

Abstract
Inhibition of vascular endothelial growth factor (VEGF) signaling can promote lymph node metastasis in preclinical models, but the mechanism is not fully understood, and successful methods of prevention have not been found. Signaling of hepatocyte growth factor (HGF) and its receptor c-Met can promote the growth of lymphatics and metastasis of some tumors. We sought to explore the contributions of c-Met signaling to lymph node metastasis after inhibition of VEGF signaling. In particular, we examined whether c-Met is upregulated in lymphatics in or near pancreatic neuroendocrine tumors in RIP-Tag2 transgenic mice and whether lymph node metastasis can be reduced by concurrent inhibition of VEGF and c-Met signaling. Inhibition of VEGF signaling by anti-VEGF antibody or sunitinib in mice from age 14 to 17 weeks was accompanied by more intratumoral lymphatics, more tumor cells inside lymphatics, and more lymph node metastases. Under these conditions, lymphatic endothelial cells - like tumor cells - had strong immunoreactivity for c-Met and phospho-c-Met. c-Met blockade by the selective inhibitor PF-04217903 significantly reduced metastasis to local lymph nodes. Together, these results indicate that inhibition of VEGF signaling in RIP-Tag2 mice upregulates c-Met expression in lymphatic endothelial cells, increases the number of intratumoral lymphatics and number of tumor cells within lymphatics, and promotes metastasis to local lymph nodes. Prevention of lymph node metastasis by PF-04217903 in this setting implicates c-Met signaling in tumor cell spread to lymph nodes.
Introduction
Metastasis to regional lymph nodes is a feature of many solid tumors. The presence of lymph node metastasis is an important prognostic factor and the basis for surgical excision and radiation of local lymph nodes (1). Lymph node metastasis occurs after tumor cells enter lymphatics within or near tumors and drain to sentinel nodes (1). Tumor-associated lymphangiogenesis promotes the process (2-4) by increasing the number of routes to lymph nodes.
Reports of recent preclinical studies indicate that tumor invasiveness and metastasis can increase after inhibition of VEGF signaling (5-8). The mechanism of the increased aggressiveness is unknown, but contributing factors are likely to include increased intratumoral hypoxia as a result of vessel pruning. Hypoxia can increase expression of c-Met (HGFR), the receptor tyrosine kinase (RTK) activated by hepatocyte growth factor (HGF) (9). Activation of c-Met can drive tumor cell motility, proliferation, invasion, and survival (10-12). The HGF/c-Met pathway is activated in a wide variety of solid tumors (12, 13), correlates with poor prognosis (14-16), and is thought to contribute to tumor aggressiveness and resistance (17). c-Met expression in tumors can increase after treatment with inhibitors of VEGF signaling that promote vascular pruning and intratumoral hypoxia (8-10). c-Met activation can drive lymphangiogenesis (18, 19), which could favor lymph node metastasis.
Metastases are more abundant in the liver of RIP-Tag2 transgenic mice after treatment with function-blocking anti-VEGFR2 antibody, sunitinib, or neutralizing anti-VEGF antibody (7, 8). The same has been found in lymph nodes of these mice after treatment with anti-VEGFR2 antibody (7), although effects of age and duration of treatment have not been examined in detail.
The present study examined the involvement of c-Met signaling in lymph node metastasis after inhibition of VEGF signaling. Specifically, we sought to learn whether the treatment increases c-Met expression and activation in the lymphatic vessels and augments lymph node metastasis, and whether inhibition of c-Met signaling can reduce tumor spread to lymph nodes. We addressed these issues by determining the effects of VEGF signaling blockade on c-Met expression in lymphatic vessels and on number of intratumoral lymphatics, tumor cells inside lymphatics, and amount of lymph node metastasis. We then determined whether inhibition of c-Met signaling reduced tumor cells inside lymphatics and lymph node metastasis.
The approach was to manipulate c-Met signaling in RIP-Tag2 mice, which are known to develop lymph node metastasis after inhibition of VEGF signaling (7), VEGF signaling was blocked by treatment with a neutralizing anti-VEGF antibody or with sunitinib, a multi-targeted RTK inhibitor of VEGFR, PDGFR, c-KIT, and related kinases (20). c-Met signaling was blocked by the selective inhibitor PF-04217903 to determine effects on lymph node metastasis (21).
The experiments revealed that inhibition of VEGF signaling increased c-Met expression in lymphatics and tumor cells and also increased the number of intratumoral lymphatics, tumor cells inside lymphatics, and metastases in local lymph nodes. Inhibition of c-Met signaling blocked the exaggerated lymph node metastasis accompanying inhibition of VEGF signaling.
Materials and Methods
Animals and Treatment
Lymph node metastasis was studied in RIP-Tag2 transgenic mice (C57BL/6 background), which develop spontaneous multi-focal, multi-stage pancreatic neuroendocrine tumors driven by expression of SV40 T-antigen in pancreatic beta cells (22). Six groups of mice (5-7 mice/group each experiment) were treated from age 14 to 17 weeks with: (i) vehicle (0.5% sodium carboxymethyl cellulose (ICN Biomedicals, Inc), 1.8% (w/v) NaCl, 0.4% (v/v) Tween-80 (Sigma Chemical), 0.9% benzyl alcohol (v/v) (Sigma-Aldrich), 5μL/g) given daily by gavage; (ii) affinity purified, function-blocking goat anti-mouse VEGF antibody (AF-493-NA, R&D Systems, 150μg in 50μL sterile phosphate-buffered saline, PBS) injected ip three times per week (MWF); (iii) sunitinib (Pfizer, 40mg/kg in 5μL vehicle) given daily by gavage; (iv) PF-04217903 (Pfizer, 30mg/kg in 5μL vehicle) given daily by gavage; (v) anti-VEGF antibody plus PF-04217903; or (vi) sunitinib plus PF-04217903. In group (vi) the agents were pre-mixed and administered together.
Body weight and survival were recorded during the treatment. Some RIP-Tag2 mice received pimonidazole hydrochloride (60mg/kg iv, Hypoxyprobe Plus Kit HP2, Chemicon) 1 hour before perfusion to detect hypoxic regions in tumors. Untreated 14-week old RIP-Tag2 mice (n=5) were used as baseline control. 14-week old wild-type C57BL/6 mice (n=5) were also examined.
In separate experiments, mice were treated from age 14 to 15 weeks with vehicle, anti-mouse VEGF antibody, or sunitinib and tumors were removed for quantitative real-time PCR analysis.
Mice were housed under barrier conditions in the animal care facility at the University of California, San Francisco (UCSF). The UCSF Institutional Animal Care and Use Committee approved all experimental procedures.
Tissue preparation, immunohistochemistry, and imaging
At the end of treatment, mice were anesthetized with ketamine (100mg/kg ip) plus xylazine (10mg/kg ip), and tissues were fixed by vascular perfusion of 1% paraformaldehyde in PBS (23). Some tissues were prepared for H&E staining. Cryostat sections (80-μm thick) were processed for immunohistochemistry using two or three primary antibodies (23): guinea pig anti-swine insulin antibody (1:100, Dako); rabbit anti-SV40 T-antigen antibody (1:500, Santa Cruz Biotechnology); rat anti-mouse LYVE-1 antibody (1:500, R&D Systems); goat anti-mouse HGFR antibody (1:500, R&D Systems); rabbit anti-mouse phosphorylated c-Met antibody (1:250, Invitrogen); goat anti-VEGFR-3 antibody (1:1000, R&D Systems); rabbit anti-PROX1 antibody (1:500, AngioBio); goat anti-mouse HGF antibody (1:500, R&D Systems); rabbit anti-VEGFR-2 antibody T014 (1:2000, from Rolf Brekken, University of Texas Southwestern Medical Center); rabbit anti-phosphohistone H3 antibody (PHH3 1:1000, Millipore); or FITC-conjugated mouse anti-pimonidazole hydrochloride (1:100, Chemicon).
