

Abstract
OBJECTIVES
Tumor microenvironment, defined by a variety of growth factors including lysophosphatidic acid (LPA), whose levels are increased in pancreatic cancer patients, plays a major role in the genesis and progression of pancreatic cancer. Since the gep proto-oncogenes, Gα12 and Gα13, are implicated in LPA-stimulated oncogenic signaling, this study is focused on evaluating the role of these proto-oncogenes in LPA-stimulated invasive migration of pancreatic cancer cells.
METHODS
Effect of LPA on the migration and proliferation of pancreatic cancer cells was assessed using BxPC3, Dan-G, MDAPanc-28, Panc-1, and PaCa-2 cell-lines. The role of Gα13 in the migration of pancreatic cancer cells was interrogated by disrupting LPAR-Gα13 interaction using CT13, a dominant negative mutant of Gα13, and by silencing the expression of Gα13.
RESULTS
Results indicate that LPA stimulates the migration of pancreatic cancer cells and such LPA-stimulated migratory response is mediated by Gα13. Furthermore, the results establish that the silencing of Gα13, but not Gα12, abrogates LPA-stimulated invasive migration of pancreatic cancer cells.
CONCLUSION
These results report for the first time a critical role for Gα13 in LPA-stimulated invasive migration of pancreatic cancer cells. These findings identify LPA-LPAR-Gα13 signaling node as a novel therapeutic target for pancreatic cancer treatment and control.
Keywords: LPA, gep, pancreatic cancer, Gα13, oncogene, metastasis
INTRODUCTION
Ductal adenocarcinomas of the pancreas are the 4th most common cause of cancer death1. The 1 and 5 year survival rates for all stages combined are currently 26% and 5% respectively with the median survival being less than 6 months. Despite remarkable progress in the fields of genetics, cancer biology and advances in surgical techniques as well as chemotherapeutics, our ability to recognize and treat patients with pancreatic cancer remains poor. This is primarily due to our relatively poor understanding of the mechanisms underlying the genesis and the progression of the disease. Recent studies have identified the crucial role of tumor microenvironment in the progression of pancreatic cancer2-4. The microenvironment of pancreatic tumors is defined by the extracellular mediators of signal transduction-growth factors, cytokines, neuropeptides, and bioactive lipids-elaborated by components of the stromal compartment in addition to the tumor itself. These extracellular mediators include growth factors such as epidermal growth factor ligands, platelet derived growth factor, fibroblast growth factor-2, interleukin-8, Hepatocyte growth factor, vasculo-endothelial growth factor, lysophosphatidic acid (LPA), cholecystokinin, somatostatin, gastrin, bombesin, thrombin, and neurotensin2,3,5-8. Of particular importance here is the G protein-coupled receptor (GPCR) ligands that appear to play a major role in tumor growth, tumor progression and metastasis by activating specific set of autocrine and paracrine signaling loops5-9. Several GPCR agonists, including LPA, along with their cognate receptors have been implicated in the oncogenic growth, progression, and metastasis of pancreatic cancer. In addition, overexpression of LPA receptors has also been observed in pancreatic cancer tissues10,11. More ominously, it has been noted that pancreatic cancer cell lines expressing higher levels of the LPA receptors show greater motility9-12. Based on these observations, it has been surmised that LPA contributes to the progression of pancreatic cancer through the promotion of a metastatic phenotype. However, the underlying mechanism has not been fully understood.
