

Abstract
Chlamydiaspecies are obligate intracellular pathogens that proliferate only within infected cells. Currently, there are no known techniques or systems that can probe the spatial distribution of metabolites of interest within intact Chlamydia-infected cells. Here we investigate the ability of Raman microscopy to probe the chemical composition of different compartments (nucleus, inclusion, and cytoplasm) of C. trachomatis-infected epithelial cells. The overall intensity of the Raman spectrum is greatest in the inclusions and lowest in the cytoplasm in fixed cells. Difference spectra generated by normalizing to the intensity of the strong 1004 cm−1 phenylalanine line show distinct differences among the three compartments. Most notably, the concentrations of adenine are greater in both the inclusions and the nucleus than in the cytoplasm, as seen by Raman microscopy. The source of the adenine was explored through a complementary approach, using two-photon microscopy imaging. Autofluorescence measurements of living, infected cells show that the adenine-containing molecules, NAD(P)H and FAD, are present mainly in the cytoplasm, suggesting that these molecules are not the source of the additional adenine signal in the nucleus and inclusions. Experiments of infected cells stained with a DNA-binding dye, Hoechst 33258, reveal that most of the DNA is present in the nucleus and the inclusions, suggesting that DNA/RNA is the main source of the additional Raman adenine signal in the nucleus and inclusions. Thus, Raman and two-photon microscopy are among the few non-invasive methods available to investigate cells infected with Chlamydia and, together, should also be useful for studying infection by other intracellular pathogens that survive within intracellular vacuoles.
Keywords: Raman microscopy, fluorescence microscopy, metabolism, Chlamydia
1. Introduction
Chlamydia trachomatis is the main cause of preventable blindness and the most common sexually transmitted bacterial species in humans, being responsible for an estimated 90 million new sexually-transmitted cases per year worldwide [1–4]. In addition, C. trachomatis infections facilitate the acquisition and transmission of human immunodeficiency virus type 1 (HIV-1) and promote the development of human papilloma virus (HPV)-induced neoplasia [5–8].
These Gram-negative bacteria are obligate intracellular pathogens that reside within a membrane-bound parasitophorous vacuole (termed an inclusion) of the eukaryotic host cell, preferentially an epithelial cell [9, 10]. In epithelial cells, the bacteria undergo a complex biphasic life cycle [9] which takes place entirely within the inclusion [10–13]. Two morphologically distinct forms of chlamydiae have been characterized during the infection cycle. Elementary bodies (EBs) are small (0.3 μm), spore-like bacteria that are infectious but are metabolically inactive and cannot replicate. The EBs differentiate intracellularly into non-infectious reticulate bodies (RBs), which are larger (1.0 μm) and are metabolically active, and multiply within the inclusion [1]. For most C. trachomatis strains, RBs are abundant at about 24 hours, and then differentiate into EBs. The entire infection cycle typically lasts two days, after which the infectious EBs are released and a new infection cycle beings in a newly-infected neighboring cell [14].
Intracellular pathogens usually rely on their hosts to provide the nutrients, amino acids, nucleotides and other metabolites necessary for survival. Chlamydiae obtain amino acids [15] and nucleotides [16, 17] from the host; however, the process whereby these metabolites cross the inclusion membrane is not well understood. In addition, it is known that the inclusion membrane is not passively permeable to fluorescent tracers as small as 520 Da, since these tracers, when introduced directly into the host-cell cytoplasm, were excluded from the chlamydial inclusion [9]. An alternative method is needed to investigate the compounds around the inclusion.
Chlamydiae are hypothesized to be “energy parasites,” whose multiplication depends on ATP and other high-energy metabolites generated by the catabolism of glucose by the host. [10] Utilizing 14C-labeled glucose, it was shown that the majority of CO2 is produced from glucose and this reaction is dependent on ATP [10, 18]. ATP is needed for the phosphorylation of glucose to glucose-6-phosphate (first committed step in glycolysis of metabolism) by a hexokinase that was considered to be, most likely, of host origin [10, 18].
We have previously used NMR as a noninvasive probe of the average concentration of ATP and other metabolites in living infected cells [19], but the NMR technique does not provide information on the intracellular localization of the metabolite. Fluorescence microscopy is used routinely to localize different antigens within fixed cells [20]. The movement of proteins tagged with green fluorescent protein (GFP) can be followed in living cells, and certain molecules such as lipids can also be tagged with fluorescent markers and visualized in real time [21, 22]. Mass spectrometry and related bioanalytical techniques can quantify the concentration of any metabolite in cells, but only after lysing the cells [23, 24]. The metabolic coenzymes, NADH and NADPH, can be imaged in living C. trachomatis-infected cells by two-photon microscopy [25]. However, general techniques for measuring concentrations of ATP and other metabolites in different intracellular compartments in an intact cell remain elusive. A powerful emerging technique for studying living cells is Raman microscopy, which combines Raman spectroscopy with a light microscope.
Raman spectroscopy has many advantages as a bioanalytical technique. The Raman spectrum of a polyatomic molecule provides a unique fingerprint for the analyte. The intensity of the Raman signal is usually linearly proportional to the concentration of the analyte, and the background signal from water is weak. When the Raman excitation and detection system is coupled to a confocal microscope, spectra can be obtained at a spatial resolution of better than 1 μm laterally and typically less than 2 μm in depth. Thus, spectra can be obtained from specific locations within cells including the cytoplasm, nucleus, or vacuoles harboring pathogens.
Recent studies have used Raman microscopy to probe glycerol and ethanol concentrations in living yeast cells under hyperosmotic stress conditions [26], to examine metabolic changes associated with spontaneous cell death in yeast [27], to observe the distribution of biochemical components in live human cells at various stages of mitosis [28], to monitor molecular changes associated with apoptosis in human cells [29, 30], to discriminate benign from malignant prostate cells [31], to determine the cell cycle phase in single living cells [32], to measure the spatial distribution of mitochondria within cells [33], and to study bacterial metabolic states [34]. Raman spectroscopy has also been used in stem cell research to monitor RNA translation in murine embryonic stem cells during differentiation [35] and to identify human embryonic stem cells [36]. To our knowledge, the only application to Chlamydia infection was a very recent study by Haider et al. [37] In that work, Raman microspectroscopy was used to differentiate between RBs and EBs of the amoeba symbiont Protochlamydia amoebophila and to demonstrate in situ labeling of the pathogen after addition of isotopically labeled phenylalanine. An unexpected result of that study was the observation that both RBs and EBs of P. amoebophila demonstrated metabolic activity outside the host cell [37].
