

Abstract
Transcription of interleukin-2 (IL-2), a pivotal cytokine in the mammalian immune response, is induced by NFAT and AP-1 transcriptional activators in stimulated T cells. NFATc2 and cJun drive high levels of synergistic human IL-2 transcription, which requires a unique interaction between the C-terminal activation domain of NFATc2 and cJun homodimers. Here we studied the mechanism by which this interaction contributes to synergistic activation of IL-2 transcription. We found that NFATc2 can recruit cJun homodimers to the −45 NFAT element, which lacks a neighboring AP-1 site. The bZip domain of cJun is sufficient to interact with the C-terminal activation domain of NFATc2 in the absence of DNA and this interaction is inhibited by AP-1 DNA. When the −45 NFAT site was replaced by either a NFAT/AP-1 composite site or a single AP-1 site the specificity for cJun homodimers in synergistically activating IL-2 transcription was lost, and cJun/cFos heterodimers strongly activated transcription. These studies support a model in which IL-2 transcriptional synergy is mediated by the unique recruitment of a cJun homodimer to the −45 NFAT site by NFATc2, where it acts as a co-activator for IL-2 transcription.
1. Introduction
In response to a foreign antigen, naive T cells undergo a process of differentiation that is initiated partly by the expression of interleukin-2 (IL-2) (Abbas and Lichtman, 2005). IL-2 is a 15 kDa cytokine that is essential in both activating and maintaining the adaptive immune response (Abbas and Lichtman, 2005; Avni and Rao, 2000). IL-2 expression requires signaling initiated by an antigen via the T cell receptor, and a co-stimulatory signal acting through a separate receptor (e.g. CD28) (Macian, 2005). Activation of the T cell receptor stimulates Ca2+ dependent signaling and subsequent activation of calcineurin, which dephosphoylates members of the NFAT (nuclear factor of activated T cells) family of transcription factors, leading to their localization in the nucleus (Hogan et al., 2003). Activation of the co-stimulatory receptor serves to stimulate MAPK pathways, which control the activities of several families of transcription factors (Karin, 1995). The co-stimulatory signal that triggers T cell activation can be mimicked in cultured Jurkat cells by treatment with ionomycin, a Ca2+ ionophore, and phorbol myristate acetate (PMA) (Flanagan and Crabtree, 1992).
IL-2 transcription requires both NFAT and AP-1 (activator protein 1) families of transcriptional activators (Abbas and Lichtman, 2005). There are five members of the NFAT family, with NFATc1 and NFATc2 being the primary members expressed in T cells (Lyakh et al., 1997). Upon T cell activation NFATc2 rapidly enters the nucleus and occupies the promoter region of the IL-2 gene, whereas NFATc1 is synthesized upon T cell stimulation, enters the nucleus, and binds the IL-2 promoter several hours later (Loh et al., 1996; Lyakh et al., 1997; Nguyen et al., 2010; Timmerman et al., 1996). NFATc1 and NFATc2 share similar overall structural motifs with the distinguishing feature being the presence of a C-terminal activation domain on NFATc2 (amino acids 688-921) that is absent from NFATc1 (Hogan et al., 2003). NFATc1 or NFATc2 bind cooperatively to DNA with dimers of the AP-1 family, comprised of Jun and Fos proteins (Chinenov and Kerppola, 2001; Ransone et al., 1989; Turner and Tjian, 1989). Specifically, cJun –which can homo- and heterodimerize – and cFos – which can only heterodimerize – function in IL-2 transcriptional activation (Ishihara and Schwartz, 2011; Nguyen et al., 2010). The IL-2 promoter spans from approximately −300 to +40 relative to the transcription start site (TSS) (Rooney et al., 1995). It is densely occupied by transcription factor binding sites, including a single NFAT site at −45 and four NFAT/AP-1 composite sites, three of which bind NFAT and AP-1 proteins in a cooperative manner (Figure 1A) (Rooney et al., 1995).
Figure 1.

The ARRE2 composite site is not required for synergistic activation from the IL-2 promoter. (A) Schematic of the human IL-2 promoter. The regulatory region spans approximately 300 bp upstream of the transcriptional start site (TSS) and contains 4 NFAT/AP-1 composite elements and a solitary NFAT site at −45, relative to the TSS. (B) Mutating the ARRE2 site in the IL-2 promoter reduces overall expression levels but does not decrease synergy between NFATc2 and cJun. Relative luciferase activity was determined by dividing the Firefly luciferase activity by the Renilla luciferase activity. The data were normalized to the average luciferase activity in the absence of overexpressed NFATc2 and cJun. Bars represent the average of three transfections and error bars represent one standard deviation.
We previously tested the response of the IL-2 promoter to different combinations of overexpressed NFAT and AP-1 proteins in Jurkat cells (Nguyen et al., 2010). The combination of NFATc2 and cJun was uniquely able to achieve high levels of transcriptional synergy, defined by the level of expression in the presence of both proteins being greater than the sum of the levels observed with either protein alone. Other combinations of NFATc1, NFATc2, cJun, and cFos were unable to achieve high levels of synergistic activation. Moreover, we found that the C-terminal activation domain of NFATc2 (amino acids 688-921) is capable of directly binding cJun homodimers, but not cJun/cFos heterodimers (Nguyen et al., 2010). This C-terminal region of NFATc2 is required for IL-2 transcriptional synergy between NFATc2 and cJun in Jurkat cells. How this interaction results in transcriptional synergy and which of the many NFAT and AP-1 sites in the IL-2 promoter are required for synergy was not known.