LPA transmits its signaling by binding to a family of LPA-receptors that are coupled to G proteins 13-15. Of the different G proteins that can be activated by LPA, the G proteins belonging to G12 family of heterotrimeric G proteins - defined by Gα12 and Gα13 -, are by far the most potent G proteins in transmitting oncogenic signals16. Gα12 and Gα13, which are characterized as gep oncogenes17-19, have been shown to be involved in the activation of similar as well as distinct set of oncogenic pathways. While Gα12 appears to be more involved in cell proliferation19, Gα13 has been shown to be specifically involved in stimulating cell migration regulated by G protein coupled receptors as well as receptor tyrosine kinases20-24. Based on these correlates, it can be hypothesized that LPA-mediated metastatic migration of pancreatic cancer cells involves Gα13. Our study presented here is focused on testing this hypothesis so as to define the critical role of Gα13 in LPA-mediated invasive migration of pancreatic cancer cells. Using a panel of pancreatic cancer cells, consisting of BxPC3, Dan-G, Panc-1, MDAPanc-28, and MIA-PaCa-2 (PaCa-2) cell lines, we demonstrate here that LPA specifically stimulates the migration of pancreatic cancer cell lines but not their proliferation. Our results also establish that the invasive migration of pancreatic cancer cells stimulated by LPA is inhibited by the expression of a competitively inhibitory minigene of Gα13 that encodes the C-terminal eleven amino acids of Gα13, which is known to disrupt receptor-Gα13 interaction25-27. Similar inhibition of LPA-stimulated migration of pancreatic cancer cells is also demonstrated by shRNA-mediated silencing of Gα13 in these cells. Together, our results points to the critical role of Gα13, a member of the gep proto-oncogene family, in transmitting signaling pathways underlying LPA-mediated invasive migration of pancreatic cancer cells. Thus our studies presented here establish for the first time a critical role for Gα13 in LPA-mediated invasive migration of pancreatic cancer cells. By demonstrating the inhibitory effect of the C-terminal eleven amino acids of Gα13, encoded by CT13, on LPA-mediated migration of pancreatic cancer cells, we also establish that LPA-LPAR-Gα13-signaling pathway as a potential target for the development of novel therapeutics for pancreatic cancer.
MATERIALS AND METHODS
Cell Lines and Cell Culture
The pancreatic cancer cell lines BxPC3 cells and PaCa-2 cells were obtained from Dr. E. Premkumar Reddy (Mount Sinai School of Medicine, New York). The Dan-G cells were kindly provided by Dr. Klaudia Giehl (Dana-Farber Cancer Institute). Panc-1 and MDAPanc-28 cell lines were kindly provided by Dr. Dan Liebermann (Fels Institute for cancer Research and Molecular Biology, Temple University School of Medicine, Philadelphia) and Dr. Paul Chiao (The University of Texas M. D. Anderson Cancer Center, Houston) respectively. MDAPanc-28 and PaCa-2 cells were maintained in Dulbecco’s Modified Eagle’s Medium (Cellgro, NJ) (DMEM) containing 10% calf serum (Life Technologies Inc., Gaithersburg MD.), 50 units/ml penicillin, and 50ug/ml streptomycin at 37% in a 5% CO2 incubator. BxPC3 and Dan-G cells were grown under similar conditions, but with 10% New Born Calf serum (Gemini Bio-Products, West Sacramento, CA) whereas Panc-1 cells were grown with 10% Fetal Calf Serum. Serum deprivation was accomplished by incubation of the cells for 24 hours in DMEM supplemented with 10 mM HEPES (pH 7.4) and 0.2% BSA. LPA was obtained from (Avanti Polar Lipids, Alabaster, AL). It was dissolved to 20 mM stock solutions in PBS containing 0.1 % fatty acid free BSA, and stored at −20° C until use.
Construction of Gα13-inhibitory CT13-pcDNA3 Vector
Vector expressing the C-terminal 12 amino acid peptide with HA-epitope tag was constructed as follows: Strands of complementary oligonucleotides encoding the C-terminal 11 amino acids of Gα13 (LHDNLKQLMLQ) were synthesized along with the flanking BamHI and HindIII sites for cloning into a pcDNA-HA-tag vector. In order to generate the double stranded DNA sequence, the following complementary strands of oligonucleotides, 5′GGTGGATCCGGGTACC CTG CAT GAC AAC CTC AAG CAG CTT ATG CTA CAG TGA AAGCTTGCG 3′ and 5′CGCAAGCTT TCA CTG TAG CAT AAG CTG CTT GAG GTT GTC ATG CAG GGTACCCGGATCCACC 3′, were mixed at 1 μg/μl, heated to 95°C and cooled slowly to anneal into a duplex DNA. The vector and adapter sequences were digested with BamHI and HindIII, sequentially and gel purified using Qiagen Gel purification kit (Qiagen, Valencia, CA). The fragments were ligated and transformed in DH5α cells. Positive clones that were identified by restriction analysis with KpnI (using a silent KpnI site that was engineered in the adapter sequence) were confirmed by DNA-sequencing.