Secondary antibodies were FITC-, Cy3- or Cy5-labeled donkey anti-goat, anti-rabbit, anti-rat, or anti-guinea pig IgG antibody (Jackson ImmunoResearch; all diluted 1:500). Cell nuclei were stained with Vectashield mounting medium containing DAPI (Vector laboratories). Specimens were examined with a Zeiss Axiophot fluorescence microscope and a Zeiss LSM 510 laser scanning confocal microscope (23).
The methods used for measuring tumor size and mRNA expression of Prox1, VEGF-A, VEGF-C and VEGF-D are presented in the Supplementary Methods.
Measurements of lymphatics and lymph node metastases
Pancreas and pancreatic lymph nodes from each mouse (5-7 mice/group) were removed, processed together. Sections of pancreas (80-μm thick) from 5-7 mice/group were examined. Intratumoral lymphatics, identified as LYVE-1-immunoreactive vessels completely surrounded by insulin-immunoreactive tumor cells, were counted in all tumors present in two 80-μm thick sections of pancreas. As tumors were larger in the vehicle and PF-04217903 groups, an average of 5 tumors were analyzed in those groups, whereas 10-15 smaller tumors were analyzed in the other treatment groups. Digital fluorescence microscopic images (5x objective, 1x Optovar) of tumors were combined as montages to show the entire tumor. The sectional area of tumors was measured, and the number of intratumoral lymphatics was expressed as lymphatics per mm2 of tumor section.
To distinguish growing lymphatics in tumors from lymphatics co-opted by invading tumor cells, we counted intratumoral lymphatics with evidence of endothelial cell proliferation, defined as one or more phosphohistone H3-positive endothelial cells. Tumors in 5 mice per group were analyzed.
The frequency of lymphatics containing tumor cells was expressed as the proportion of LYVE-1-positive lymphatics with one or more SV40 T-antigen-positive tumor cells within the lumen per section of pancreas. A total of 25-30 intratumoral lymphatics were examined in each mouse in all groups.
Lymph node metastases were identified by the presence of SV40 T-antigen-positive tumor cells in pancreatic lymph nodes. The incidence of metastasis was determined in 5-7 mice/group and was expressed as the proportion of lymph nodes that contained SV40 T-antigen-positive cells in a survey of at least four 80-μm-thick sections cut at multiple levels (average 240-μm increment). Each section contained 3-5 lymph nodes.
Assessment of HGF, c-Met, and phospho-c-Met immunoreactivities
The abundance of HGF immunoreactive cells in tumors or lymph nodes was estimated as the area density of HGF staining in digital fluorescence microscopic images (10x objective, 1x Optovar) of RIP-Tag2 tumors or lymph nodes. Tumor cells were marked in the same sections by staining for SV40 T-antigen or insulin. Lymph nodes were identified by the distinctive LYVE-1 staining. The area density was measured at a predetermined threshold with ImageJ (23).
The proportion of c-Met or phospho-c-Met-positive lymphatic vessels was expressed as the proportion of LYVE-1-positive intratumoral lymphatics that had c-Met or phospho-c-Met immunoreactivity in 80-μm sections of pancreas. The abundance of tumor cells with c-Met or phospho-c-Met immunoreactivity was expressed as the proportion of SV40 T-antigen or insulin immunoreactive cells that colocalized with c-Met or phospho-c-Met.
Statistical analysis
Differences among groups were assessed by ANOVA followed by Fisher’s Least Significant Difference Test for multiple comparisons. Error bars show means ± SEM. Fisher’s Exact Test was used to compare the incidence of lymph node metastasis. P<0.05 was considered significant.
Results
Increased lymph node metastasis after inhibition of VEGF signaling
To build on the published report of exaggeration of lymph node metastasis by inhibition of VEGF signaling in RIP-Tag2 mice (7), we sought to obtain confirmatory data after treatment with anti-VEGF antibody or sunitinib before attempting to block it. As an indicator of lymph node metastasis, we used the presence of SV40 T-antigen-immunoreactive tumor cells in pancreatic lymph nodes. These lymph nodes include gastric, pancreatic-duodenal, and colic lymph nodes (24). The lymph nodes were removed, processed, and examined together with the pancreas.
At least one of the pancreatic lymph nodes contained SV40 T-antigen-positive cells in all RIP-Tag2 mice examined at age 17 weeks, regardless of the treatment (Figure 1A), but the proportion of positive nodes varied with the treatment. Only 50% of pancreatic lymph nodes had metastases after vehicle for 3 weeks, but 78% had metastases after anti-VEGF antibody, and 81% had metastases after sunitinib (Figure 1B). Most metastatic tumor cells were within the LYVE-1-positive cortical sinus of the lymph node (Figure 1A).
Figure 1.
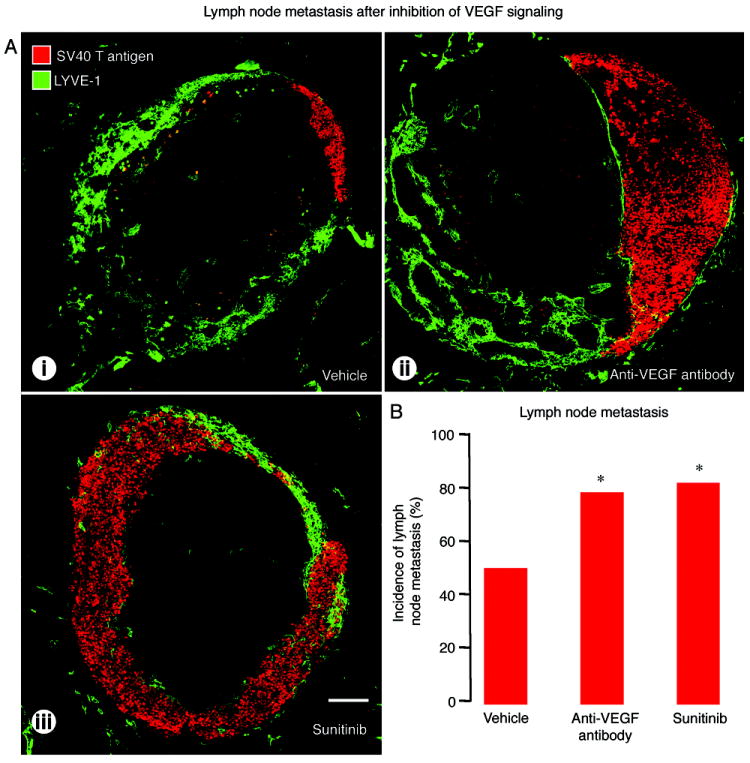
Effect of VEGF signaling inhibition on metastases in pancreatic lymph nodes of RIP-Tag2 mice treated from age 14 to 17 weeks. A: Confocal micrographs of lymph nodes (LYVE-1, green) with metastases (SV40 T-antigen, red) after treatment with (i) vehicle, (ii) anti-VEGF antibody, or (iii) sunitinib. B: Proportion of lymph nodes with metastases showing significantly higher incidence of metastasis after anti-VEGF antibody or sunitinib. *P<0.05 compared to vehicle. Scale bar in A-iii applies to all images: 200μm.