The fundamental molecular mechanisms resulting in transcriptional synergy have been documented, giving rise to common themes observed at mammalian promoters. For example, repeats of cis-elements bound by sequence-specific transcriptional activators can act in a synergistic manner by recruiting coactivators or the general transcription machinery (Carey et al., 1990; Lin et al., 1988; Lin et al., 1990). Synergy can also be achieved by protein-protein interactions between two or more different transcriptional activators that results in cooperative DNA binding at composite elements (Carey, 1998). The IL-2 promoter utilizes both of these modes to drive activation but they are not sufficient to explain the transcriptional synergy observed with overexpressed NFATc2 and cJun. Indeed if cooperative DNA binding by NFATc2 and cJun was the predominant mechanism of synergistic activation then we would expect NFATc1 and cJun/cFos to achieve high levels of synergy as well since they can bind cooperatively to composite elements in the IL-2 promoter (Macian et al., 2000; Rao et al., 1997; Sun et al., 1997). Hence, the unique ability of NFATc2 and cJun to achieve the observed levels of activation must be driven by another mode of synergy.
Here, we studied the mechanism by which NFATc2 and cJun achieve high levels of transcriptional synergy at the IL-2 promoter by asking how cis elements in the promoter, nuclear architecture, and protein-nucleic acid and/or protein-protein interactions contribute to synergy. Collectively, our data support a model in which the stimulation of T cells triggers NFATc2 to occupy the −45 NFAT site in the IL-2 promoter and recruit cJun homodimers to this site via its C-terminal activation domain. Since this protein-protein interaction does not occur with cJun/cFos heterodimers, NFATc2 has the unique ability to impart specificity for the recruitment of cJun homodimers in the absence of an AP-1 site. This model suggests that at the −45 NFAT site, cJun functions as a co-activator to mediate high levels of transcriptional synergy.
2. Methods
2.1. Plasmid construction
The pBS-IL2-Luciferase reporter has been previously described (Ferguson et al., 2001; Nguyen et al., 2010; Weaver et al., 2007). Mutations in the pBS-IL2-Luciferase reporter were introduced by QuickChange site-directed mutagenesis and verified by sequencing. Plasmids pcDNA-HA-cJUN, pcDNA-HA-cFos, pcDNA-HA-NFATc2, and pGEX-NFATc2(688-921) have been previously described (Nguyen et al., 2010). pET-6His-cJun(1-255) was made by amplifying this region of cJun cDNA by PCR and digesting with NdeI and HindIII. The fragment was subsequently ligated into pET-21a with 6His between HindIII and XhoI and the construct was verified by seqeuncing. pET-6His-NFATc2(DBD) was made by digesting pVL-GST-NFATc2(DBD), previously described (Kim et al., 2000), and ligating the fragment into a pET vector. pET-cJun(bZip) has been previously described (Lively et al., 2001).
2.2. Protein Purification
Purification of full length NFATc2 from insect cells has been previously described (Nguyen et al., 2010). Purification of NFATc2-DBD, GST-NFATc2(688-921), full length cJun, and cJun-bZip from E coli have been previously described (Chen et al., 1995; Chen et al., 1998; Chen et al., 1998; Lively et al., 2001; Nguyen et al., 2010). cJun activation domain (1-255) was expressed in Escherichia colistrain BL-21. Cultures transformed with pET-6His-cJun(1-255) were grown in the presence of 100 μg/ml ampicillin in Luria-Bertani broth at 37 °C to an optical density of 0.4 at 600 nm before expression was induced by the addition of isopropylthio-β-D-galactoside at a final concentration of 0.5 mM. After 2 hr, cells were harvested and the cell pellet was resuspended in a buffer containing 20 mM Tris (pH 7.9), 1 mM EDTA, 100 mM NaCl, 1 mM DTT, 0.2 mM phenylmethylsulfonyl fluoride, and sonicated 4 times for 15 s. Samples were centrifuged for 30 min at 15,000 rpm and 4° C. Precipitated material containing c-Jun was resuspended in 10 ml of 20 mM Tris (pH 7.9), 1 mM EDTA, 100 mM NaCl, 5 mM DTT, 0.2 mM phenylmethylsulfonyl fluoride and sonicated 2 times for 30 s. Samples were centrifuged for 10 min at 15,000 rpm and 4 °C. The pellet was resuspended in 5 ml of buffer A (20 mM Tris (pH 7.9), 1 mM EDTA, 5 mM DTT, and 7 M urea, 0.1 M NaCl) containing 20 mM imidazole. Soluble material was loaded onto a Ni-NTA agarose column (Qiagen) and washed with 5 column volumes of buffer A containing 20 mM imidazole followed by 5 column volumes of buffer A containing 40 mM imidazole. cJun(1-255) was eluted with buffer A containing 100 mM imidazole and subjected to three sequential dialyses in buffer B (20 mM Tris (pH 7.9), 0.1 mM EDTA, 10% glycerol, 5 mM DTT) containing the following additions: 1) 1 M urea and 1 M NaCl; 2) 1 M NaCl; 3) 0.1 M NaCl. After dialysis, the purified c-Jun(1-255) was separated into aliquots and stored at −80 °C.