In the recent study of P. amoebophila by Haider et al. [37], all of the Raman experiments were performed on EBs and RBs released from lysed host cells. However, Raman microspectroscopy also has the potential to measure differences in the concentration of ATP and other metabolites in intact cells. In the present work we explore the possibility of utilizing non-invasive Raman microscopy to investigate the concentrations of DNA, RNA and metabolites such as ATP in cervical epithelial cells infected by C. trachomatis. As a complementary approach, we use cellular autofluorescence imaging of NAD(P)H and FAD by two-photon microscopy and DNA staining to confirm the results of Raman microscopy.
2. Materials and Methods
2.1. Cells and Materials
The Chlamydia species used here, the LGV/L2 strain of C. trachomatis [lymphogranuloma venereum (LGV/L2)] was obtained from American Type Culture Collection (ATCC, Manassas, VA). HeLa cells (HeLa 229) from ATCC were maintained in a humidified incubator at 37 °C with 5% CO2. The cells were both cultured and maintained in Dulbecco’s modified Eagle’s medium (Invitrogen by Gibco, Carlsbad, CA) supplied with 10% heat inactivated fetal bovine serum (Invitrogen) and 2 mM (4.5 ml/L) L-glutamine. The substrates for Raman microscopy are commercial quality flat, S1-UV grade fused silica windows, 1.0 mm thick by 12.7 mm diameter (Esco Products, Oak Ridge, NJ).
2.2. Preparation of Chlamydiae
The chlamydiae were cultured in infected HeLa cell monolayer cultures in a standard manner as described [19, 38]. Briefly, infected HeLa cells were cultured on multiple 9-cm Petri culture dishes and harvested at 48 h post-infection. The cells and supernatant were combined and centrifuged for 60 min at 12,000 rpm in a Sorvall type GSA rotor. The pellet was resuspended in ice-cold sucrose/phosphate/glucose buffer (SPG), and the cells were sonicated on ice for 30 s. The resulting suspension was centrifuged for 10 min at 2,000 rpm in a Sorvall SS34 rotor to remove unbroken cells. To collect the bacteria, the new supernatant was centrifuged again for 30 min at 15,000 rpm at 4 °C. The pellet was resuspended in ice-cold SPG with a 21-gauge 2-ml syringe to dissociate aggregates, giving the final suspension of EBs used in subsequent infection experiments. This suspension was aliquoted and stored at −80 °C until ready for use.
2.3. Cell Culture and Infection on Fused Silica
HeLa cells cultivated as described above were trypsinized and plated in 3 separate wells of a sterile 12-well plate (at 4.8×105 cells per 1 ml of standard growth medium described above) by Costar (Corning Incorporated, Corning, NY) containing the fused silica substrates. The plate was gently swirled and placed in the CO2 incubator. The cells with the silica substrates remained in the incubator for 6–8 hours allowing the cells to attach to the silica before infection or other treatment.
Cells were infected with C. trachomatis (L2) at a multiplicity of infection (MOI) of 1 for the indicated time. The infection was stopped by removing the media and fixing the cells with cold methanol (> 99.8% A.C.S reagent, from Sigma) or formalin for 15 minutes, before being transferred to the Raman microscope.
2.4. Raman Microscopy
The Raman spectra were recorded using a Jobin-Yvon T64000 triple spectrometer equipped with a liquid-N2-cooled charge-coupled-device detector and a confocal Raman microprobe based on an Olympus BX-41 microscope. A 100x objective was used for all experiments. Laser excitation (~5 mW at 488 nm) was provided by a Coherent I90-C argon-ion laser. Each spectrum was obtained as an average of three accumulations of 60 seconds each. The focus was placed very slightly above the surface of the cell to avoid cellular damage. Background signal from the substrate was removed by subtracting a user-defined background. The entrance slit to the spectrograph was set to 100 μm, corresponding to a spectral resolution of about 5 cm−1 at the detector. Spectra were calibrated by reference to the peak positions of cyclohexane. Different cells from the same microscope field were analyzed. Spectra show representative results from at least three separate experiments.
2.5. Hoechst 33258 Staining of Living and Fixed Cells
HeLa cells were grown on cover glass in 50 mm tissue culture dishes and infected with C. trachomatis as described above. For two-photon microscopy studies, cells were incubated with 1 μg/ml Hoechst 33258 for 5 minutes and cover glasses were transferred to a MiniCeM chamber for microscopy (JenLab, Jena, Germany). For fluorescence microscopy imaging, cells were fixed in ice-cold methanol for 5 minutes and incubated with 0.5 μg/ml Hoechst 33258 solution for 15 minutes and washed twice with PBS. The cover glasses were mounted on glass slides for imaging.
2.6. Two-photon Microscopy Imaging of NAD(P)H and FAD
The two-photon microscope (DermaInspect; Jenlab) was equipped with a Chroma 640DCSPXR dichroic mirror (AHF analysentechnik AG, Tübingen, Germany), a blue emission filter (BG39, Schott AG, Mainz, Germany) and a 40x/1.3 Plan-Apochromat oil-immersion objective (Zeiss, Göttingen, Germany). A tunable infrared titanium-sapphire femtosecond-laser (710–920 nm tuning range; MaiTai; Spectra Physics, Darmstadt, Germany) was used as an excitation source at 730 nm excitation for NAD(P)H and at 890nm for FAD.
2.7. Fluorescence microscopy and image analysis
Images of fixed cells were obtained using the BZ-9000 fluorescence microscope (Keyence, Osaka, Japan). Pseudo-color images were created using the ImageJ software (NIH, Bethesda, MD).