Establishing CT13-expressing Pancreatic Cancer Cell Lines
CT13-pcDNA3 vectors encoding HA-epitope tagged CT13 constructs were transfected into MDAPanc-28 cells using the FuGENE 6 reagent (Roche, Indianapolis, IN) according to the manufacturer’s protocol. Briefly, 9 μL FuGENE reagent was mixed with 738 μL DMEM supplemented with 12.5 mM HEPES. 3 μg DNA was added to this solution and incubated for 20 minutes at room temperature. This solution was added to a 100 mm culture dish of the indicated cell line grown to approximately 40 % confluence. After 24 hours, the cells were inspected for any signs of cytotoxicity, and changed to fresh media containing 10 % NBCS. Stable clones were selected from the transfectants using a G418 antibiotic selection protocol following previously published procedures28. Expression of the CT13 was verified by immunoblot analysis. Establishing Gα13-silenced Panc-1 Cancer Cell Line pLKO.1 vector encoding human shRNA targeting Gα13, namely RHS3979-NM_9604293, was obtained from Open Biosystems (Huntsville, AL) and the control non-Target shRNA Control Vector (SHC0020 was from Sigma Aldrich). Panc-1 cells were transfected with pLKO.1-shRNA/Gα13 or sh-Control vector using Amaxa Nuclearfector (Lonza, Walkersville, MD). To select for stably transfected clones of shRNA-Gα13 cells, puromycin (2 μg/ml; MP Biomedicals, Solon, Ohio) was added 24 hours post-transfection. Single clones were cored and the silencing of Gα13 expression was by western blotting.
In vitro wound healing assay
The protocols for wound healing assays were adapted from previously published procedures23. Briefly, 1×106 BxPC3, Dan-G, MDAPanc-28 and PaCa-2 pancreatic cancer cells were seeded in a 35 mm plate. After 24 hours, these cultures were washed twice with 5 mL of PBS and serum starved for 24 hrs. Two hours prior to wounding, these cells were treated with 10 μg/mL Mitomycin C, which inhibits cell proliferation. Cell-free space was created by scraping the monolayer with a 200 μL pipette tip. Cells were washed twice with PBS to remove debris, then stimulated with either serum free media, serum free media with 2 μM LPA, or 10 % NBCS. NBCS was used as a positive control since it contains many growth- as well as migration-promoting ligands including LPA. For each assay triplicate fields were photographed at T=0hr and T=24hr. These assays were quantified by estimating the percentage of wound closure at 24 hrs time-point. This calculation was made using the formula: (Width of the wound at 0 hr – Width of the wound at 24 hr)/(Width of the wound at 0 hr).
Transwell migration assay
Cell migration was also monitored using a chamber assay as described previously22. The cell culture inserts (#353097 PET membrane with 8.0 μm pores, BD Biosciences, Franklin Lakes, NJ) containing 5 × 105 cells suspended in 200 μL serum-free media were placed in the well of the transwell insert. Each well contained 500 μL media containing serum-free media control, serum-free media containing 2 μM LPA or 10 % NBCS (10 % FCS in the case of Panc-1 cells). The cells were incubated for 24 hours. Non-migrating cells on the proximal side of the inserts were removed with a cotton swab and the migrated cells on the distal side of the insert were fixed and stained with Hemacolor (EMD Chemicals, Inc., Gibbstown, NJ). To monitor invasive migration, similar procedures were carried out with the culture inserts (polyethylene terephthalate membrane with 8.0 μm pores #353097, BD Biosciences, Franklin Lakes, NJ), which were coated with rat-tail collagen, type 1 (BD Biosciences) as previously described19.
Images were obtained of random fields of view at 100 X magnification and the number of migrated cells was enumerated. The collagen-coated cell culture inserts containing 4 × 105 cells suspended in 200 μL serum-free media were placed in the well of the companion plate. Each well contained 500 μL media containing serum-free media control, serum-free media containing 2 μM LPA or 10 % NBCS or FCS (Panc-1 cells). The cells were incubated for 20 hours. Non-migrating cells on the proximal side of the inserts were removed with a cotton swab and the migrated cells on the distal side of the insert were fixed and stained with Hemacolor (EMD Chemicals, Inc., Gibbstown, NJ). Images were obtained of random fields of view at 100X magnification and the number of migrated cells was enumerated.