Increased intratumoral lymphatics after inhibition of VEGF signaling
To explore these findings further, we asked whether the higher incidence of lymph node metastasis after inhibition of VEGF signaling was accompanied by more intratumoral lymphatics, more invasion of tumor cells into peritumoral lymphatics, or both (1, 3, 25, 26). We found that intratumoral lymphatics detected by LYVE-1 immunoreactivity were more numerous after inhibition of VEGF signaling from age 14 to 17 weeks (Figure 2A).
Figure 2.
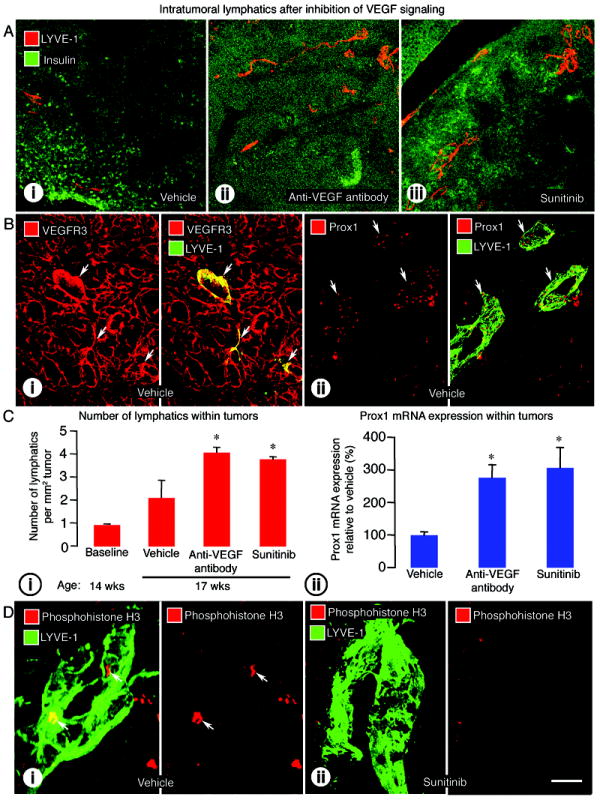
Effect of VEGF signaling inhibition on lymphatics inside RIP-Tag2 tumors. A: Confocal micrographs of intratumoral lymphatics (LYVE-1, red) in tumors (insulin, green) after treatment with (i) vehicle, (ii) anti-VEGF antibody, or (iii) sunitinib from age 14 to 17 weeks. B: (i) VEGFR-3 immunoreactivity (red) in abundant tumor blood vessels and a few lymphatic vessels (LYVE-1, green, arrows); (ii) Prox1 immunoreactivity (red) in lymphatic vessels (LYVE-1, green, arrows) in vehicle-treated RIP-Tag2 tumors. C: (i) Measurements showing significantly higher density of lymphatics inside tumors treated with anti-VEGF antibody or sunitinib from age 14 to 17 weeks; (ii) qRT-PCR measurements showing significantly higher Prox1 mRNA expression in tumors treated with anti-VEGF antibody or sunitinib from age 14 to 15 weeks. D: Confocal images illustrating the presence of proliferating cells (phosphohistone H3, red, arrows) in a lymphatic vessel (LYVE-1, green) after vehicle (i) but not after sunitinib (ii). The colocalization of phosphohistone H3 stained nuclei and LYVE-1 stained lymphatic cells was confirmed by examination of individual optical slices. *P<0.05 compared to vehicle. Scale bar in D-ii applies to all images: 200μm for A; 50μm for B; 7μm for D.
We assessed the effectiveness of LYVE-1 as a marker of lymphatics in RIP-Tag2 tumors by comparing the staining for VEGFR-3, podoplanin, and Prox1 (27). VEGFR-3 immunoreactivity in RIP-Tag2 tumors was found in lymphatics and blood vessels (Figure 2B-i) (23). Podoplanin immunoreactivity was detected in lymphatics and epithelial cells of pancreatic ducts (data not shown), but most Prox1 immunoreactivity was in lymphatic endothelial cell nuclei (Figure 2B-ii). By comparison, VEGFR-2 immunoreactivity was in blood vessels and lymphatics of tumors and lymph nodes (Supplemental Figure 1A).
Intratumoral lymphatics were more abundant in tumors of mice treated with vehicle from age 14 to 17 weeks than in untreated RIP-Tag2 mice at age 14 weeks (onset control) (Figure 2C-i). After anti-VEGF antibody or sunitinib, intratumoral lymphatics were 93% or 79% more numerous than after vehicle (Figure 2C-i). Consistent with the greater abundance of intratumoral lymphatics, expression of Prox1 mRNA was 2.5 times greater in tumors after anti-VEGF antibody and 3 times greater after sunitinib, from age 14 to 15 weeks (Figure 2C-ii).
Expression of the lymphangiogenic factors VEGF-C and VEGF-D in untreated RIP-Tag2 tumors was much lower than VEGF-A (Supplemental Figure 1B-i), which is known to be highly expressed in RIP-Tag2 tumor cells (28). However, expression of both VEGF-C and VEGF-D was increased after treatment with sunitinib from age 14 to 15 weeks (Supplemental Figure 1B-ii-iii).
The contribution of lymphangiogenesis to the increase in intratumoral lymphatics after anti-VEGF therapy was assessed by determining the frequency of proliferating lymphatic endothelial cells detected by phosphohistone H3 immunoreactivity. In vehicle-treated mice, an average of 5 lymphatics had one or more phosphohistone H3-positive endothelial cells per section of pancreas (5-6 tumors analyzed in each of 5 mice) (Figure 2D-i). By comparison, after sunitinib none of the tumors had phosphohistone H3-positive cells that colocalized with LYVE-1 staining (10-13 tumors analyzed in each of 5 mice) (Figure 2D-ii).
Because we found no evidence for lymphatic proliferation after sunitinib, we asked whether inhibition of VEGF signaling was accompanied by tumor cell invasion into peritumoral lymphatics. Tumors of mice treated with sunitinib had the appearance of being more invasive, in that their border was more irregular and penetrated the acinar pancreas (Figure 3A-i-ii). After sunitinib, tumors were also accompanied by more LYVE-1-positive lymphatics (Figure 3B-i-ii and Figure 3C-i-ii), and more lymphatics had intraluminal tumor cells (Figure 3D-i-ii). The proportion of lymphatics with SV40 T-antigen-positive tumor cells inside was 58% higher after sunitinib than after vehicle (Figure 3D-iii).
Figure 3.
