008.07071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Adler LE, MylesWorsley M, Nagamoto HT, Miller C, Kisley M, McRae K, Cawthra E, Waldo M. Inhibitory gating of an evoked response to repeated auditory stimuli in schizophrenic and normal subjects - Human recordings, computer simulation, and an animal model. Archives of General Psychiatry. 1996;53:1114–1121. doi: 10.1001/archpsyc.1996.01830120052009. [DOI] [PubMed] [Google Scholar]
- Gallowitsch-Puerta M, Tracey KJ. Immunologic role of the cholinergic anti-inflammatory pathway and the nicotinic acetylcholine alpha 7 receptor. Human Immunology: Patient-Based Research. 2005;1062:209–219. doi: 10.1196/annals.1358.024. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: Intrinsic immuneffector cell of the brain. Brain Research Reviews. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Gejman PV, Sanders AR, Kendler KS. Genetics of Schizophrenia: New Findings and Challenges. Annu Rev Genomics Hum Genet. 2011;12:121–144. doi: 10.1146/annurev-genom-082410-101459. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Heath PR, Eastwood SL, Burnet PWJ, McDonald B, Pearson RCA. The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: Selective mRNA vulnerability and comparison with their encoded proteins. Neuroscience Letters. 1995;200:151–154. doi: 10.1016/0304-3940(95)12102-a. [DOI] [PubMed] [Google Scholar]
- Heckers S, Konradi C. Hippocampal neurons in schizophrenia. Journal of Neural Transmission. 2002;109:891–905. doi: 10.1007/s007020200073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du HP, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano Si, Nwulia E, Niwa M, Chen Y, Sawa A, Cascella N. Altered MHC class I expression in dorsolateral prefrontal cortex of nonsmoker patients with schizophrenia. Neuroscience Research. 2011;71:289–293. doi: 10.1016/j.neures.2011.07.1818. [DOI] [PubMed] [Google Scholar]
- Khandaker GM, Zimbron J, Dalman C, Lewis G, Jones PB. Childhood infection and adult schizophrenia: A meta-analysis of population-based studies. Schizophrenia Research. 2012;139:161–168. doi: 10.1016/j.schres.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DJ, Kim WY, Yoon SJ, Go HJ, Choi BM, Jun TY, Kim YK. Effect of Risperidone on Serum Cytokines. International Journal of Neuroscience. 2001;111:11. doi: 10.3109/00207450108986549. [DOI] [PubMed] [Google Scholar]
- Kolluri N, Sun ZX, Sampson AR, Lewis DA. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. American Journal of Psychiatry. 2005;162:1200–1202. doi: 10.1176/appi.ajp.162.6.1200. [DOI] [PubMed] [Google Scholar]
- Krishnadas R, Cavanagh J. Depression: an inflammatory illness? Journal of Neurology, Neurosurgery & Psychiatry. 2012;83:495–502. doi: 10.1136/jnnp-2011-301779. [DOI] [PubMed] [Google Scholar]
- Law AJ, Kleinman JE, Weinberger DR, Weickert CS. Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum Mol Genet. 2007;16:129–141. doi: 10.1093/hmg/ddl449. [DOI] [PubMed] [Google Scholar]
- Leonard S, Adler LE, Benhammou K, Berger R, Breese CR, Drebing C, Gault J, Lee MJ, Logel J, Olincy A, Ross RG, Stevens K, Sullivan B, Vianzon R, Vernich DE, Waldo M, Walton K, Freedman R. Smoking and mental illness. Pharmacol Biochem Behav. 2001;70:561–570. doi: 10.1016/s0091-3057(01)00677-3. [DOI] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Healy DJ. Glutamate receptor expression in schizophrenic brain. Brain Research Reviews. 2000;31:288–294. doi: 10.1016/s0165-0173(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Mexal S, Berger R, Adams CE, Ross RG, Freedman R, Leonard S. Brain pH has a significant impact on human postmortem hippocampal gene expression profiles. Brain Res. 2006;1106:1–11. doi: 10.1016/j.brainres.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Mexal S, Frank M, Berger R, Adams CE, Ross RG, Freedman R, Leonard S. Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. Molecular Brain Research. 2005;139:317–332. doi: 10.1016/j.molbrainres.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biological Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J Exp Med. 1997;185:305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton SS, Dow A, Terwilliger R, Duman R. A simplified method for combined immunohistochemistry and in-situ hybridization in fresh-frozen, cryocut mouse brain sections. Brain research Brain research protocols. 2002;9:214–219. doi: 10.1016/s1385-299x(02)00148-4. [DOI] [PubMed] [Google Scholar]