Cell proliferation assays
Determination of cell proliferation by enumeration was carried out as follows: Equal number of cells (3 × 105) were seeded in 60 mm culture dishes with media containing 10 % NBCS and allowed to adhere overnight. Cells were then incubated in serum-free media for 24 hours after washing twice with PBS. Cells were then stimulated with 2 μM LPA dissolved in serum-free media, serum-free media alone (unstimulated control), or media containing 10% NBCS as indicated in the appropriate experiment. At the indicated time-points, triplicate samples were harvested by incubation with 0.25 % Trypsin EDTA solution and gentle agitation for 2 minutes. Cell solutions were resuspended with media containing 0.2 BSA, and counted with a hemocytometer. Cells were counted in this manner at immediately before stimulation (0 hour), and at 24, 48, and 72 hours.
RT-PCR analysis for the expression of LPA3
Total RNA from cells grown to approximately 80% confluence was isolated using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. RT-PCR reaction was carried out with the ThermoScript System (Invitrogen, Carlsbad, CA) using a 5 μg aliquot of total RNA for cDNA synthesis. 2 μL of cDNA solution was subjected to PCR amplification using Taq PCR Master Mix Kit (Qiagen, Valencia, CA). The following primers were used for the PCR reactions for expression of LPA-receptor, LPA329.
LPA3-specific forward (5′-TTAGCTGCTGCCGATTTCTT-3′), and reverse (5′-ATGATGAGGAAGGCCATGAG-3′).
The PCR reaction conditions were carried out with 30 cycles at 94° C (3 minutes and 30 seconds), 55° C (2 minutes), 72°C (11 minutes). The GAPDH (Glyceraldehyde-3-Phosphate Dehydrogenase) specific forward (5′-GTGAAGGTCGGTTGTGAACGG-3′) and reverse (5′-GATGCAGGGATGATGTTCTG-3′) primers were used as a loading control. GAPDH was amplified with 33 cycles at 94° C (30 seconds), 58° C (1 minute), 72° C (1 minute). The amplification products were analyzed by 1 % agarose gel electrophoresis.
Immunoblot analysis
Immunoblot analyses with specific antibodies were carried out following our previously published procedures19. The following antibodies were used for immunoblot analyses. For LPAR analyses, LPA1 (#AP6138a) and LPA2 (#AP6140a) antibodies were obtained from Abgent (San Diego, CA). GAPDH antibody (#4300) was purchased from Ambion (Austin, TX). For Gα-subunit analyses, Gα12 (sc-409), Gαs (sc-823), Gαi (sc-1521), and Gαq (sc-393) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). For probing the expression of the Hemagglutinin (HA)-tagged CT13, HA-antibody (sc-805) was also purchased from Santa Cruz Biotechnology. Gα13 antibody (AS1-89-2) was raised in rabbit against the C-terminus of Gα1322. Peroxidase-conjugated anti-rabbit IgG (W401B) and anti-mouse IgG (NA93IV) were purchased from Promega (Madison, WI) and GE Healthcare (Buckinghamshire, UK), respectively.
Statistical Analysis
Statistical analyses were conducted and displayed graphically using GraphPad Prism version 4.0 (La Jolla, CA). All of the statistical data presented were derived from multiple independent experiments, each performed with triplicate samples unless otherwise indicated. Statistical significance was determined using Student’s t-tests (* indicates P values < 0.05).