2.3. Transfection assays
Jurkat cells were maintained at 37 °C and 5% CO2 in RPMI containing 10% FBS, 100 U/ml Penicillin, 100 μg/ml Streptomycin, 2 mM GlutaMax, and 50 μM 2-Mercaptoethanol. On the day of the transfection, Jurkat cells were seeded into a 24-well plate (5.0 × 105 cells/well) in antibiotic-free RPMI. Cells were transfected with X-tremeGENE HP DNA transfection reagent (Roche Applied Science) according to manufacturers instructions. Briefly, 100 ng of each protein expression construct or pcDNA3.1(+) vector, IL2-firefly-Luc reporter, and pRL-null-Renilla-luciferase (Promega) were incubated with X-tremeGENE HP in OptiMEM for 30 min before adding to cells. pcDNA3.1(+) was used to maintain an equal amount of plasmid DNA for each reaction. After 24 hr incubation at 37 °C, cells were stimulated with 1 μM ionomycin and 20 ng/ml PMA for 6 hours. Cells were then harvested and lysed with 150 μl Passive Lysis Buffer (Promega). Firefly and Renilla luciferase activities were determined using the Dual-luciferase kit (Promega). Firefly luciferase was normalized to renilla luciferase for each reaction and the data presented are the average of three independent biological replicates.
2.4. DNA pull-down assay
The -45 NFAT recognition element was created using a 5′ biotinylated DNA hairpin 5′-GATAATATTTTTCCAGAATTACACAAATTCTGGAAAAATATTATC- 3′ (the NFAT site is underlined). 4 pmol DNA was immobilized on 10μl streptavidin agarose beads (Novagen) per manufacturers instructions. Beads containing immobilized NFAT DNA were equilibrated in TGEM(0.15) (20 mM Tris-HCl (pH 7.9), 10% glycerol, 5 mM MgCl2, 0.1% NP-40, 1 mM DTT, 0.2 mM PMSF, 150 mM NaCl, 1 mM EDTA). 15 pmol of either full length NFATc2 or NFATc2 DBD were added to immobilized DNA and incubated at 4 C for 1 hr with nutation. For the sample without NFAT protein an equal volume of TGEM(0.15) was added and the sample was processed in parallel. Beads were washed twice with TGEM(0.15). 16 pmol purified cJun and 16 pmol mutant AP-1 DNA used as a competitor were added to each sample and incubated at room temperature for 45 min with nutation. Beads were washed three times with TGEM(0.15), eluted with SDS sample buffer and heat, and bound protein was subjected to Western blot analysis using α-cJun antibody (sc45; Santa Cruz).
2.5. Protein-protein interaction assays
GST and GST-NFATc2(688-921) were immobilized by incubating cell extracts with glutathione-Sepharose beads for 2 hr at 4°C. Immobilized proteins were washed two times in 10-bead volumes of TGEMD buffer containing 1 M NaCl (TGEMD(1 M NaCl)) followed by three washes in 10-bead volumes of TGEMD buffer containing 0.1 M NaCl (TGEMD(0.1 M NaCl)). Prior to use in interaction assays, the amount of each protein on the beads was estimated and approximately 0.5 μg of total immobilized protein was added to each interaction assay. Input proteins were added to the immobilized GST fusion proteins and nutated for 2 hr at 4°C. After washing four times with 10-bead volumes of TGEMD(0.1 M NaCl), the bound fraction was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS- PAGE) and silver staining, coomassie staining or immunoblotting (as indicated in the figure legends). For the AP-1 DNA competition (Figure 6C) 15 pmol cJun was added to immobilized GST or GST-NFATc2(688-921). WT AP-1 DNA
Figure 6.

The bZip domain of cJun is sufficient for binding the C-terminal activation domain of NFATc2 and binding is inhibited by AP-1 DNA. (A) Schematic of cJun showing the activation domain region (AD) and the bZip, which is responsible for dimerization and DNA binding. (B) The bZip domain of cJun is sufficient to bind the C-terminal activation domain of NFATc2. Proteins were subjected to SDS-PAGE and visualized with coomassie stain. (C)AP-1 DNA blocks the interaction between cJun and the C-terminal activation domain of NFATc2. GST-NFATc2(688-921) and control GST were immobilized and incubated with 15 pmol of purified cJun. AP-1 DNA or mutant DNA (10 or 20 pmol) were added to the reactions, bound cJun was resolved by SDS-PAGE and visualized by silver stain.
5′-GTCCATGACTCAGAAGAGACACACTCTTCTGAGTCATGGAC- 3′ (AP-1 sequence underlined) or mutant AP-1 DNA
5′-GTCCAAATCTCAGAAGAGACACACTCTTCTGAGATTTGGAC- 3′ (mutant AP-1 sequence underlined) was added at 10 or 20 pmol.
3. Results
3.1. The -45 NFAT site is essential for synergistic IL-2 activation
To understand the mechanism of synergistic IL-2 activation by NFATc2 and cJun, we performed transient transfection assays in Jurkat cells using a reporter plasmid containing the human IL-2 promoter region from −326 to +45 upstream of the Firefly luciferase gene. Like the endogenous IL-2 gene, this reporter requires co-stimulation by PMA and ionomycin to be actively transcribed (Nguyen et al., 2010). Expression from the IL-2 reporter was measured in the absence of overexpressed protein and in the presence of overexpressed NFATc2 and cJun, both individually and in combination (Figure 1B, white bars). Synergistic activation is apparent since the luciferase level due to the combined overexpression of NFATc2 and cJun is substantially greater than the sum of luciferase levels obtained when the proteins were overexpressed individually.