3. Results
The fixed cells were first viewed optically with a light microscope (an alternative mode of operation of the Raman microscope) to identify the nucleus, the cytoplasm, and the chlamydial inclusion. In a single microscope field, both infected and uninfected cells were identified by the presence of inclusions in the infected cells and a clear distinction could be made between the nuclei and inclusions (Fig. 1). The excitation laser was then brought into the microscope and the focused beam was positioned in the cell compartment of interest for collection of Raman spectra.
Fig. 1.
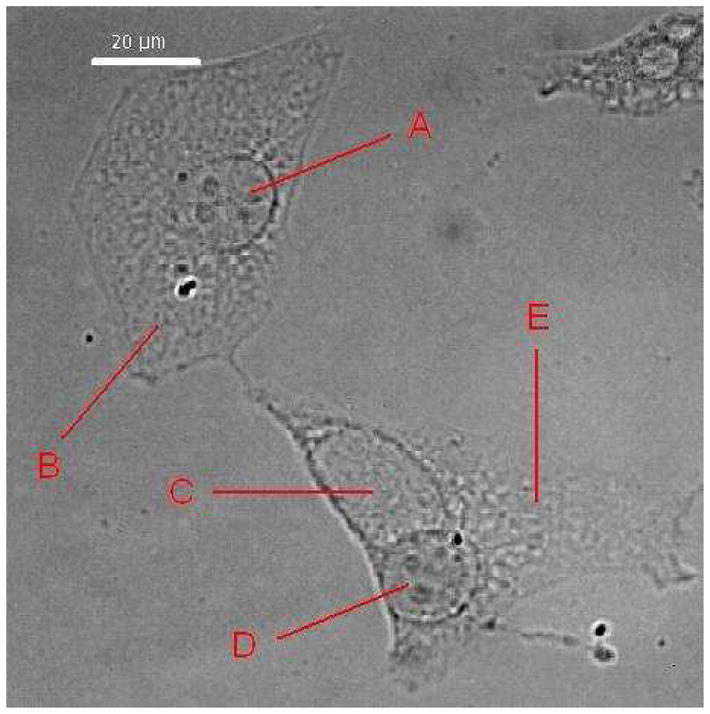
Light microscopy of epithelial cells cultured on fused silica. Methanol-fixed HeLa cells, uninfected or infected with C. trachomatis at an MOI of 1 for 24 hrs. The nucleus “A” and the cytoplasm “B” of an uninfected cell (upper left) contain no chlamydiae. The infected cell (lower right) contains an inclusion “C” and a nucleus “D” surrounded by the cytoplasm “E”.
HeLa cells growing on silicon cover slips were infected at an MOI = 1.0 with C. trachomatis LGV2 for 36 hours (when inclusions are slightly larger than at 24 hours and RBs are differentiating back to EBs [39]) and then fixed with methanol. Raman spectra of separate components of infected cells are shown in Fig. 2. Single Raman scans from the cytoplasm, nucleus, and inclusion of independent cells give very reproducible Raman spectra from cell to cell. The most obvious difference among the compartments is the much greater intensity of the Raman spectra obtained from the inclusions relative to the nucleus or cytoplasm. This result was also highly reproducible over different sets of cells infected during separate experiments.
Fig. 2.

Raman spectra of individual cells. Single Raman scans of the inclusion (black), nucleus (red) and cytoplasm (blue) regions of four separate infected cells. Spectra have been displaced vertically for clarity.
Because of the limited signal-to-noise ratio obtained from single scans on individual cells, multiple spectra of each region of five different cells were averaged to obtain a typical spectrum of each compartment (Fig. 3). These Raman spectra show a number of well-defined peaks, most of which can be assigned by reference to prior work [31, 35, 40]. A relatively intense and very sharp peak around 1004 cm−1 in each spectrum is assigned to phenylalanine (Phe).
Fig. 3.
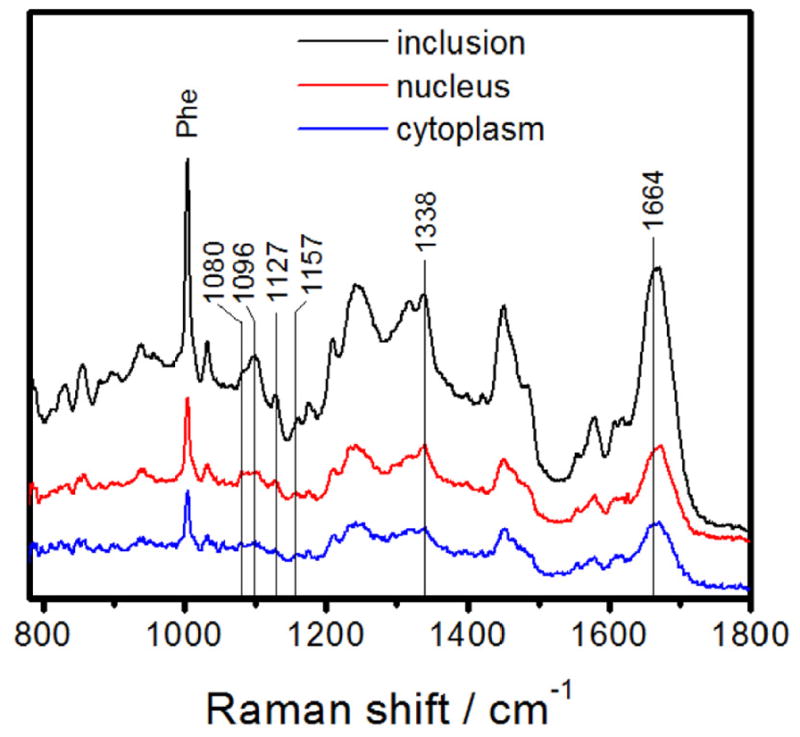
Raman spectra averaged over multiple scans of each of five cells for the inclusion (black), nucleus (red) and cytoplasm (blue). Spectra have been displaced vertically for clarity. The labeled peaks correspond to frequencies that appear both as resolved peaks in one or more of the averaged cell compartment spectra and in the difference spectra of Fig. 4 (below). “Phe” labels the sharp phenylalanine peak used for normalization in composing the difference spectra.