RESULTS
Effect of LPA on the Migration and Proliferation of Pancreatic Cancer Cells
LPA has been shown to be a potent mitogenic factor in inducing cell proliferation and/or migration in a variety of normal and cancer cell types including vascular smooth muscles, astrocytes, as well as breast, ovarian, prostate, and colorectal cancer cells30-39. To investigate the role of LPA in proliferation and/or migration in pancreatic cancer cells, we first monitored the mitogenic potential of LPA in a panel of pancreatic cancer cells, consisting of BxPC3, Dan-G, MDAPanc-28, and PaCa-2. After monitoring the expression profiles of LPA receptors (Fig 1A & 1B) and their cognate G protein α-subunits (Fig 1C) in these cells, we assessed the ability of LPA to stimulate proliferation by stimulating the cells with 2 μM of LPA for 96 hours. Stimulation with 10% New Born Calf Serum was used as a positive control. As shown in Figure 2, LPA failed to stimulate the proliferation of these cells except in the case of BxPC3 cells. Next, we examined the ability of LPA to stimulate cell migration using wound-healing and trans-well based migration assays. In the wound-healing assay, equal number of BxPC3, Dan-G, MDAPanc-28 and PaCa-2 pancreatic cancer cells (1×106) were seeded in a 35 mm plate and serum-starved for 16 hrs. Cell-free space created by scraping the monolayer with a 200 μL pipette tip was allowed to “heal” by cells migrating in response to 2μM LPA, or 10% NBCS along with appropriate controls as shown in the case of MDAPanc-28 cells (Figure 3A & B). For each assay, triplicate fields were photographed at 0- and 24-hr and quantified as described under Methods. Results from such analysis indicated that LPA potently stimulated migration in all of the pancreatic cancer cells that were tested (Figure 3). To further quantify, a series of transwell migration assays were carried out following previously published methods19,22. As shown in Figure 4, results from such analyses confirmed the data obtained with the wound-healing assay by demonstrating the potent stimulation of migration in BxPC3, MDAPanc-28, Dan-G, and PaCa-2 by 2 μM LPA (Fig 4 A-E).
Figure 1. Expression Profiles of LPA-receptors and Gα-subunits.
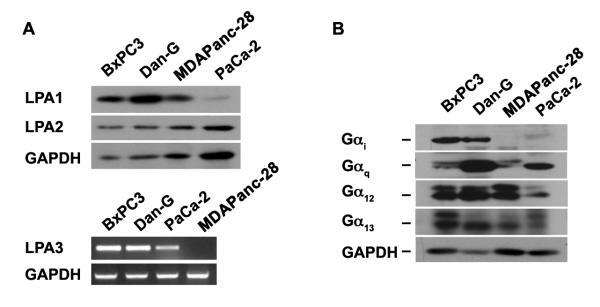
Expression of LPA1 and LPA2 in BxPc3, Dan-G, MDAPanc-28, and PaCa-2 was monitored by immunoblot analysis (A). Lysates (50 μg) from the respective cells were collected, separated by 10% SDS-PAGE and subjected to immunoblot analysis using antibodies specific to LPA-receptors, LPA1, LPA2, or GAPDH (loading control). The experiment was repeated 4 times, and a representative immunoblot is presented (Upper Panel). Expression of LPA3 was monitored by RT-PCR analysis RNA from these cells using primers specific to LPA3 and GAPDH (positive control), as described in Materials and Methods. The experiment was repeated 3 times, and a representative RT-PCR analysis is presented (Lower Panel). Expressions of LPA-responsive Gα-subunits were monitored in these cells by immunoblot analysis (B). Lysates from these cells (50 μg), separated by 10 % SDS-PAGE were probed with antibodies specific to Gαi, Gαq, Gα12, or Gα13, Equal protein loading was monitored by reprobing this blot with antibodies specific to GAPDH. Results are from a typical experiment (n = 3).
Figure 2. Effect of LPA on the Proliferation of Pancreatic Cancer Cells.

Equal number of cells (3× 105) BxPC3 (A), Dan-G (B), MDAPanc-28 (C), or PaCa-2 (D) were seeded in 60 mm culture dishes, allowed to adhere overnight and serum-deprived for 24 hours. Cells were then stimulated with 2 μM LPA dissolved in serum-free media or 10 % NBCS. At 24 hour intervals following stimulation, cell were trypsinized and counted on a hemocytometer. Results are expressed as total cell numbers (Mean ± SEM; n = 3).
Figure 3. Effect of LPA on the Migration of Pancreatic Cancer Cells.

(A) Equal number of MDAPanc-28 cells were seeded in triplicate into 35 mm culture dishes. Cells were washed with PBS and incubated in serum-free media for 24 hours. Two hours prior to wounding, these cells were treated with 10 μg/mL Mitomycin C, following which a linear scratch wound was made across the cell monolayer as described in the Materials and Methods. The cells were washed with serum-free media to remove cellular debris and imaged at 100X-magnification. The cells were then incubated with serum-free media containing 2 μM LPA, or 10% NBCS along with untreated control group After 24 hours incubation, the fields of view were identified and re-imaged. (B) Wound closure was calculated in triplicate along the length of the field according to the formula: (Width of the wound at 0 hr – Width of the wound at 24 hr)/(Width of the wound at 0 hr). (n = 3, Mean ± SEM). Similar image analyses followed by quantification (n = 3, Mean ± SEM) were carried out for BxPc3 (C) and PaCa-2 cells (D).