We reasoned that mutating known NFAT and AP-1 elements within the IL-2 promoter would reveal which cis-elements are responsible for synergistic activation, thereby providing insight into the mechanism of NFATc2 and cJun driven transcriptional synergy. The Antigen Receptor Response Element 2 (ARRE2) contains a NFAT/AP-1 composite site that is crucial for inducing IL-2 expression in response to T cell activation (Durand et al., 1988). To ask whether this site is required for synergistic activation we mutated both the NFAT and AP-1 elements and determined if overexpressed NFATc2 and cJun could still exhibit synergistic activation. As expected, the overall expression level of the ARRE2 mutant reporter was lower than the wild type reporter (Figure 1B, gray bars). NFATc2 and cJun, however, were able to achieve 6.8 fold synergistic activation on the ARRE2 mutant reporter compared to 8.6 fold synergy on the wild type reporter, showing that the ARRE2 element is not required for synergistic transcriptional activation.
We next individually mutated the NFAT and AP-1 sites within the remaining three composite elements in the IL-2 promoter to determine which, if any, of these sites were required for synergistic activation by NFATc2 and cJun in transient transfection assays. As shown in Figure 2A, individually mutating either the NFAT or AP-1 site within any of the composite elements did not decrease synergy between NFATc2 and cJun. The mutations did decrease overall activation as expected, with the exception of mutation of the -90 NFAT site, which resulted in higher levels of activation and synergy. Unexpectedly, none of the four AP-1 elements within the IL-2 promoter were required for NFATc2 and cJun dependent synergistic activation.
Figure 2.

Of the NFAT and AP-1 sites in the IL-2 promoter, only the −45 NFAT site is required for synergy. (A) Mutating NFAT and AP-1 sites within three of the IL-2 promoter composite elements does not decrease transcriptional synergy. The data were normalized to the average luciferase activity in the absence of overexpressed NFATc2 and cJun. Bars represent the average of three transfections and error bars represent one standard deviation. (B) Mutation of the −45 NFAT site abolishes NFATc2 and cJun transcriptional synergy. Bars represent the average of three transfections and error bars represent one standard deviation.
The only NFAT element that remained to be mutated was the −45 site, which lacks an adjacent AP-1 element for cooperative DNA binding. DNase I footprinting of the IL-2 promoter sequence have shown that this site is sufficient for high affinity binding by NFATc2 (Kim et al., 2000). We mutated the −45 NFAT element and were surprised to find that this abolished synergy (Figure 2B); overexpression of NFATc2 and cJun together did not increase activation above the levels observed with each protein individually overexpressed. Taken together, these data show that although the NFAT/AP-1 composite elements are important for overall expression levels, only the −45 NFAT site is required for synergistic activation by NFATc2 and cJun.
3.2. IL-2 synergistic activation is sensitive to the helical phasing between the −45 NFAT site and the transcriptional start site
We hypothesized that factors bound at the −45 site must uniquely interact with the core transcription machinery to mediate transcriptional synergy. If so, the spacial orientation between the −45 NFAT site and the core promoter would be important for synergy to occur. To test this we inserted either 5 bp or 10 bp at position −32 in the IL-2 luciferase reporter, which is positioned between the −45 NFAT site and the TATA box (Figure 3A schematics). Insertion of 5 bp rotates the regulatory region upstream of −45 one half of a helical turn, thereby changing its orientation with respect to the core promoter. Insertion of an additional 5 bp (for a total of 10 bp) restores the helical orientation of the −45 NFAT site with respect to the IL-2 core promoter.
Figure 3.

Altering the helical phasing between the −45 NFAT site and the core promoter region decreases NFATc2 and cJun synergy. (A) 5 bp to invert helical phasing or 10 bp to restore helical phasing were inserted at position −32 in the IL-2 luciferase reporter. For each reporter, the data were normalized to the activity in the absence of overexpressed protein. Bars represent the average of three transfections and error bars represent one standard deviation. (B) Changing the helical phasing by insertion of 5 bp upstream of the −45 NFAT site (at −60) does not reduce IL-2 activation with overexpressed NFATc2 and cJun. Data were normalized to wild type activity with overexpressed NFATc2 and cJun.
We tested these two helical phasing mutants for their ability to respond synergistically to overexpressed NFATc2 and cJun in Jurkat cells. Disrupting the helical phasing by inserting 5 bp reduced synergy, resulting in approximately half maximal activation in the presence of overexpressed NFATc2 and cJun (Figure 3A). Restoring helical phasing with the 10 bp insertion restored synergistic activation to the wild type level. To test whether the reduction in synergy by changing helical phasing was specific to the orientation between the −45 NFAT site and the core promoter, we inserted 5 bp at −60 and observed no significant difference in luciferase activity with overexpressed NFATc2 and cJun when compared to the wild type promoter (Figure 3B). These data show that NFATc2 and cJun driven transcriptional synergy is sensitive to the helical orientation of the −45 NFAT site with respect to the core promoter. This suggests that interaction between the factors bound at −45 and the core transcription machinery requires a specific architecture.
3.3. NFATc2 can recruit cJun to the −45 NFAT site in the absence of an AP-1 site
The interactions between the DNA binding domains of NFAT and AP-1 proteins that facilitate cooperative DNA binding have been widely investigated (Chen et al., 1998; Macian et al., 2000; Peterson et al., 1996), however, how the interaction between the C-terminal activation domain of NFATc2 and cJun homodimers promotes transcriptional synergy remains unclear (Figure 4A). Since no known AP-1 site within the IL-2 promoter region was required for synergy, we hypothesized that via its C-terminal region, NFATc2 bound to the −45 site could recruit cJun homodimers. To test this hypothesis we performed a DNA pull-down assay using the isolated −45 NFAT element sequence. We used a biotinylated ssDNA oligonucleotide designed to form a hairpin containing the NFAT site in the double stranded region (Figure 4B), which minimized the amount of DNA beyond that protected by NFATc2 in DNase I footprinting assays (Kim et al., 2000). As shown in Figure 4C, cJun was recovered on immobilized DNA only in the presence of full length NFATc2 (lane 3). Importantly, the NFATc2 DNA binding domain (DBD) did not pull down cJun over the amounts observed with DNA only (compare lanes 2 and 4). Since the NFATc2 DNA-binding domain contains the minimal region necessary for cooperative DNA binding with AP-1 proteins (Chen et al., 1998) we conclude that the recruitment of cJun to the −45 NFAT site by NFATc2 is not due to cooperative DNA binding. Instead, the data in Figure 4C suggest that the interaction between the C-terminal region of NFATc2 and cJun homodimers can recruit cJun to the −45 NFAT element in the absence of an AP-1 site.