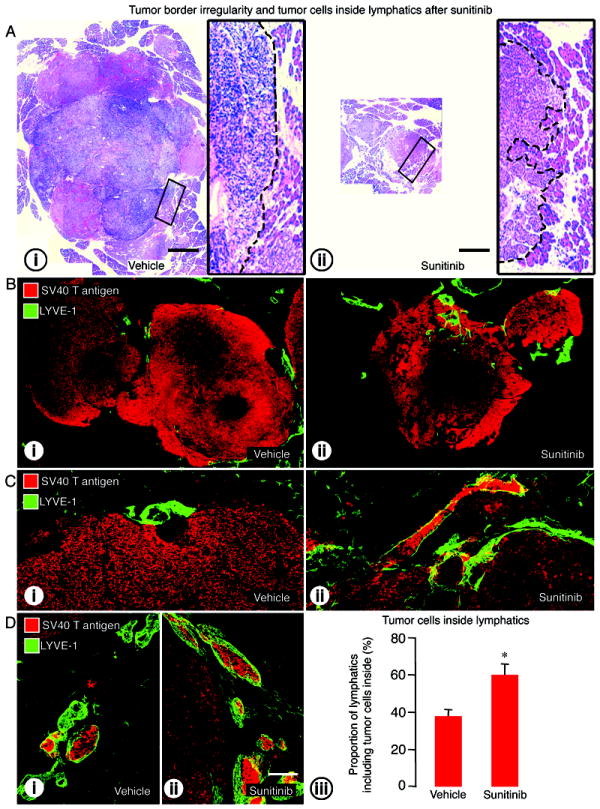
Effect of VEGF signaling inhibition on tumor border irregularity and tumor cells inside lymphatics in RIP-Tag2 mice treated from age 14 to 17 weeks. A: Tumor sections stained with hematoxylin and eosin (H&E) comparing tumor size and tumor edges after treatment with (i) vehicle or (ii) sunitinib. Composite images of tumor sections (B) and confocal micrographs (C) showing the irregularity of tumor border (SV40 T-antigen, red) and tumor cells inside lymphatics (LYVE-1, green) after treatment with (i) vehicle or (ii) sunitinib. D: Tumor cells inside lymphatics after (i) vehicle or (ii) sunitinib. (iii) Measurements showing significantly higher proportion of lymphatics with tumor cells in the lumen after sunitinib. *P<0.05 compared to vehicle. Scale bar in A applies to Ai and Aii: 1.3mm for the large images and 200μm for inserts. Scale bar in D-ii applies to B-D: 800μm for B; 120μm for C; 200μm for D.
These differences in RIP-Tag2 mice treated from age 14 to 17 weeks accompanied conspicuous treatment-related differences in tumor size, indicated by mean sectional area: 19.2±6.3mm2 after vehicle, 4.3±0.8mm2 after anti-VEGF antibody (78% less than vehicle), and 3.7±0.8mm2 after sunitinib (81% less than vehicle) (8).
Increased c-Met expression after inhibition of VEGF signaling
The involvement of c-Met signaling in the changes in lymphatics and lymphatic metastases was examined by immunohistochemical assessment of amount of the receptor and receptor activation reflected by c-Met and phospho-c-Met immunoreactivities. In the normal pancreas of wild-type mice, c-Met immunoreactivity was not detected in pancreatic islets or in lymphatics (Figure 4A-i). In RIP-Tag2 mice treated with vehicle from age 14 to 17 weeks, c-Met immunoreactivity was strong in some blood vessels, but was weak or absent in tumor cells and lymphatics (Figure 4A-ii and Supplemental Figure 2A-i). By comparison, after treatment with sunitinib, strong c-Met immunoreactivity was found in tumor cells, lymphatics (Figure 4A-iii and Supplemental Figure 2A-ii-iii), and blood vessels (Supplemental Figure 2B).
Figure 4.
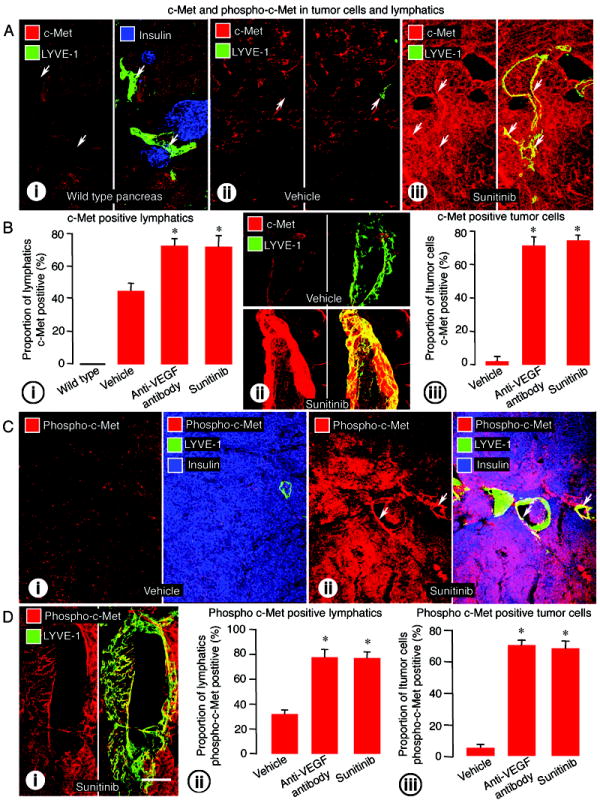
c-Met and phospho-c-Met immunoreactivities in tumor cells and lymphatics of RIP-Tag2 mice treated from age 14 to 17 weeks. A: (i) Wild-type pancreas (normal islets, insulin, blue) with lymphatic vessels (LYVE-1, green, arrows) lacking c-Met immunoreactivity (red). ii-iii: c-Met (red) in intratumoral lymphatics (LYVE-1, green, arrows) in tumors treated with (ii) vehicle or (iii) sunitinib. B: (i) Measurements showing significantly higher proportion of lymphatic vessels with c-Met immunoreactivity after anti-VEGF antibody or sunitinib. (ii) High magnification confocal images showing the difference in intensity of c-Met staining in lymphatics after vehicle and sunitinib treatment. (iii) Measurements showing higher proportion of tumor cells with c-Met immunoreactivity after anti-VEGF antibody or sunitinib. C: Comparison of phospho c-Met immunoreactivity (red) in tumor cells (insulin, blue) or intratumoral lymphatics (LYVE-1, green, arrows) after treatment with (i) vehicle or (ii) sunitinib. D: (i) Phospho c-Met staining (red) in the endothelium of an intratumoral lymphatic (LYVE-1, green) after treatment with sunitinib. (ii-iii) Bar graphs showing significantly higher proportion of phospho-c-Met immunoreactive (ii) lymphatic vessels and (iii) tumor cells after anti-VEGF antibody or sunitinib. *P<0.05 compared to vehicle. Scale bar in D-i applies to all images: 50μm for A and C; 8μm for C and D.
The proportion of lymphatics with c-Met immunoreactivity, which was zero in wild-type islets, was 43% in tumors after vehicle - although weak in intensity, and was significantly greater (73-75%) after treatment with anti-VEGF antibody or sunitinib (Figure 4B-i). The intensity of c-Met immunoreactivity in lymphatics was also greater after anti-VEGF therapy (Figure 4B-ii). The proportion of tumor cells with c-Met immunoreactivity increased from 2% after vehicle to 73% after anti-VEGF antibody and 74% after sunitinib (Figure 4B-iii).
The increase in c-Met immunoreactivity after sunitinib was accompanied by reduced vascular density (8) and more severe intratumoral hypoxia, assessed by pimonidazole staining (Supplemental Figure 2C-i). However, staining for c-Met was present throughout tumors, though variable in intensity, and was not restricted to regions of pimonidazole staining (Supplemental Figure 2C-ii).
Phospho-c-Met immunoreactivity was detected in scattered cells in tumors treated with vehicle (Figure 4C-i), but was stronger and more widespread after sunitinib (Figure 4C-ii). Staining for phospho-c-Met was found in lymphatic endothelial cells as well as in tumor cells (Figure 4D-i and Supplemental Figure 3A). As evidence of specificity, this staining was absent when the primary antibody was omitted (Supplemental Figure 3B). The proportion of phospho-c-Met immunoreactive lymphatics and tumor cells was significantly greater after anti-VEGF antibody or sunitinib (Figure 4D-ii-iii). After sunitinib, some tumor cells inside lymphatics had immunoreactivity for c-Met and for phospho-c-Met (Supplemental Figure 3D).