Differences in the compositions of the three compartments were examined by constructing difference spectra, inclusion minus cytoplasm, and nucleus minus cytoplasm. The cytoplasm spectrum was multiplied by a constant selected to make the sharp Phe line disappear in the difference spectrum. This should effectively cancel the contribution from amino acid residues to the difference spectra to the extent that the proteins in the different cell compartments have comparable abundances of Phe. The resulting spectra, shown in Fig. 4, contain positive-going peaks for compounds whose abundance relative to Phe is greater in the inclusion (or nucleus) than in the cytoplasm, and negative-going peaks representing compounds that are present in greater abundance relative to Phe in the cytoplasm than in the inclusion (or nucleus). The difference spectra are noisy and contain a number of features at frequencies that do not clearly correspond to peaks in the original spectra. While these may represent true Raman features that become apparent only when the difference spectra are constructed, we have chosen to report only those frequencies that appear both in the difference spectra and in the original spectra of one or more cell compartments.
Fig. 4.

Raman difference spectra from the averaged spectra of Fig. 3. Left: subtraction of 1.5 times the cytoplasm spectrum from the nucleus spectrum. Right: subtraction of 3.5 times the cytoplasm spectrum from the inclusion spectrum. In both cases the subtraction parameter was chosen to remove the sharp 1004 cm−1 Phe line. The labeled peaks correspond to frequencies that appear both in the difference spectra and as resolved peaks in one or more of the averaged cell compartment spectra of Fig. 3.
The nucleus minus cytoplasm difference spectrum shows positive peaks (more in nucleus) at 1084, 1130, and 1336 cm−1. The 1336 cm−1 band is assigned to the strongest band of adenine [31, 41] although protein C-H modes also appear near this frequency [35]. The 1130 and 1084 cm−1 features may be the 1116–1129 cm−1 band of ATP and the ~1085 cm−1 band of ADP, respectively [42–44]. However, other assignments are also possible. The 1130 cm−1 line could also originate from protein C-N stretching [31, 35] if the protein backbone contribution to the spectrum is incompletely subtracted, and lines near 1085 cm−1 can arise not only from the symmetric PO2 stretch of ADP but also from phospholipids and carbohydrates [33, 35].
The inclusion minus cytoplasm difference spectrum shows positive peaks (more in inclusions) at ~1098, 1336, and 1661 cm−1. The 1661 cm−1 band is difficult to assign, as this region around 1660 cm−1 contains not only the protein amide I vibration [31, 35, 40] but also modes of thymine [31] and C=C stretches of unsaturated lipids [30, 31, 35, 45]. The 1336 cm−1 adenine peak is present as it was in the nucleus minus cytoplasm spectrum. The ~1098 cm−1 feature is close to the 1085–1097 cm−1 phosphate stretching band of ADP but there is no strong feature at 1120–1130 cm−1, suggesting that the adenine is not primarily from ATP. There is also a negative peak at 1156 cm−1 for which we do not have a good assignment. Raman bands near this frequency in cells are usually assigned as protein C-C stretching [35], but we do not expect significant protein contributions to our difference spectra.
Difference spectra between the cytoplasm at 36 hr after infection and the cytoplasm at 0 hr were also examined with the goal of revealing modification of host cell metabolite resources by C. trachomatis, but these difference spectra showed no reproducible features to within the signal-to-noise ratio of the data.
Adenine is found not only in ATP and ADP but also in DNA, RNA, and metabolic coenzymes such as NAD(H), NADP(H), FAD(H), and signaling molecules such as cAMP. NAD(P)H (reduced form), and FAD (oxidized form) can be imaged in living cells through their cellular autofluorescence [46]. Two photon microscopy imaging of NAD(P)H at 730 nm excitation and of FAD at 890 nm excitation in living C. trachomatis-infected HeLa cells shows that the concentrations of these coenzymes are highest in the cytoplasm and in the inclusion, and lowest in the nucleus (Fig. 5A). It was previously shown that the highest intensity NAD(P)H signal in the cytoplasm originates from the mitochondria in C. trachomatis-infected Hep2 cells [25]. FAD autofluorescence shows a similar distribution to NAD(P)H. The Raman adenine signal that is highest in the nucleus and in the chlamydial inclusion does not correspond to the distribution of NAD(P)H and FAD. Consequently, the source of adenine in the inclusion and nucleus is mainly DNA/RNA. The DNA binding fluorochrome, Hoechst 33258, preferentially binds to adenine and thymine containing DNA sequences [47]. In living cells that were incubated with Hoechst 33258, two photon microscopy imaging can visualize the cellular adenine that originates from DNA in the nucleus; however, as the inclusion membrane is not permeable to Hoechst 33258, the chlamydial DNA in the inclusion cannot be visualized (Fig. 5B). Nevertheless, in fixed cells, Hoechst 33258 staining clearly shows chlamydial DNA in the inclusion and cellular DNA in the nucleus, the most likely source of increased Raman adenine signal in these compartments (Fig. 5C).
Fig. 5.