Figure 4.

Full length NFATc2 can recruit cJun to the −45 site in the absence of an AP-1 element. (A) Schematic of NFATc2; AD is activation domain. (B)DNA -bound full length NFATc2, but not the minimal DNA binding domain (DBD) of NFATc2, can recruit cJun homodimers to DNA. Immobilized DNA containing the −45 NFAT recognition element was incubated with equal molar amounts of either full length NFATc2 or the NFATc2 DBD followed by washing to remove unbound protein. A control sample without NFAT was processed in parallel. Each sample was incubated with cJun, complexes were washed, eluted, and cJun was visualized by Western blot analysis with α-cJun antibody.
3.4. Replacing the −45 NFAT site with an NFAT/AP-1 composite element allows cJun/cFos to synergize with NFATc2
The C-terminal region of NFATc2 does not interact with cJun/cFos heterodimers in vitro, and when overexpressed with NFATc2, cJun/cFos cannot achieve the same levels of synergistic activation from the IL-2 promoter compared to cJun homodimers (Nguyen et al., 2010). Indeed, when cFos was overexpressed along with cJun and NFATc2 it drove down synergy of the IL-2 reporter. We hypothesized that the inability of cJun/cFos to interact with the C-terminal activation domain of NFATc2 precludes its recruitment to the −45 element, thereby preventing cJun/cFos from synergizing with NFATc2. Replacing the −45 NFAT site with an NFAT/AP-1 composite element would allow overexpressed cJun/cFos to be recruited to this region of the promoter and perhaps allow the heterodimer to synergistically activate transcription with overexpressed NFATc2.
We mutated the −45 NFAT site to an NFAT/AP-1 composite element, with a sequence and orientation similar to the ARRE2 element (Figure 5). This mutant reporter was tested in Jurkat cells alongside the wild type reporter, overexpressing different combinations of NFATc2, cJun, and cFos proteins. As expected, when NFATc2, cJun, or cJun/cFos were overexpressed individually, the mutant reporter responded similarly to the wild type reporter. Importantly, the mutant reporter showed equivalent activation to the wild type reporter in the presence of NFATc2 and cJun, providing evidence that recruitment of cJun to the −45 promoter region significantly contributes to synergistic activation. Interestingly, on the mutant reporter overexpressed cJun/cFos and NFATc2 were able to achieve the same level of activation as overexpressed cJun and NFATc2. This was not the case on the wild-type promoter. These data suggest that the absence of an AP-1 site neighboring the −45 NFAT site in the wild-type promoter provides specificity for NFATc2 to recruit cJun homodimers and not cJun/cFos heterodimers. However, upon artificial recruitment of AP-1 proteins to the −45 promoter region via interaction with an inserted AP-1 site, cJun homodimers and cJun/cFos heterodimers are both capable of achieving the same level of activation.
Figure 5.

Replacing the −45 NFAT site with a NFAT/AP-1 composite element results in high synergistic activation with NFATc2 and cJun/cFos. For the mutant reporter, a NFAT/AP-1 composite element replaced the single NFAT site at −45 while maintaining the orientation of the NFAT site relative to the TSS. Overexpression of cJun or cJun/cFos in combination with NFATc2 resulted in high synergistic activation on the mutant NFAT/AP-1 composite reporter. Bars are the average of three transfections and error bars represent one standard deviation.
3.5. The bZip domain of cJun is sufficient to interact with the C-terminal region of NFATc2 and this interaction is blocked by AP-1 DNA
We next wanted to identify the region of cJun that interacts with the C-terminal region of NFATc2. We expressed and purified the cJun activation domain (amino acids 1-255) and the bZip domain (amino acids 255-317), the latter of which contains the regions responsible for DNA binding and dimerization (Figure 6A) (Glover and Harrison, 1995; Junius et al., 1996). Both domains of cJun were incubated with immobilized GST-NFATc2(688-921) or control GST. As shown in Figure 6B, the bZip domain was sufficient for binding GST-NFATc2(688-921) while the cJun activation domain was unable to bind.
Since the bZip domain of cJun contains the DNA binding domain, we asked whether the interaction between cJun and the C-terminal region of NFATc2 could be blocked by AP-1 DNA. To determine this we conducted a similar interaction assay with purified cJun and immobilized GST-NFATc2(688-921), adding either a wild type AP-1 DNA sequence or a mutant DNA sequence as a control. As shown in Figure 6C, AP-1 DNA, but not mutant DNA, blocked the interaction between cJun and GST-NFATc2(688-921). Collectively, these data suggest that the C-terminal region of DNA-bound NFATc2 interacts with the DNA binding domain of cJun/cJun to recruit the homodimer to the −45 NFAT site in the absence of a neighboring AP-1 site. Moreover, this interaction cannot occur when cJun is bound to DNA, therefore it is the unique protein-protein interaction that gives rise to synergistic transcriptional activation.