Decreased intratumoral lymphatics after inhibition of c-Met and VEGF signaling
To determine whether the number of intratumoral lymphatics in RIP-Tag2 mice treated with anti-VEGF antibody or sunitinib could be reduced by inhibiting c-Met, we examined the effects of the selective inhibitor PF-04217903 given from age 14 to 17 weeks. Tumors treated with PF-04217903 together with anti-VEGF antibody or sunitinib had significantly fewer lymphatics than after either VEGF inhibitor alone and were comparable to the vehicle (Figure 5A). Intratumoral lymphatics were 53% less abundant after PF-04217903 plus anti-VEGF antibody and 75% less after PF-04217903 plus sunitinib (Figure 5B). With both drug combinations, c-Met-positive lymphatics were less than half as numerous as after the anti-VEGF antibody or sunitinib alone (Supplemental Figure 2C-i), and phospho-c-Met-positive lymphatics were reduced essentially to zero (Supplemental Figure 3C-i). Tumors treated with PF-04217903 alone tended to have fewer intratumoral lymphatics than after vehicle, but the difference was not statistically significant (Figure 5A).
Figure 5.
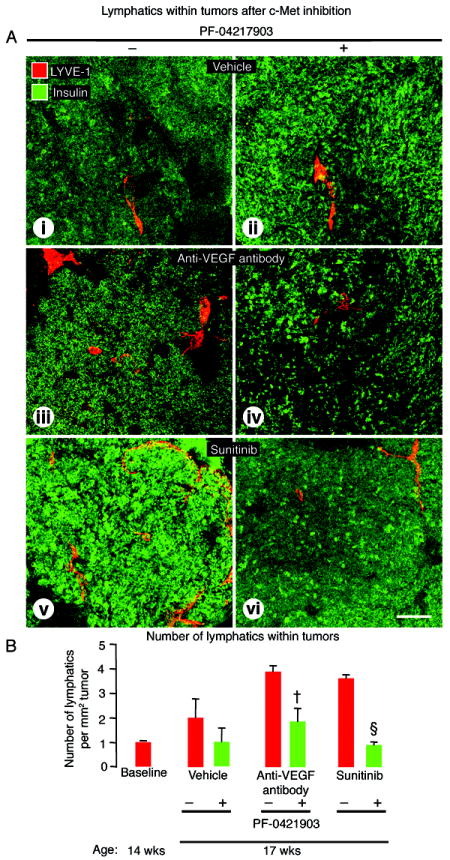
Effect of c-Met inhibition on lymphatics within RIP-Tag2 tumors after treatment from age 14 to 17 weeks. A: Confocal micrographs of intratumoral lymphatics (LYVE-1, red) in tumors (insulin, green) after treatment with (i-ii) vehicle, (iii-iv) anti-VEGF antibody, or (v-vi) sunitinib without (-) or with (+) c-Met inhibition by PF-04217903. B: Measurements showing significantly fewer lymphatics in tumors treated with PF-04217903. P<0.05 compared to anti-VEGF antibody (†) or sunitinib (§) without PF-04217903. Scale bar in A-vi applies to all images: 125μm.
Decreased tumor cells in lymphatics after inhibition of c-Met and VEGF signaling
Blockade of c-Met by PF-04217903 together with VEGF signaling inhibition by sunitinib reduced tumor cell invasion into lymphatics and into the acinar pancreas. Addition of the c-Met inhibitor to sunitinib did not lead to further reduction in tumor size, but did change tumor borders from an irregular, invasive phenotype to a smoother contour (Figure 6A). Intratumoral and peritumoral lymphatics, which were abundant and contained tumor cells after sunitinib, were less numerous and had few or no tumor cells when PF-04217903 was also given (Figure 6B, C).
Figure 6.
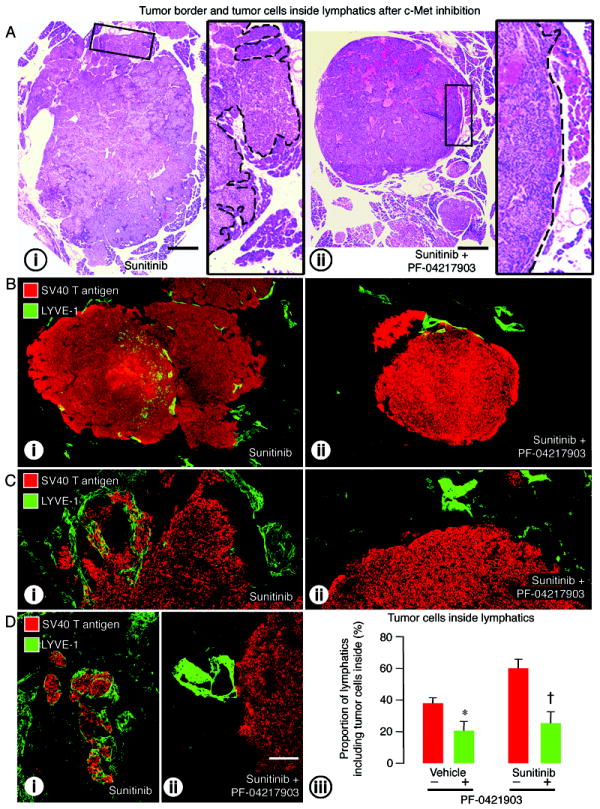
Effect of c-Met inhibition on tumor border irregularity and lymphatics in RIP-Tag2 mice treated from age 14 to 17 weeks. A: Tumor sections stained with H&E comparing tumor size and border contour after treatment with (i) sunitinib alone or (ii) sunitinib plus PF-04217903. Composite images of tumor sections (B) and confocal micrographs (C) showing the tumor border (SV40 T-antigen, red) and lymphatic vessels (LYVE-1, green) after treatment with (i) sunitinib or (ii) sunitinib plus PF-04217903. D: Tumor cells inside lymphatics after (i) sunitinib alone (-) or (ii) sunitinib with PF-04217903 (+). (iii) Measurements showing significantly smaller proportion of lymphatics with tumor cells in the lumen after PF-04217903 administered with vehicle or sunitinib. P<0.05 compared to vehicle (*) or sunitinib (†) without PF-04217903. Scale bar in A applies to A-i and A-ii: 900μm for the large images and 200μm for inserts. Scale bar in D-ii applies to B-D: 500μm for B; 120μm for C; 200μm for D.
The proportion of lymphatics with tumor cells inside was 58% less after PF-04217903 plus sunitinib than after sunitinib alone (Figure 6D-i-ii-iii) and was 46% less after PF-04217903 alone than after vehicle (Figure 6D-iii). Fewer tumor cells stained for c-Met (Supplemental Figure 2D-ii) or phospho-c-Met (Supplemental Figure 3C-ii) after treatment with PF-04217903 alone or with anti-VEGF antibody or sunitinib.
HGF immunoreactivity was found in scattered cells in tumors and lymph nodes (HGF area density 4% in tumors and 20% in lymph nodes) of vehicle-treated RIP-Tag2 mice (Supplemental Figure 4A-i-ii). The amount of HGF immunoreactivity was less after PF-04217903 alone but was similar in all other treatment groups (Supplemental Figure 4B-i-ii).