- Olincy A, Young DA, Freedman R. Increased levels of the nicotine metabolite cotinine in schizophrenic smokers compared to other smokers. Biological Psychiatry. 1997;42:1–5. doi: 10.1016/S0006-3223(96)00302-2. [DOI] [PubMed] [Google Scholar]
- Pantelis C, Yucel M, Wood SJ, Velakoulis D, Sun D, Berger G, Stuart GW, Yung A, Phillips L, McGorry PD. Structural brain imaging evidence for multiple pathological processes at different stages of brain development in schizophrenia. Schizophr Bull. 2005;31:672–696. doi: 10.1093/schbul/sbi034. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Neuroscience. Maternal effects on schizophrenia risk. Science. 2007;318:576–577. doi: 10.1126/science.1150196. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol. 2000;59:137–150. doi: 10.1093/jnen/59.2.137. [DOI] [PubMed] [Google Scholar]
- Rosoklija G, Derkits E, Serafimova T, Dika A, Mancevski B, Stankov A, Davceva N, Jakovski Z, Pavlovski G, Todorova L, Todorov B, Duma A, Dwork AJ. Post mortem studies of dendritic abnormalities in schizophrenia and mood disorders. Schizophrenia Bulletin. 2007;33:271–272. [Google Scholar]
- Schmeisser MJ, Baumann B, Johannsen S, Vindedal GF, Jensen V, Hvalby ØC, Sprengel R, Seither J, Maqbool A, Magnutzki A, Lattke M, Oswald F, Boeckers TM, Wirth T. IκB Kinase/Nuclear Factor κB-Dependent Insulin-Like Growth Factor 2 (Igf2) Expression Regulates Synapse Formation and Spine Maturation via Igf2 Receptor Signaling. The Journal of Neuroscience. 2012;32:5688–5703. doi: 10.1523/JNEUROSCI.0111-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of Lipofuscin-like Autofluorescence in Fluorescently Labeled Tissue. Journal of Histochemistry & Cytochemistry. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Shatz CJ. Neural activity, immune genes and synaptic remodeling in brain development. FASEB Journal. 2002;16:A378–A378. [Google Scholar]
- Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron. 2009;64:40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, Kendler KS, Freedman R, Gejman PV. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith IA, Knezevic BR, Ammann JU, Rhodes DA, Aw D, Palmer DB, Mather IH, Trowsdale J. BTN1A1, the Mammary Gland Butyrophilin, and BTN2A2 Are Both Inhibitors of T Cell Activation. The Journal of Immunology. 2010;184:3514–3525. doi: 10.4049/jimmunol.0900416. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Borglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Bottcher Y, Olesen J, Breuer R, Moller HJ, Giegling I, Rasmussen HB, Timm S, Mattheisen M, Bitter I, Rethelyi JM, Magnusdottir BB, Sigmundsson T, Olason P, Masson G, Gulcher JR, Haraldsson M, Fossdal R, Thorgeirsson TE, Thorsteinsdottir U, Ruggeri M, Tosato S, Franke B, Strengman E, Kiemeney LA, Melle I, Djurovic S, Abramova L, Kaleda V, Sanjuan J, de FR, Bramon E, Vassos E, Fraser G, Ettinger U, Picchioni M, Walker N, Toulopoulou T, Need AC, Ge D, Yoon JL, Shianna KV, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Carracedo A, Arango C, Costas J, Jonsson EG, Terenius L, Agartz I, Petursson H, Nothen MM, Rietschel M, Matthews PM, Muglia P, Peltonen L, St CD, Goldstein DB, Stefansson K, Collier DA. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stone WS, Hsi X. Declarative memory deficits and schizophrenia: Problems and prospects. Neurobiology of Learning and Memory. 2011;96:544–552. doi: 10.1016/j.nlm.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Stone WS, Seidman LJ. Toward a Model of Memory Enhancement in Schizophrenia: Glucose Administration and Hippocampal Function. Schizophrenia Bulletin. 2008;34:93–108. doi: 10.1093/schbul/sbm041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang HC, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha 7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Zhang XY, Zhou DF, Cao LY, Zhang PY, Wu GY, Shen YC. Changes in serum interleukin-2, -6, and -8 levels before and during treatment with risperidone and haloperidol: relationship to outcome in schizophrenia. The Journal of clinical psychiatry. 2004;65:940–947. doi: 10.4088/jcp.v65n0710. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.