3.6. An AP-1 DNA element can replace the −45 NFAT site to mediate transcriptional synergy
If the role of NFATc2 bound to the −45 site is to recruit cJun/cJun to drive synergy, then replacing the −45 NFAT site on the IL-2 luciferase reporter with a canonical AP-1 element should result in a promoter that still synergistically responds to NFATc2 and cJun (Figure 7). In this model, NFATc2 and cJun bound to the upstream composite elements would function with cJun/cJun bound at the engineered −45 AP-1 site to drive full activation of the IL-2 promoter. This mutant reporter was tested alongside the wild type reporter with different combinations of overexpressed NFATc2, cJun, and cFos proteins. Unlike the wild type IL-2 reporter, both overexpressed cJun and cJun/cFos proteins were able to significantly activate transcription in the absence of overexpressed NFATc2. Importantly, the combination of NFATc2 and cJun achieved similar levels of activation on both reporters, while NFATc2 and cJun/cFos achieved substantially higher levels of activation on the mutant reporter. These data indicates that having an AP-1 site at −45 results in similar overall levels of transcription in the presence of NFATc2 and AP-1 proteins consisting of either cJun homodimers or cJun/cFos. In contrast, the NFAT site at −45 confers specificity for NFATc2 to recruit cJun/cJun homodimers to achieve high levels of transcription.
Figure 7.

Replacing the −45 NFAT site with an AP-1 element in the IL-2 promoter provides an alternative mechanism for recruitment of AP-1 proteins and synergistic activation. The data were normalized to the activity of the wild type reporter in the absence of overexpressed protein. Bars are the average of three transfections and error bars represent one standard deviation.
4. Discussion
Here, we studied the mechanism by which NFATc2 and cJun achieve high levels of transcriptional synergy on the IL-2 promoter. Systematic mutation of each of the known NFAT and AP-1 elements within the IL-2 promoter revealed that only the −45 NFAT element is essential for synergy. Considering that synergy requires overexpression of cJun we were surprised to find that none of the AP-1 sites in the IL-2 promoter were required. Additionally, alterations in the helical orientation of the −45 NFAT site relative to the TSS showed that full synergy requires a specific promoter architecture, perhaps allowing interactions of the factors bound at the −45 site with coactivators or the general transcription machinery bound at the core promoter. Given that the interaction between the C-terminal activation domain and cJun can occur in the absence of DNA, we hypothesized that this interaction could mediate recruitment of cJun to DNA-bound NFATc2. By immobilizing the −45 NFAT site we demonstrated that DNA-bound full length NFATc2 was able to recruit cJun, whereas the NFATc2 DNA binding domain was not. The bZip domain of cJun was sufficient to bind the C-terminal activation domain of NFATc2 and this interaction could be inhibited with AP-1 DNA. Additionally, cJun could be recruited to the −45 region of the IL-2 promoter via an engineered AP-1 DNA element to drive synergistic levels of transcription.
Prior to this work, the mechanism by which specific combinations of NFAT proteins and AP-1 dimers synergistically activate transcription apart from cooperative DNA binding, and how specificity between these factors is achieved was not well understood. Our studies lead to a model by which in activated T cells, NFATc2 occupies the −45 site of the IL-2 promoter and through its C-terminal activation domain recruits cJun homodimers (Figure 8A), which substantially contributes to IL-2 activation. Our model suggests that occupancy of cJun at the −45 promoter region is important for synergy, and the absence of an AP-1 site adjacent to the −45 NFAT site functions to impart specificity for cJun homodimers over cJun/cFos heterodimers (Figure 8B). We mutated the −45 NFAT site on the IL-2 reporter into either a NFAT/AP-1 composite element or a consensus AP-1 site giving it the ability to recruit both cJun homodimers and cJun/cFos heterodimers. We reasoned that while overexpression of cFos decreased the observed synergy between NFATc2 and cJun on the wild type reporter, artificial recruitment of cJun/cFos heterodimers by either a −45 composite site or consensus AP-1 site could attenuate this observed decrease. Indeed, when AP-1 proteins were recruited to the −45 region by an engineered AP-1 site, the synergistic specificity for cJun/cJun was lost.
Figure 8.
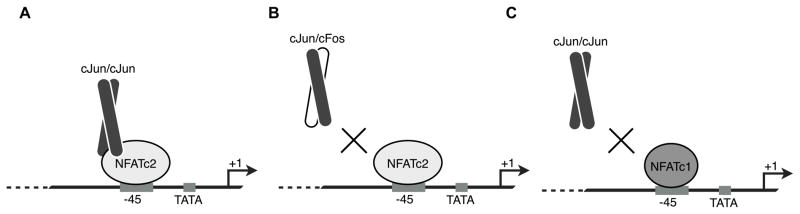
Model for synergistic activation of IL-2 by NFATc2 and cJun. (A) NFATc2, via the C-terminal activation domain, recruits cJun homodimers to the −45 promoter region in the absence of an AP-1 site. (B) cJun/cFos heterodimers, which do not interact with the C-terminal activation domain of NFATc2, cannot be recruited to the −45 promoter region and therefore cannot drive high levels of synergistic activation. (C) NFATc1, lacking the C-terminal activation domain present on NFATc2, is unable to recruit cJun homodimers.