Decreased lymph node metastasis after inhibition of c-Met and VEGF signaling
The pronounced effect of PF-04217903 on intratumoral lymphatics and on lymphatics that had tumor cells inside led us to determine whether lymph node metastases were also reduced. We found significantly fewer lymph node metastases in mice treated with PF-04217903 as a single agent (80% reduction) (Figure 7A-i-ii, 7B). Importantly, lymph node metastases were also less numerous when PF-04217903 was given together with anti-VEGF antibody (75% reduction) or sunitinib (75% reduction) (Figure 7A-iii-iv-v-vi, 7B).
Figure 7.
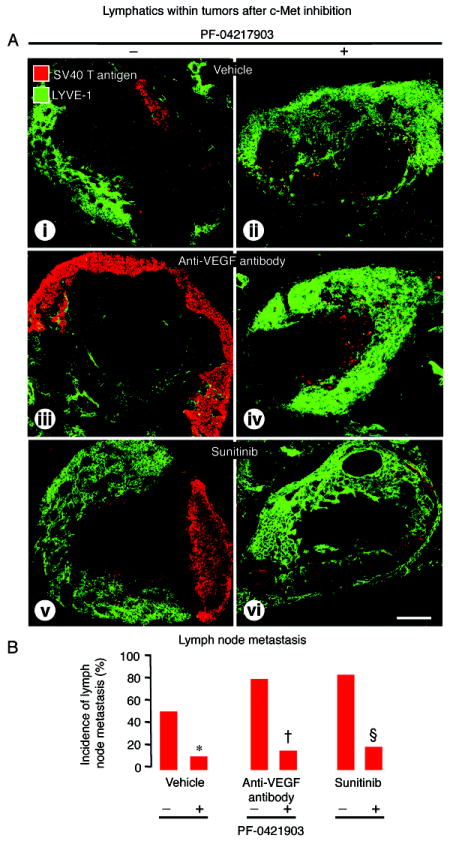
Effect of c-Met inhibition on pancreatic lymph node metastases in RIP-Tag2 mice treated from age 14 to 17 weeks. A: Confocal micrographs of lymph nodes (LYVE-1, green), some of which have metastases (SV40 T-antigen, red), after treatment with (i-ii) vehicle, (iii-iv) anti-VEGF antibody, or (v-vi) sunitinib without (-) or with (+) c-Met inhibition by PF-04217903. B: Measurements showing significantly smaller proportion of lymph nodes with metastases after PF-04217903 in all three groups. P<0.05 compared to vehicle (*), anti-VEGF antibody (†), or sunitinib (§) without PF-04217903. Scale bar in D-ii applies to all images: 220μm.
Discussion
The present study sought to determine the involvement of c-Met signaling in lymph node metastasis after inhibition of VEGF signaling in RIP-Tag2 mice. We found that the number of intratumoral lymphatics, proportion of lymphatics containing tumor cells, and incidence of lymph node metastasis were all increased after inhibition of VEGF signaling by a function blocking anti-VEGF antibody or sunitinib. However, this was not found when c-Met was blocked by concurrent administration of the selective inhibitor PF-04217903. The addition of the c-Met blocker significantly reduced intratumoral lymphatics, tumor cells inside lymphatics, and lymph node metastases. Together, the results indicate that inhibition of c-Met in this model can reduce lymph node metastasis in the presence of VEGF blockade.
Lymph node metastasis after VEGF inhibition
Most angiogenesis inhibitors used for cancer therapy block VEGF signaling (29). This action stops sprouting angiogenesis, prunes tumor vessels, normalizes surviving tumor vessels, and reduces tumor growth (23, 30, 31).
Vascular pruning in tumors can lead to hypoxia, upregulation of hypoxia inducible factor (HIF)-1α, increase of c-Met expression, and promotion of cell proliferation, motility, and invasion (9, 10, 32, 33). Increased tumor invasiveness found after inhibition of VEGF signaling could reflect this mechanism (5-8).
Treatment of RIP-Tag2 mice with anti-VEGF antibody or sunitinib from age 14 to 17 weeks increased c-Met and phospho-c-Met immunoreactivities in lymphatic endothelial cells. Intratumoral lymphatics and lymph node metastases were also more abundant. The increase in lymph node metastasis fits with the known relationship between the abundance of intratumoral lymphatics and incidence of lymph node metastasis (1, 3, 25, 26, 34, 35).
In the present study four different markers were used to identify lymphatic vessels: LYVE-1, Prox1, VEGFR-3, and podoplanin. All are considered reliable markers of lymphatics, but some also label other cells. Prox1 is present in some colorectal cancer cells (36). VEGFR-3 and podoplanin marked lymphatic endothelial cells in RIP-Tag2 tumors but also marked some blood vessels and other cell types. However, LYVE-1 and Prox1 immunoreactivities in RIP-Tag2 tumors were mainly associated with lymphatic endothelial cells.
Treatment of RIP-Tag2 mice with anti-VEGF antibody or sunitinib from age 14 to 17 weeks led to a 75% or greater decrease in tumor size but a 4-fold increase in number of intratumoral lymphatics. The increase in intratumoral lymphatics could result from growth of new lymphatics, from tumor cell co-option of peritumoral lymphatics, or both. The anti-VEGF antibody blocks VEGF-A, but not VEGF-C or VEGF-D, and sunitinib blocks the signaling of all VEGF receptors. Lymphangiogenesis is generally thought to be driven by activation of VEGFR-2 and/or VEGFR-3 signaling by VEGF-C, VEGF-D, or in some cases VEGF-A (37-39). VEGF-C and VEGF-D are expressed by alpha cells of the islets of Langerhans in human pancreas (40) but were relatively low in untreated RIP-Tag2 tumors. However, VEGF-C and VEGF-D were significantly increased in tumors after treatment with sunitinib.
Lymphatic endothelial cell growth and lymph node metastasis are reduced in a breast cancer model after inhibition of VEGFR-2 and VEGFR-3 by antibodies or sunitinib (41, 42). Overexpression of VEGF-A in the skin of transgenic mice can promote lymphangiogenesis at sites of wound healing, but systemic blockade of VEGFR-2 prevents the formation of lymphatics (43). Sunitinib inhibits lymphangiogenesis in tumors under some conditions (7). Differences between our findings and some published data could reflect the tumor model, stage of tumor progression, and duration of treatment.
Proliferating endothelial cells marked by phosphohistone H3 immunoreactivity were found in intratumoral lymphatics of mice treated with vehicle but not with sunitinib. This finding fits better with co-option of peritumoral lymphatics than with lymphangiogenesis as the source of more abundant intratumoral lymphatics after sunitinib.
Effects of inhibition of VEGF signaling on lymph node metastasis appears to be context- and time-dependent. Promotion of lymph node metastasis by sunitinib was not evident in RIP-Tag2 mice treated from age 10 to 15 weeks (7), unlike the findings in our mice treated for age 14 to 17 weeks. This difference could reflect tumor stage or treatment duration. Promotion of tumor invasion and metastasis by VEGF inhibition has been reported in some studies (6-8, 44), but not in others (45, 46). In a model of non-melanoma skin cancer, inhibition of VEGFR-2 by a blocking antibody stopped angiogenesis and prevented invasion of malignant cells (47). These effects could depend on multiple factors, including tumor stage, tumor microenvironment, and nature, dose and schedule of the inhibitor.