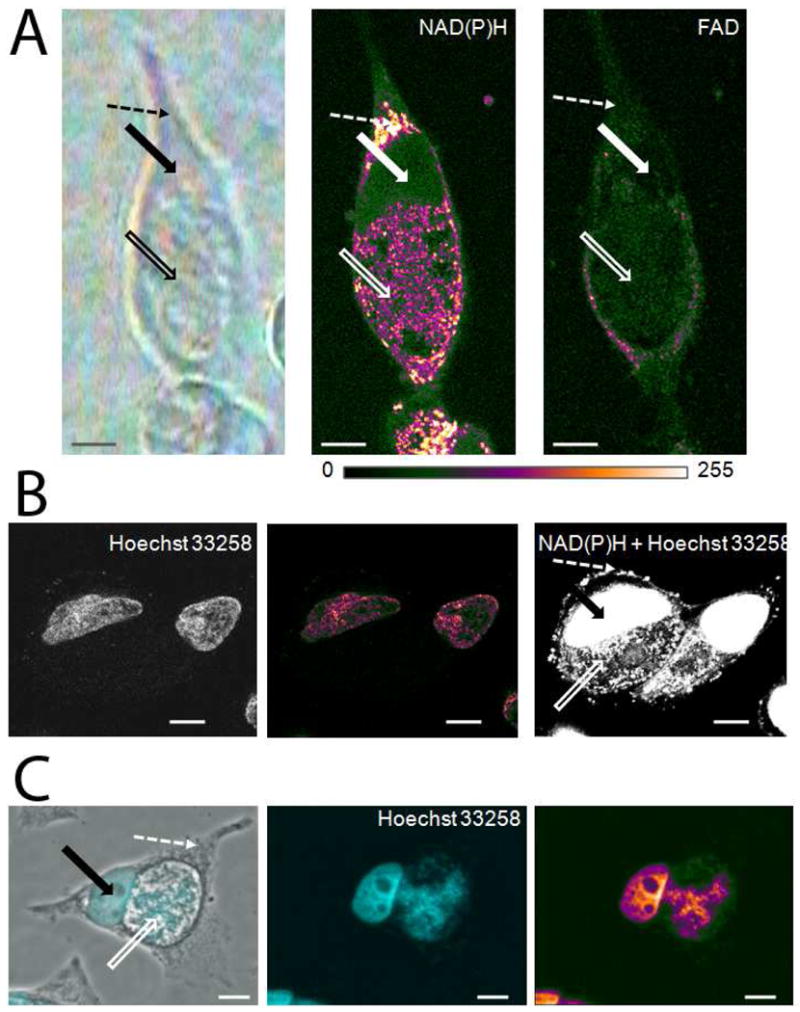
(A) Imaging the autofluorescence of the adenine containing metabolic coenzymes NAD(P)H and FAD in living cells by two-photon microscopy. Bright field microscopy image (left picture) of Hela cells infected with C. trachomatis for 36 hours. NAD(P)H autofluorescence intensities (middle picture) are depicted in pseudo-colors. NAD(P)H was excited at 730 nm by a laser power of 12 mW by two-photon microscopy. At 2.3 mW excitation, no NAD(P)H autofluorescence signal can be detected. FAD autofluorescence intensities (right picture) are depicted in pseudo-colors.
(B). Hoechst 33258 staining of living C. trachomatis-infected HeLa cells. Imaging of living cells at 730 nm excitation with a low laser power (2.3mW) visualizes Hoechst staining of the nucleus (left picture). Fluorescence intensities of Hoechst are depicted in pseudo-colors (middle picture). Higher laser power (12mW) causes the simultaneous excitation of Hoechst fluorescence and NAD(P)H autofluorescence due to their very similar excitation spectra (right picture).
(C). Hoechst 33258 staining of fixed C. trachomatis-infected HeLa cells. Hoechst staining of fixed cells visualizes nuclear and chlamydial DNA. Overlay of phase contrast image and Hoechst staining image (left picture). Solid arrows point to the nucleus of cells, open arrows point to the inclusion and dashed arrows point to selected cytoplasmic regions of C. trachomatis-infected cells. Intensities of Hoechst staining of chlamydial and nuclear DNA (middle picture) are depicted in pseudo colors (right picture). Scale bars represent 10 μm.
4. Discussion
The most striking feature of the Raman spectra shown in Figs. 2 and 3 is the greatly increased overall intensity of the inclusion spectra relative to those obtained from the nucleus or cytoplasm. The sharp band at 1004 cm−1, clearly assigned to Phe, is 2–4 times stronger in the inclusion spectra than in the cytoplasm or nucleus spectra. It is unlikely that the phenylalanine concentration in the inclusions is actually 2–4 times greater than in the other cell compartments. More likely, this is a result of deformation of the cells when they are fixed with cold methanol. If the cytoplasmic and nuclear regions of the cells “flatten out” more than the inclusions upon fixation, the optical path length through the inclusions will be greater and they will generate more Raman scattering. This effect is exaggerated by the placement of the laser focus slightly above the cell: the laser is more tightly focused on the inclusions than on the other cell compartments and, perhaps more importantly, the Raman scattering from the inclusions is collected more efficiently by the objective.
The signal-to-noise ratio of these Raman difference spectra is not sufficient to provide clear conclusions about the chemical composition of the inclusions. The most prominent band in the inclusion difference spectrum, near 1660 cm−1, most likely indicates a greater concentration of unsaturated lipids in the inclusions. The alternative assignment to the protein amide I band is less likely because of the protein-normalizing subtraction protocol employed. The positive feature at the frequency of the strongest Raman line of adenine (1337 cm−1) [48] suggests that the inclusions, like the nuclei, contain relatively more adenine than does the cytoplasm. However, there are no apparent features near 1120–1125 or 983 cm−1, where ATP also has fairly strong lines [49]. As the line of adenine can be also attributed to NAD, FAD or DNA, we performed further investigations by fluorescence microscopy.
To circumvent artifacts associated with methanol fixation, we used two-photon fluorescence imaging of C. trachomatis-infected cells. One of the advantages of two-photon microscopy is that it enables the imaging of living cells by minimizing photo-induced damage. There are a limited number of molecules that are autofluorescent; among them are certain amino-acids, lipids, vitamins and the adenine-containing metabolic coenzymes, NAD(P)H and FAD [50]. By visualizing NAD(P)H and FAD in living C. trachomatis-infected cells, we could show that the autofluorescence intensities of NAD(P)H and FAD were highest in the cytoplasm and in the inclusion (Fig. 5A), representing mitochondrial and chlamydial energy metabolism. Therefore we could exclude these metabolic coenzymes as the source of the increased Raman adenine signal in the nucleus. To visualize the DNA in living and fixed cells, we studied Hoechst 33258 staining by two-photon and fluorescence microscopy. As the inclusion membrane is not permeable to Hoechst 33258 in living cells, we could label only DNA in the host cell nucleus (Fig. 5B). NAD(P)H and Hoechst 33258 have similar excitation spectra, which enabled the simultaneous visualization of nuclear DNA and NAD(P)H in the chlamydial inclusion and the host cell cytoplasm. After methanol fixation, the inclusion membrane also becomes permeable for Hoechst 33258. Cellular autofluorescence imaging by two-photon microscopy indicates that DNA and/or RNA, and not NAD(P)H and FAD, are the major sources of the Raman adenine signal in the host cell nucleus and inclusion.