Our data indicate that transcriptional synergy between NFATc2 and cJun requires specific architectural constraints between −45 and the core transcription machinery. We altered the helical phasing between the −45 NFAT site and the TATA box by making a 5 bp insertion, which inverted the orientation of the −45 site relative to the TSS. This reduced synergy in the presence of overexpressed NFATc2 and cJun. This was specific for the −45 site; a 5 bp insertion at −60 did not impact activation levels. Higher order structure at other promoters has been shown to be important for achieving high levels of transcription (Thanos and Maniatis, 1995). The nature of the contacts that confer architectural specificity between factors bound at the −45 site in the IL-2 promoter and its core promoter are not yet understood. We have previously characterized interactions between cJun and TAF1 (the largest subunit of TFIID) and NFATc2 and the TAF4 subunit of TFIID that are important for transcriptional activation (Lively et al., 2001; Lively et al., 2004; Nguyen et al., 2010); however, the precise interplay between these interactions and the NFATc2/cJun complex bound at the −45 promoter element remain to be determined.
Previous studies have shown that the cJun homodimer can function as a coactivator at the interleukin-1β (IL-1β) promoter (Behre et al., 1999; Grondin et al., 2007). In this case cJun/cJun is recruited by DNA-bound PU.1 and C/EBPβ and synergistically activates transcription. Interestingly, residues within the basic region of cJun that contact DNA are responsible for mediating the protein-protein interactions with PU.1 and C/EBPβ (Grondin et al., 2007). These results are similar to our observations that cJun binding to DNA and binding to the NFATc2 C-terminal activation domain are mutually exclusive. AP-1 DNA, but not a mutant AP-1 DNA sequence, was capable of inhibiting the interaction between the C-terminal activation domain of NFATc2 and cJun. These data suggest that cJun homodimers recruited by NFATc2 in the absence of an AP-1 element can function as a coactivator to drive IL-2 synergistic activation during the early stages of T cell activation.
The occupancy of NFATc2 and cJun at the IL-2 promoter shortly after stimulation coincides with a rapid production of IL-2 mRNA, suggesting these proteins are critical for the initial induction of transcription (Nguyen et al., 2010). After several hours of stimulation lower levels of IL-2 transcriptional activity are observed (Ishihara and Schwartz, 2011; Nguyen et al., 2010). Coincident with this, NFATc1 can be detected at the IL-2 promoter (Ishihara and Schwartz, 2011; Nguyen et al., 2010). We have previously shown that NFATc1 is not capable of synergistically activating IL-2 transcription with cJun nor interacting with cJun homodimers off of DNA, which is consistent with the fact that NFATc1 lacks a C-terminal activation domain (Nguyen et al., 2010). We propose that inability of NFATc1 to recruit cJun homodimers to the −45 site is the cause of the lower IL-2 transcription observed several hours after T cells are first stimulated (Figure 8C).
The mechanism of synergy we have studied here allows for a naive T cell, upon stimulation, to rapidly synthesize IL-2 mRNA, in part due to the unique interaction between NFATc2 and cJun homodimers. The specificity of this interaction allows different combinations of NFAT and AP-1 proteins to tune the level of IL-2 transcription at different points during T cell stimulation. It will be interesting to determine whether this mechanism of transcriptional synergy is used at other genes induced during T cell activation.
Highlights.
Only a single NFAT element at −45 is essential for transcriptional synergy by NFATc2 and cJun.
IL-2 transcriptional synergy is sensitive to the helical phasing between the −45 NFAT element and the transcriptional start site.
NFATc2 can recruit cJun to the −45 NFAT site in the absence of an AP-1 site.
The bZip domain of cJun is sufficient to interact with the C-terminal activation domain of NFATc2 and this interaction is blocked by AP-1 DNA.
An AP-1 DNA element can replace the −45 NFAT site to mediate transcriptional synergy.
Acknowledgments
This research was supported by Public Health Service Grant R01 GM55235 from the National Institute of General Medical Sciences. R.D.W. was supported in part by NIH Predoctoral Training Grant T32 GM08759. We would like to thank Lea Witkowsky for her insightful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas AK, Lichtman AH. Cellular and Molecular Immunology. W.B. Saunders Company; Philadelphia, PA: 2005. [Google Scholar]
- Avni O, Rao A. T cell differentiation: a mechanistic view. Curr Opin Immunol. 2000;12:654–659. doi: 10.1016/s0952-7915(00)00158-8. [DOI] [PubMed] [Google Scholar]
- Behre G, Whitmarsh AJ, Coghlan MP, Hoang T, Carpenter CL, Zhang DE, Davis RJ, Tenen DG. c-Jun is a JNK-independent coactivator of the PU. 1 transcription factor. J Biol Chem. 1999;274:4939–4946. doi: 10.1074/jbc.274.8.4939. [DOI] [PubMed] [Google Scholar]
- Carey M. The enhanceosome and transcriptional synergy. Cell. 1998;92:5–8. doi: 10.1016/s0092-8674(00)80893-4. [DOI] [PubMed] [Google Scholar]
- Carey M, Lin YS, Green MR, Ptashne M. A mechanism of synergistic activation of a mammalian gene by GAL4 derivatives. Nature. 1990;345:361–364. doi: 10.1038/345361a0. [DOI] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–48. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- Chen L, Oakley MG, Glover JN, Jain J, Dervan PB, Hogan PG, Rao A, Verdine GL. Only one of the two DNA-bound orientations of AP-1 found in solution cooperates with NFATp. Curr Biol. 1995;5:882–889. doi: 10.1016/s0960-9822(95)00178-3. [DOI] [PubMed] [Google Scholar]
- Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;20:2438–2452. doi: 10.1038/sj.onc.1204385. [DOI] [PubMed] [Google Scholar]
- Durand DB, Shaw JP, Bush MR, Replogle RE, Belagaje R, Crabtree GR. Characterization of antigen receptor response elements within the interleukin-2 enhancer. Mol Cell Biol. 1988;8:1715–1724. doi: 10.1128/mcb.8.4.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson HA, Kugel JF, Goodrich JA. Kinetic and mechanistic analysis of the RNA polymerase II transcription reaction at the human interleukin-2 promoter. J Mol Biol. 2001;314:993–1006. doi: 10.1006/jmbi.2000.5215. [DOI] [PubMed] [Google Scholar]
- Flanagan WM, Crabtree GR. In vitro transcription faithfully reflecting T-cell activation requirements. J Biol Chem. 1992;267:399–406. [PubMed] [Google Scholar]
- Glover JN, Harrison SC. Crystal structure of the heterodimeric bZIP transcription factor cFos-cJun bound to DNA. Nature. 1995;373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- Grondin B, Lefrancois M, Tremblay M, Saint-Denis M, Haman A, Waga K, Bédard A, Tenen DG, Hoang T. c-Jun homodimers can function as a context-specific coactivator. Mol Cell Biol. 2007;27:2919–2933. doi: 10.1128/MCB.00936-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Ishihara S, Schwartz RH. Two-step binding of transcription factors causes sequential chromatin structural changes at the activated IL-2 promoter. J Immunol. 2011;187:3292–3299. doi: 10.4049/jimmunol.1003173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junius FK, O’Donoghue SI, Nilges M, Weiss AS, King GF. High resolution NMR solution structure of the leucine zipper domain of the c-Jun homodimer. J Biol Chem. 1996;271:13663–13667. doi: 10.1074/jbc.271.23.13663. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Kim LJ, Ferguson HA, Seto AG, Goodrich JA. Characterization of DNA binding, transcriptional activation, and regulated nuclear association of recombinant human NFATp. BMC Immunol. 2000;1:1. doi: 10.1186/1471-2172-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YS, Carey M, Ptashne M, Green MR. How different eukaryotic transcriptional activators can cooperate promiscuously. Nature. 1990;345:359–361. doi: 10.1038/345359a0. [DOI] [PubMed] [Google Scholar]
- Lin YS, Carey MF, Ptashne M, Green MR. GAL4 derivatives function alone and synergistically with mammalian activators in vitro. Cell. 1988;54:659–664. doi: 10.1016/s0092-8674(88)80010-2. [DOI] [PubMed] [Google Scholar]
- Lively TN, Ferguson HA, Galasinski SK, Seto AG, Goodrich JA. c-Jun binds the N terminus of human TAF(II)250 to derepress RNA polymerase II transcription in vitro. J Biol Chem. 2001;276:25582–25588. doi: 10.1074/jbc.M100278200. [DOI] [PubMed] [Google Scholar]
- Lively TN, Nguyen TN, Galasinski SK, Goodrich JA. The basic leucine zipper domain of c-Jun functions in transcriptional activation through interaction with the N terminus of human TATA-binding protein-associated factor-1 (human TAF(II)250) J Biol Chem. 2004;279:26257–26265. doi: 10.1074/jbc.M400892200. [DOI] [PubMed] [Google Scholar]
- Loh C, Carew JA, Kim J, Hogan PG, Rao A. T-cell receptor stimulation elicits an early phase of activation and a later phase of deactivation of the transcription factor NFAT1. Mol Cell Biol. 1996;16:3945–3954. doi: 10.1128/mcb.16.7.3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyakh L, Ghosh P, Rice NR. Expression of NFAT-family proteins in normal human T cells. Mol Cell Biol. 1997;17:2475–2484. doi: 10.1128/mcb.17.5.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Macian F, Garcia-Rodriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 2000;19:4783–4795. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Kim LJ, Walters RD, Drullinger LF, Lively TN, Kugel JF, Goodrich JA. The C-terminal region of human NFATc2 binds cJun to synergistically activate interleukin-2 transcription. Mol Immunol. 2010;47:2314–2322. doi: 10.1016/j.molimm.2010.05.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BR, Sun LJ, Verdine GL. A critical arginine residue mediates cooperativity in the contact interface between transcription factors NFAT and AP-1. P Natl Acad Sci USA. 1996;93:13671–13676. doi: 10.1073/pnas.93.24.13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransone LJ, Visvader J, Lamph WW, Sassone-Corsi P, Verma IM. fos and jun interaction: the role of the leucine zipper. Int J Cancer Suppl. 1989;4:10–21. [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Rooney JW, Sun YL, Glimcher LH, Hoey T. Novel NFAT sites that mediate activation of the interleukin-2 promoter in response to T-cell receptor stimulation. Mol Cell Biol. 1995;15:6299–6310. doi: 10.1128/mcb.15.11.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LJ, Peterson BR, Verdine GL. Dual role of the nuclear factor of activated T cells insert region in DNA recognition and cooperative contacts to activator protein 1. P Natl Acad Sci USA. 1997;94:4919–4924. doi: 10.1073/pnas.94.10.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. Virus induction of human IFNb gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 1996;383:837–840. doi: 10.1038/383837a0. [DOI] [PubMed] [Google Scholar]
- Turner R, Tjian R. Leucine repeats and an adjacent DNA binding domain mediate the formation of functional cFos-cJun heterodimers. Science. 1989;243:1689–1694. doi: 10.1126/science.2494701. [DOI] [PubMed] [Google Scholar]
- Weaver JR, Good K, Walters RD, Kugel JF, Goodrich JA. Characterization of the sequence and architectural constraints of the regulatory and core regions of the human interleukin-2 promoter. Mol Immunol. 2007;44:2813–2819. doi: 10.1016/j.molimm.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]