Figure 4. LPA-stimulated Migration of Pancreatic Cancer Cells.

(A) MDAPanc-28 cells (5 × 105) suspended in 200 μL serum-free media were placed in the upper well of the transwell insert. Each well of the companion plate contained 500 μL media containing serum-free media control or serum-free media containing 2 μM LPA or 10% NBCS. The cells were incubated for 24 hours. Non-migrating cells on the proximal side of the inserts were removed with a cotton swab and the migrated cells on the distal side of the insert were fixed stained with Hemacolor. Images were obtained of random fields of view at 100 X magnification. The images shown are representative of the three independent experiments, each performed with triplicate fields of view. (B) The number of migrated cells in each field was enumerated and the results are presented (n = 3, Mean ± SEM). Similar analyses (Mean ± SEM; n = 3) were carried out with Dan-G (C), PaCa-2 (D), and BxPc3 cells (E).
LPA-stimulated migration of Pancreatic Cancer Cells Involves Gα13
Signaling by LPA involves the activation of specific LPA-receptors that can couple to the α-subunits of Gi, Gq and G12 family of G proteins. Of these α-subunits, primarily Gαi or Gα13 has been shown to mediate LPA-stimulated migratory response in many different cancer cells12,15,22,23,31,32. However, the observation that MDAPanc-28 and PaCa-2 that exhibit potent migratory response to LPA in spite of their low expression levels of Gαi (Figures 1 & 3) suggests a dominant role for either Gα12 or Gα13 in LPA-stimulated migratory response of pancreatic cancer cells. Since previous studies from several laboratories including ours have established a critical role for Gα13 in cell migration22-24, we investigated the effect of inhibiting Gα13 in LPA-stimulated migration of Pancreatic cancer cells. Based on the observation that the C-terminal eleven amino acids of Gα13 is critical for its interactions with the cognate receptors26,27, a minigene construct encoding this peptide (LHDNLKQLMLQT) has been used as a dominant negative mutant of Gα13 that can competitively inhibit Gα13-receptor interaction. To test, we utilized wound-healing assay to evaluate whether the expression of CT13 had any effect on LPA-stimulated migration of MDAPanc-28 cell. Results obtained from this assay demonstrated an extensive abrogation of the LPA- or serum stimulated migratory response in MDAPanc-28 cells expressing CT13 (Figure 5).
Figure 5. Attenuation of LPA- and Serum-stimulated Migration of MDAPanc-28 cells by CT13, the Inhibitory Mutant of Gα13.

(A) Wound healing assays were conducted as described in the Materials and Methods. MDAPanc-28-PCDNA3.1 and MDAPanc-28-CT-13 cells were then incubated with serum-free media, serum free media containing 2 μM LPA, or 10% NBCS. After 24 hours of incubation, the fields of view were identified and re-imaged. (B) Wound closure was calculated in triplicate along the length of the field according to the formula: (Width of the wound at 0 hr – Width of the wound at 24 hr) / (Width of the wound at 0 hr). The inset shows the immunoblot analysis of the lysates from these cells to verify the expression of Hemagglutinin (HA)-tagged CT13 in lysates from the cells expressing pcDNA3 vector (Vector Control) or HA-tagged CT13 (CT13) using antibodies specific to HA-epitope (n = 3, Mean ± SEM).
Next, we sought to investigate whether LPA stimulates invasive migration of pancreatic cancer cells and if so, whether such invasive migration involves Gα13. This was carried out by analyzing the invasion of MDAPanc-28 cells through the synthetic membrane coated with type-I rat-tail collagen19. Results from these studies, as shown in Figure 6 indicate that LPA promotes potent invasive migration in the presence of type-1 collagen. Such LPA-stimulated invasive migration was also observed in BxPC3, PaCa-2, and DAN-G cells (data not shown). However, the expression of CT13 in MDAPanc-28 cells, in addition to inhibiting the basal level of migratory response, drastically inhibited LPA as well as stimulated migration in type-1 collagen matrix (Figure 6A & 6B).