Suppression of lymph node metastasis by c-Met inhibition
Although c-Met was not detected by immunohistochemistry in normal pancreatic islets and was limited to blood vessels in vehicle-treated RIP-Tag2 tumors, c-Met immunoreactivity was widespread in tumor cells, blood vessels, and lymphatics after treatment with anti-VEGF antibody or sunitinib. c-Met immunoreactivity was also found in tumor cells inside lymphatics after sunitinib. Treatment with the c-Met selective RTK inhibitor PF-04217903 reduced tumor invasion, intratumoral lymphatics, and lymph node metastasis in RIP-Tag2 mice treated with vehicle, anti-VEGF antibody, or sunitinib from age 14 to 17 weeks. As the amount of HGF did not change under these conditions, c-Met activation could be partially ligand-independent (17).
In humans, c-Met is overexpressed, constitutively active, mutated, or otherwise amplified in many aggressive tumors (11, 12), and high c-Met activity is associated with poor prognosis (14-16). Consistent with c-Met activation, inhibitors of c-Met signaling, including anti-HGF and anti-c-Met antibodies, soluble decoy receptors, and small molecule RTK inhibitors, have shown promise, particularly in combination with inhibitors of VEGF or epidermal growth factor (EGF) signaling, in preclinical tumor models (8, 12, 48-52) and in some clinical trials (53-59).
Multiple mechanisms are likely to be involved in the anti-metastatic action of PF-04217903. Many intratumoral and peritumoral lymphatics had strong c-Met immunoreactivity after treatment with sunitinib or anti-VEGF antibody, but c-Met staining was not detected in lymphatics of the normal pancreas and was found in fewer than half of the lymphatics in untreated RIP-Tag2 tumors. Similarly, c-Met is strongly expressed by activated or growing lymphatics at sites of inflammation or wound healing but not in normal skin (19).
The absence of proliferating intratumoral lymphatics after sunitinib suggests that factors other than lymphangiogenesis promote the increase in intratumoral lymphatics and lymph node metastasis. Tumor borders were conspicuously smoother after treatment with PF-04217903, consistent with an anti-invasive action (8, 60). PF-04217903 could decrease intratumoral lymphatics by reducing tumor invasion into peritumoral lymphatics and by pruning lymphatics with high c-Met expression.
Effects of inhibiting c-Met and VEGF signaling together
We previously reported that inhibition of c-Met and VEGF signaling together reduced liver metastasis and prolonged survival of RIP-Tag2 mice (8). The present study expands these actions to include the reduction of lymph node metastasis. Inhibition of c-Met by PF-04217903 not only reduced lymph node metastasis when given as a single agent, but also prevented the increase in intratumoral lymphatics, tumor cell trafficking into lymphatics, and lymph node metastasis that occurred during treatment with anti-VEGF antibody or sunitinib. Together, the findings are consistent with the involvement of c-Met signaling in metastasis to liver and lymph nodes in this model and with the efficacy of blocking c-Met signaling in reducing metastasis.
In conclusion, inhibition of VEGF signaling by function-blocking antibody or by sunitinib increases intratumoral lymphatics, invasion of tumor cells into lymphatics, and lymph node metastasis in RIP-Tag2 mice with late stage tumors. Co-option of existing lymphatics by invading tumor cells is likely to contribute to the population of intratumoral lymphatics and perhaps to promotion of lymph node metastasis. Inhibition of c-Met by PF-04217903 reduces intratumoral lymphatics and lymph node metastasis under these conditions.
Supplementary Material
Acknowledgments
We thank Peter Baluk, Casey Williamson and Grace Laidlaw for critical review of the manuscript and Debbie Chen for genotyping the RIP-Tag2 mice. This work was supported in part by an Overseas Research Fellowship from Ehime University (to T. Ishiguro-Oonuma) and by the National Heart, Lung, and Blood Institute grants HL24136 and HL59157, the National Cancer Institute grant CA82923, a grant from Pfizer Global Research and Development, and funding from AngelWorks Foundation (to D.M. McDonald).
Footnotes
Disclosure of Potential Conflicts of Interest
J. Christensen: employee of Pfizer Global Research and Development. D.M. McDonald: research funding from a grant to University of California- San Francisco from Pfizer Global Research and Development. The other authors had no potential conflicts of interest to disclose.
References
- Cao Y. Opinion: emerging mechanisms of tumour lymphangiogenesis and lymphatic metastasis. Nat Rev Cancer. 2005;5:735–43. doi: 10.1038/nrc1693. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, et al. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J. 2001;20:672–82. doi: 10.1093/emboj/20.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–8. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186–91. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012;2:1–18. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–61. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- Benvenuti S, Comoglio PM. The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol. 2007;213:316–25. doi: 10.1002/jcp.21183. [DOI] [PubMed] [Google Scholar]
- Christensen J, Anderes K. Beyond VEGF: targeting tumor growth and angiogenesis via alternative mechanisms. Adv Exp Med Biol. 2008;610:43–53. doi: 10.1007/978-0-387-73898-7_4. [DOI] [PubMed] [Google Scholar]
- Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- Graveel CR, DeGroot JD, Su Y, Koeman J, Dykema K, Leung S, et al. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc Natl Acad Sci U S A. 2009;106:12909–14. doi: 10.1073/pnas.0810403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoussoub RA, Dillon DA, D’Aquila T, Rimm EB, Fearon ER, Rimm DL. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;82:1513–20. doi: 10.1002/(sici)1097-0142(19980415)82:8<1513::aid-cncr13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Kong DS, Song SY, Kim DH, Joo KM, Yoo JS, Koh JS, et al. Prognostic significance of c-Met expression in glioblastomas. Cancer. 2009;115:140–8. doi: 10.1002/cncr.23972. [DOI] [PubMed] [Google Scholar]
- Sawada K, Radjabi AR, Shinomiya N, Kistner E, Kenny H, Becker AR, et al. c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007;67:1670–9. doi: 10.1158/0008-5472.CAN-06-1147. [DOI] [PubMed] [Google Scholar]
- Lai AZ, Abella JV, Park M. Crosstalk in Met receptor oncogenesis. Trends Cell Biol. 2009;19:542–51. doi: 10.1016/j.tcb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Cao R, Bjorndahl MA, Gallego MI, Chen S, Religa P, Hansen AJ, et al. Hepatocyte growth factor is a lymphangiogenic factor with an indirect mechanism of action. Blood. 2006;107:3531–6. doi: 10.1182/blood-2005-06-2538. [DOI] [PubMed] [Google Scholar]
- Kajiya K, Hirakawa S, Ma B, Drinnenberg I, Detmar M. Hepatocyte growth factor promotes lymphatic vessel formation and function. EMBO J. 2005;24:2885–95. doi: 10.1038/sj.emboj.7600763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JG. A preclinical review of sunitinib, a multitargeted receptor tyrosine kinase inhibitor with anti-angiogenic and antitumour activities. Ann Oncol. 2007;18(Suppl 10):x3–10. doi: 10.1093/annonc/mdm408. [DOI] [PubMed] [Google Scholar]