Although the results obtained here are not definitive, Raman microscopy does show considerable promise for detecting the relative concentration of metabolites in different particular regions of intact (and potentially living) infected and non-infected cells. In order to improve the results from this study, two challenges must be overcome. First, the signal-to-noise ratio of the spectra of each compartment must be improved. In principle this can be achieved by averaging spectra of a larger number of cells, although this is very tedious. (Signal averaging for a longer period of time on each cell is of doubtful value because of the possibility of damage to the cells during prolonged laser irradiation.) It should also be possible to improve the signal-to-noise ratio by using a more efficient light collection system. The triple spectrograph employed in these experiments provides excellent stray light rejection but has high optical losses. A fast single spectrograph with a notch filter is a more appropriate instrument for experiments performed at a single, fixed excitation wavelength where detection at low Raman shifts is not required. Second, the Raman spectra of cells are extremely rich and there is almost no vibrational frequency that is unique to a particular compound. Isotopic labeling of specific compounds is often necessary to definitively assign a given Raman peak to a particular compound.
A common alternative approach to increasing the strength of weak Raman signals is the use of surface-enhanced Raman scattering (SERS) [51]. Compounds located at or near the surface of gold or silver nanoparticles undergo enormous enhancements in their Raman scattering signals, allowing detection of very small quantities of material, sometimes down to the single-molecule level. SERS is most often used as an in vitro analytical tool and has been employed, for example, in the detection of DNA sequences specific to Chlamydia [52]. SERS has been demonstrated inside living cells [53], but its utility for probing the distribution of metabolites in Chlamydia-infected cells is questionable for several reasons. First, SERS enhancements tend to be highly nonlinear with concentration and strongly dependent on chemical and physical interactions between the analyte of interest and the surface of the nanoparticle, and only certain analytes are readily detectable. We are not aware that SERS has ever been used to distinguish between ATP and other adenine-containing compounds, even in vitro. Second, the low permeability of the chlamydial inclusions [9] suggests that harsh, membrane-disrupting methods would have to be employed in order to get nanoparticles into the inclusions. For these reasons we believe that ordinary, unenhanced Raman microscopy remains a more promising tool for this type of in situ cellular analysis.
Concluding Remarks
Different metabolites are localized in diverse cellular compartments due to their involvement in defined cellular functions. Techniques such as two-photon microscopy and Raman spectroscopy that provide information about the spatial distribution of molecules in different cellular compartments provide an extra level of information compared to traditional biochemical methods that do not consider the different subcellular concentrations of molecules. In addition, these techniques will enable the separate analysis of microbial and host activity during diverse obligate intracellular infections that were almost impossible to characterize until now. These noninvasive analytical techniques for characterizing metabolism and the intracellular distribution of metabolites within cells should be valuable for investigating infections with other pathogens that also survive within vacuoles (inclusions or parasitophorous vacuoles) in the host cell, such as the causative agents of tuberculosis, leprosy or leishmaniasis.
Acknowledgments
Two-photon microscopy imaging was performed at the Institute of Biomedical Optics (University of Lübeck, Germany), with the expert assistance of Lisa Krapf. Raman microscopy was performed at the University of California, Merced. These studies were funded partially with the support from a University of California Presidential Chair and the University of Lübeck, Germany (grant number: E01-2011).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rockey DD, Lenart J, Stephens RS. Genome sequencing and our understanding of chlamydiae. Infect Immun. 2000;68:5473–5479. doi: 10.1128/iai.68.10.5473-5479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schachter J. Infection and disease epidemiology. In: Stephens RS, editor. Chlamydia: Intracellular Biology, Pathogenesis, and Immunity. ASM Press; Washington, D.C: 1999. pp. 139–169. [Google Scholar]
- 3.Schachter J, Dawson CR. The epidemiology of trachoma predicts more blindness in the future. Scand J Infect Dis. 1990;69:55–62. [PubMed] [Google Scholar]
- 4.Stamm WE. Chlamydia trachomatis infections: progress and problems. J Infect Dis. 1999;179(Suppl 2):S380–S383. doi: 10.1086/513844. [DOI] [PubMed] [Google Scholar]
- 5.Plummer FA, Simonsen JN, Cameron DW, Ndinya-Achola JO, Kreiss JK, Gakinya MN, Waiyaki P, Cheang M, Piot P, Ronald AR. Cofactors in male-female sexual transmission of human immunodeficiency virus type 1. J Infect Dis. 1991;163:233–239. doi: 10.1093/infdis/163.2.233. [DOI] [PubMed] [Google Scholar]
- 6.Peeling RW, Mabey D, Herring A, Hook EWr. Why do we need quality-assured diagnostic tests for sexually transmitted infections? Nature Rev Microbiol. 2006;4:909–921. doi: 10.1038/nrmicro1555. [DOI] [PubMed] [Google Scholar]