- Timofeevski SL, McTigue MA, Ryan K, Cui J, Zou HY, Zhu JX, et al. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors. Biochemistry. 2009;48:5339–49. doi: 10.1021/bi900438w. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–22. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, et al. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165:35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Broeck W, Derore A, Simoens P. Anatomy and nomenclature of murine lymph nodes: Descriptive study and nomenclatory standardization in BALB/cAnNCrl mice. J Immunol Methods. 2006;312:12–9. doi: 10.1016/j.jim.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Dadras SS, Lange-Asschenfeldt B, Velasco P, Nguyen L, Vora A, Muzikansky A, et al. Tumor lymphangiogenesis predicts melanoma metastasis to sentinel lymph nodes. Mod Pathol. 2005;18:1232–42. doi: 10.1038/modpathol.3800410. [DOI] [PubMed] [Google Scholar]
- Hall FT, Freeman JL, Asa SL, Jackson DG, Beasley NJ. Intratumoral lymphatics and lymph node metastases in papillary thyroid carcinoma. Arch Otolaryngol Head Neck Surg. 2003;129:716–9. doi: 10.1001/archotol.129.7.716. [DOI] [PubMed] [Google Scholar]
- Baluk P, McDonald DM. Markers for microscopic imaging of lymphangiogenesis and angiogenesis. Ann N Y Acad Sci. 2008;1131:1–12. doi: 10.1196/annals.1413.001. [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–44. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Mass RD, Campa C, Kim R. Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu Rev Med. 2007;58:491–504. doi: 10.1146/annurev.med.58.061705.145635. [DOI] [PubMed] [Google Scholar]
- Bruns CJ, Liu W, Davis DW, Shaheen RM, McConkey DJ, Wilson MR, et al. Vascular endothelial growth factor is an in vivo survival factor for tumor endothelium in a murine model of colorectal carcinoma liver metastases. Cancer. 2000;89:488–99. [PubMed] [Google Scholar]
- Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–37. [PubMed] [Google Scholar]
- Eckerich C, Zapf S, Fillbrandt R, Loges S, Westphal M, Lamszus K. Hypoxia can induce c-Met expression in glioma cells and enhance SF/HGF-induced cell migration. Int J Cancer. 2007;121:276–83. doi: 10.1002/ijc.22679. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Sakata M, Takeda T, Tahara M, Yamamoto T, Okamoto Y, et al. Up-regulation of c-met protooncogene product expression through hypoxia-inducible factor-1alpha is involved in trophoblast invasion under low-oxygen tension. Endocrinology. 2005;146:4682–9. doi: 10.1210/en.2005-0416. [DOI] [PubMed] [Google Scholar]
- Maula SM, Luukkaa M, Grenman R, Jackson D, Jalkanen S, Ristamaki R. Intratumoral lymphatics are essential for the metastatic spread and prognosis in squamous cell carcinomas of the head and neck region. Cancer Res. 2003;63:1920–6. [PubMed] [Google Scholar]
- Lee K, Park do J, Choe G, Kim HH, Kim WH, Lee HS. Increased intratumoral lymphatic vessel density correlates with lymph node metastasis in early gastric carcinoma. Ann Surg Oncol. 2010;17:73–80. doi: 10.1245/s10434-009-0707-y. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Nykanen A, Norrmen C, Ivanov KI, Andersson LC, Haglund C, et al. Transcription factor PROX1 induces colon cancer progression by promoting the transition from benign to highly dysplastic phenotype. Cancer Cell. 2008;13:407–19. doi: 10.1016/j.ccr.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med. 2005;201:1089–99. doi: 10.1084/jem.20041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohela M, Bry M, Tammela T, Alitalo K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154–65. doi: 10.1016/j.ceb.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–76. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- Partanen TA, Arola J, Saaristo A, Jussila L, Ora A, Miettinen M, et al. VEGF-C and VEGF-D expression in neuroendocrine cells and their receptor, VEGFR-3, in fenestrated blood vessels in human tissues. FASEB J. 2000;14:2087–96. doi: 10.1096/fj.99-1049com. [DOI] [PubMed] [Google Scholar]
- Roberts N, Kloos B, Cassella M, Podgrabinska S, Persaud K, Wu Y, et al. Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. Cancer Res. 2006;66:2650–7. doi: 10.1158/0008-5472.CAN-05-1843. [DOI] [PubMed] [Google Scholar]
- Kodera Y, Katanasaka Y, Kitamura Y, Tsuda H, Nishio K, Tamura T, et al. Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res. 2011;13:R66. doi: 10.1186/bcr2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YK, Lange-Asschenfeldt B, Velasco P, Hirakawa S, Kunstfeld R, Brown LF, et al. VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB J. 2004;18:1111–3. doi: 10.1096/fj.03-1179fje. [DOI] [PubMed] [Google Scholar]
- Maione F, Capano S, Regano D, Zentilin L, Giacca M, Casanovas O, et al. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J Clin Invest. 2012;122:1832–48. doi: 10.1172/JCI58976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Couto SS, Forrest WF, Lima A, Cheng JH, Molina R, et al. Anti-VEGF antibody therapy does not promote metastasis in genetically engineered mouse tumor models. J Pathol. 2012;227:417–30. doi: 10.1002/path.4053. [DOI] [PubMed] [Google Scholar]
- Chung AS, Kowanetz M, Wu X, Zhuang G, Ngu H, Finkle D, et al. Differential drug-class specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J Pathol. 2012;227:404–16. doi: 10.1002/path.4052. [DOI] [PubMed] [Google Scholar]
- Skobe M, Rockwell P, Goldstein N, Vosseler S, Fusenig NE. Halting angiogenesis suppresses carcinoma cell invasion. Nat Med. 1997;3:1222–7. doi: 10.1038/nm1197-1222. [DOI] [PubMed] [Google Scholar]
- Mazzone M, Basilico C, Cavassa S, Pennacchietti S, Risio M, Naldini L, et al. An uncleavable form of pro-scatter factor suppresses tumor growth and dissemination in mice. J Clin Invest. 2004;114:1418–32. doi: 10.1172/JCI22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–22. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68:4360–8. doi: 10.1158/0008-5472.CAN-07-5960. [DOI] [PubMed] [Google Scholar]
- Puri N, Ahmed S, Janamanchi V, Tretiakova M, Zumba O, Krausz T, et al. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13:2246–53. doi: 10.1158/1078-0432.CCR-06-0776. [DOI] [PubMed] [Google Scholar]
- Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10:2298–308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- Hussain M, Smith MR, Sweeney C, Corn PG, Elfiky A, Gordon MS, et al. Cabozantinib (XL184) in metastatic castration-resistant prostate cancer (mCRPC): Results from a phase II randomized discontinuation trial. J Clin Oncol. 2011;29(suppl) Abstract 4516. [Google Scholar]
- Lennerz JK, Kwak EL, Ackerman A, Michael M, Fox SB, Bergethon K, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011;29:4803–10. doi: 10.1200/JCO.2011.35.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catenacci DV, Henderson L, Xiao SY, Patel P, Yauch RL, Hegde P, et al. Durable complete response of metastatic gastric cancer with anti-Met therapy followed by resistance at recurrence. Cancer Discov. 2011;1:573–9. doi: 10.1158/2159-8290.CD-11-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surati M, Patel P, Peterson A, Salgia R. Role of MetMAb (OA-5D5) in c-MET active lung malignancies. Expert Opin Biol Ther. 2011;11:1655–62. doi: 10.1517/14712598.2011.626762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RJ, Smith MR. Targeting MET and vascular endothelial growth factor receptor signaling in castration-resistant prostate cancer. Cancer J. 2013;19:90–8. doi: 10.1097/PPO.0b013e318281e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor K. Cabozantinib approved for advanced medullary thyroid cancer. Am J Health Syst Pharm. 2013;70:88. doi: 10.2146/news130005. [DOI] [PubMed] [Google Scholar]
- Santoro A, Rimassa L, Borbath I, Daniele B, Salvagni S, Van Laethem JL, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14:55–63. doi: 10.1016/S1470-2045(12)70490-4. [DOI] [PubMed] [Google Scholar]
- You WK, Sennino B, Williamson CW, Falcon B, Hashizume H, Yao LC, et al. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71:4758–68. doi: 10.1158/0008-5472.CAN-10-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.