- 7.Trottier H, Franco EL. The epidemiology of genital human papillomavirus infection. Vaccine. 2006;24:S1–S15. doi: 10.1016/j.vaccine.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 8.Simonetti AC, Melo JH, de Souza PR, Bruneska D, de Lima Filho JL. Immunological’s host profile for HPV and Chlamydia trachomatis, a cervical cancer cofactor. Microbes Infect. 2009;11:435–442. doi: 10.1016/j.micinf.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Heinzen RA, Hackstadt T. The Chlamydia trachomatis parasitophorous vacuolar membrane is not passively permeable to low-molecular-weight compounds. Infect Immun. 1997;65:1088–1094. doi: 10.1128/iai.65.3.1088-1094.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moulder JW. Interaction of chlamydiae and host cells in vitro. Microbiol Rev. 1991;55:143–190. doi: 10.1128/mr.55.1.143-190.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grieshaber S, Swanson JA, Hackstadt T. Determination of the physical environment within the Chlamydia trachomatis inclusion using ion-selective ratiometric probes. Cell Microbiol. 2002;4:273–283. doi: 10.1046/j.1462-5822.2002.00191.x. [DOI] [PubMed] [Google Scholar]
- 12.Friis RR. Interaction of L cells and Chlamydia psittaci: entry of the parasite and host responses to its development. J Bacteriol. 1972;180:706–721. doi: 10.1128/jb.110.2.706-721.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rockey DD, Scidmore MA, Bannantine JP, Brown WJ. Proteins in the chlamydial inclusion membrane. Microbes Infect. 2002;4:333–340. doi: 10.1016/s1286-4579(02)01546-0. [DOI] [PubMed] [Google Scholar]
- 14.Bavoil PM, Hsia Rc, Ojcius DM. Closing in on Chlamydia and its intracellular bag of tricks. Microbiol. 2000;146:2723–2731. doi: 10.1099/00221287-146-11-2723. [DOI] [PubMed] [Google Scholar]
- 15.Hatch TP. Competition between Chlamydia psittaci and L cells for host isoleucine pools: a limiting factor in chlamydial multiplication. Infect Immun. 1975;12:211–220. doi: 10.1128/iai.12.1.211-220.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatch TP. Utilization of L-cell nucleoside triphosphates by Chlamydia psittaci for ribonucleic acid synthesis. J Bacteriol. 1975;122:393–400. doi: 10.1128/jb.122.2.393-400.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClarty G, Tipples G. In situ studies on incorporation of nucleic acid precursors into Chlamydia trachomatis DNA. J Bacteriol. 1991;173:4922–4931. doi: 10.1128/jb.173.16.4922-4931.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vender J, Moulder JW. Initial step in catabolism of glucose by the meningopneumonitis agent. J Bacteriol. 1967;94:867–869. doi: 10.1128/jb.94.4.867-869.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ojcius DM, Degani H, Mispelter J, Dautry-Varsat A. Enhancement of ATP levels and glucose metabolism during an infection by Chlamydia. J Biol Chem. 1998;273:7052–7058. doi: 10.1074/jbc.273.12.7052. [DOI] [PubMed] [Google Scholar]
- 20.Ojcius DM, Niedergang F, Subtil A, Hellio R, Dautry-Varsat A. Immunology and the confocal microscope. Res Immunol. 1996;147:175–188. doi: 10.1016/0923-2494(96)83169-5. [DOI] [PubMed] [Google Scholar]
- 21.Hackstadt T, Scidmore MA, Rockey DD. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc Natl Acad Sci USA. 1995;92:4877–4881. doi: 10.1073/pnas.92.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuste R. Fluorescence microscopy today. Nature Methods. 2005;2:902–904. doi: 10.1038/nmeth1205-902. [DOI] [PubMed] [Google Scholar]
- 23.Dunn WB. Current trends and future requirements for the mass spectrometric investigation of microbial, mammalian and plant metabolomes. Phys Biol. 2008;5:11001. doi: 10.1088/1478-3975/5/1/011001. [DOI] [PubMed] [Google Scholar]
- 24.Gomase VS, Changbhale SS, Patil SA, Kale KV. Metabolomics. Curr Drug Metab. 2008;9:89–98. doi: 10.2174/138920008783331149. [DOI] [PubMed] [Google Scholar]
- 25.Szaszák M, Steven P, Shima K, Orzekowsky-Schröder R, Hüttmann G, König IR, Solbach W, Rupp J. Fluorescence lifetime imaging unravels C. trachomatis metabolism and its crosstalk with the host cell. PLoS Pathog. 2011;7:e1002108. doi: 10.1371/journal.ppat.1002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh GP, Creely CM, Volpe G, Grötsch H, Petrov D. Real-time detection of hyperosmotic stress response in optically trapped single yeast cells using Raman microspectroscopy. Anal Chem. 2005;77:2564–2568. doi: 10.1021/ac048359j. [DOI] [PubMed] [Google Scholar]
- 27.Naito Y, Toh-e A, Hamaguchi Ho. In vivo time-resolved Raman imaging of a spontaneous death process of a single budding yeast cell. J Raman Spectrosc. 2005;36:837–839. [Google Scholar]
- 28.Matthäus C, Boydston-White S, Miljković M, Romeo M, Diem M. Raman and infrared microspectral imaging of mitotic cells. Appl Spectrosc. 2006;60:1–8. doi: 10.1366/000370206775382758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verrier S, Notingher I, Polak JM, Hench LL. In situ monitoring of cell death using Raman microspectroscopy. Biopolymers. 2004;74:157–162. doi: 10.1002/bip.20063. [DOI] [PubMed] [Google Scholar]
- 30.Uzunbajakava N, Lenferink A, Kraan Y, Volokhina E, Vrensen G, Greve J, Otto C. Nonresonant confocal Raman imaging of DNA and protein distribution in apoptotic cells. Biophys J. 2003;84:3968–3981. doi: 10.1016/S0006-3495(03)75124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taleb A, Diamond J, McGarvey JJ, Beattie JR, Toland C, Hamilton PW. Raman microscopy for the chemometric analysis of tumor cells. J Phys Chem B. 2006;110:19625–19631. doi: 10.1021/jp061981q. [DOI] [PubMed] [Google Scholar]
- 32.Swain RJ, Jell G, Stevens MM. Non-invasive analysis of cell cycle dynamics in single living cells with Raman micro-spectroscopy. J Cell Biochem. 2008;104:1427–1438. doi: 10.1002/jcb.21720. [DOI] [PubMed] [Google Scholar]
- 33.Matthaus C, Chemenko T, Newmark JA, Warner CM, Diem M. Label-Free detection of mitrochondrial distribution in cells by nonresonant Raman microspectroscopy. Biophys J. 2007;93:668–673. doi: 10.1529/biophysj.106.102061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Escoriza MF, Vanbriesen JM, Stewart S, Maier J. Studying bacterial metabolic states using Raman spectroscopy. Appl Spectrosc. 2006;60:971–976. doi: 10.1366/000370206778397290. [DOI] [PubMed] [Google Scholar]
- 35.Notingher I, Bisson I, Bishop AE, Randle WL, Polak JMP, Hench LL. In situ spectral monitoring of mRNA translation in embryonic stem cells during differentiation in vitro. Anal Chem. 2004;76:3185–3193. doi: 10.1021/ac0498720. [DOI] [PubMed] [Google Scholar]
- 36.Chan JW, Lieu DK, Huser T, Li RA. Label-free separation of human embryonic stem cells and their cardiac derivatives using Raman spectroscopy. Anal Chem. 2009;81:1324–1331. doi: 10.1021/ac801665m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haider S, Wagner M, Schmid MC, Sixt BS, Christian JG, Hacker G, Pichler P, Mechtler K, Muller A, Baranyi C, Toenshoff ER, Montanaro J, Horn M. Raman microspectroscopy reveals long-term extracellular activity of chlamydiae. Mol Microbiol. 2010;77:687–700. doi: 10.1111/j.1365-2958.2010.07241.x. [DOI] [PubMed] [Google Scholar]
- 38.Perfettini JL, Ojcius DM, Andrews CW, Korsmeyer SJ, Rank RG, Darville T. Role of proapoptotic BAX in propagation of Chlamydia muridarum (the mouse pneumonitis strain of Chlamydia trachomatis) and the host inflammatory response. J Biol Chem. 2003;278:9496–9502. doi: 10.1074/jbc.M211275200. [DOI] [PubMed] [Google Scholar]
- 39.Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, Beatty WL, Caldwell HD. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci USA. 2003;100:8478–8483. doi: 10.1073/pnas.1331135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maiti NC, Apetri MM, Zagorski MG, Carey PR, Anderson VE. Raman spectroscopic characterization of secondary structure in natively unfolfed proteins: α-synuclein. J Am Chem Soc. 2004;126:2399–2408. doi: 10.1021/ja0356176. [DOI] [PubMed] [Google Scholar]
- 41.Toyama A, Hanada N, Abe Y, Takeuchi H, Harada I. Assignment of adenine ring in-plane vibrations in adenosine on the basis of 15N and 13C isotopic frequency shifts and UV resonance Raman enhancement. J Raman Spectrosc. 1994;25:623–630. [Google Scholar]
- 42.Jenkins RH, Tuma R, Juuti JT, Bamford DH, Thomas GJ., Jr A novel Raman spectrophotometric method for quantitative measurement of nucleoside triphosphate hydrolysis. Biospectroscopy. 1999;5:3–8. doi: 10.1002/(SICI)1520-6343(1999)5:1<3::AID-BSPY2>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 43.Lanir A, Yu NT. A Raman spectroscopic study of the interaction of divalent metal ions with adenine moiety of adenosine 5′-triphosphate. J Biol Chem. 1979;254:5882–5887. [PubMed] [Google Scholar]
- 44.Takeuchi H, Murata H, Harada I. Interaction of ademosine 5′-triphosphate with Mg2+: Vibrational study of coordination sites by use of 18O-labeled triphosphates. J Am Chem Soc. 1988;110:392–397. [Google Scholar]
- 45.Onogi C, Motoyama M, Hamaguchi H-o. High concentration trans form unsaturated lipids detected in a HeLa cell by Raman microspectroscopy. J Raman Spectrosc. 2008;39:555–556. [Google Scholar]
- 46.Huang S, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J. 2002;82:2811–2825. doi: 10.1016/S0006-3495(02)75621-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Latt SA, Stetten G. Spectral studies on 33258 Hoechst and related bisbenzimidazole dyes useful for fluorescent detection of deoxyribonucleic acid synthesis. J Histochem Cytochem. 1976;24:24–33. doi: 10.1177/24.1.943439. [DOI] [PubMed] [Google Scholar]
- 48.Toyama A, Hanada N, Abe Y, Takeuchi H, Harada I. Assignment of adenine ring in-plane vibrations in adenosine on the basis of 15N and 13C isotopic frequency shifts and UV resonance Raman enhancement. J Raman Spectrosc. 1994;25:623–630. [Google Scholar]
- 49.Lanir A, Yu NT. A Raman spectroscopic study of the interaction of divalent metal ions with adenine moiety of adenosine 5′-triphosphate. J Biol Chem. 1979;254:5882–5887. [PubMed] [Google Scholar]
- 50.Wagnieres GA, Star WM, Wilson BC. In vivo fluorescence spectroscopy and imaging for oncological applications. Photochem Photobiol. 1998;68:603–632. [PubMed] [Google Scholar]
- 51.Stiles PL, Dieringer JA, Shah NC, Van Duyne RP. Surface-enhanced Raman spectroscopy. Ann Rev Anal Chem. 2008;1:601–626. doi: 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- 52.Monaghan PB, McCarney KM, Ricketts A, Littleford RE, Docherty F, Smith WE, Graham D, Cooper JM. Bead-based DNA diagnostic assay for Chlamydia using nanoparticle-mediated surface-enhanced resonance Raman scattering detetction within a lab-on-a-chip format. Anal Chem. 2007;79:2844–2849. doi: 10.1021/ac061769i. [DOI] [PubMed] [Google Scholar]
- 53.Kneipp J, Kneipp H, McLaughlin M, Brown D, Kneipp K. In vivo molecular probing of cellular compartments with gold nanoparticles and nanoaggregates. Nano Lett. 2006;6:2225–2231. doi: 10.1021/nl061517x. [DOI] [PubMed] [Google Scholar]