Figure 6. Inhibition of LPA-stimulated Invasive Migration by CT13.

(A) MDAPanc-28-PCDNA3.1 and MDAPanc-28-CT-13 cells (5 × 105) suspended in 200 μL serum-free media were placed in the upper well of the transwell insert. Each well of the companion plate contained 500 μL media containing serum-free media (control), serum-free media containing 2 μM LPA (LPA) or 10% NBCS. The cells were incubated for 24 hours. Non-migrating cells on the proximal side of the inserts were removed with a cotton swab and the migrated cells on the distal side of the insert were fixed stained with Hemacolor. Images were obtained of random fields of view at 100 X magnification. The images shown are representative of the three independent experiments, each performed with triplicate fields of view using clone #4 of MDAPanc-28-CT13 cells. (B) Migrated cells per field were quantified for two different clones, namely clones #4 and #5, as well as for a pool of CT13-expressing MDAPanc-28 cells (Pool CT13). Results are presented as Migrated cells / field for the non-treated (control), LPA-treated (LPA), and serum-treated (NBCS) group (n = 3, Mean ± SEM).
Silencing Gα13 inhibits LPA-mediated invasive migration of pancreatic cancer cells
Although CT13, which is also denoted as Gα13-minigene, has been widely used as a specific inhibitor of receptor-Gα13 interaction downstream signaling events26,27, it could be argued that the inhibitory effect is due to the attenuation of signaling from the closely related Gα1240. Therefore, we decided to further confirm the specificity of this response. First we investigated whether the silencing of Gα12, which show s more than 60% amino acid identity with Gα1340, had any effect on LPA-mediated invasive migration of pancreatic cancer cells. Using Panc-1 pancreatic cancer cells in which the expression of Gα12 was partially silenced by shRNA strategy, we monitored the migration of these cells in response to LPA or FCS using a trans-well migration assay. Results indicated that the silencing of Gα12 rather promoted the migratory response of these cells (Figure 7). Next, we investigated the effect of silencing Gα13 on LPA-stimulated migratory response of pancreatic cancer cells. Trans-well migration assay was used to monitor the migration of Panc-1 cells in which the expression of Gα13 was silenced by Gα13-specific shRNA. These cells were stimulated with LPA or FCS and the invasive migration of these cells along with the control group was monitored. Our results from such analysis indicated that the silencing of Gα13 attenuated the migratory response of these cells to LPA and FCS by 85 and 90 % respectively (Figure 8), thus confirming the critical role of Gα13 in LPA as well as serum-stimulated migration of pancreatic cancer cells. In summary, our findings presented here, firmly establish that Gα13 is specifically involved in LPA-mediated invasive migratory response.
Figure 7. Effect of Silencing Gα12 on LPA- or Serum-stimulated Invasive Migration of Panc-1 Cells.

(A) Panc-1 cells in which the expression of Gα12 was silenced by shRNA to Gα12 (sh12) or cells expressing control shRNA (sh-Control) were monitored for their migratory response to LPA (2 μM), 10% FCS, or serum-free medium (control) on type-1 collagen treated transwell system as described under Materials and Methods. At 24 hrs following LPA stimulation, images were obtained of random fields of view at 100X magnification. The images shown are representative of three independent experiments, each performed with triplicate fields of view. (B) Cell migration profiles were quantified by enumerating the migrated cells in a minimum of three fields. Results are presented as the number of migrated cells per field and the bars represent the mean ± SEM from three independent experiments. (C) Silencing of endogenous Gα12 was monitored by immunoblot analysis using antibodies to Gα12, probing the lysates from cells expressing control non-specific shRNA (sh-C) and cells in which the expression of Gα12-was silenced using Gα12-specific shRNA (sh12). The blot was stripped and re-probed with antibodies to GAPDH to monitor equal loading of protein.
Figure 8. Effect of Silencing Gα13 on LPA- and Serum-stimulated Invasive Migration of Panc-1 Cells.

(A) Panc-1 cells in which the expression of Gα13 was silenced by shRNA to Gα13 (sh13) or cells expressing control shRNA (sh-Control) were monitored for their invasive migratory response to LPA (20 μM), 10% FCS, or serum-free medium (control) on type-1 collagen treated transwell system as described under Materials and Methods. At 20 hrs following LPA stimulation, images were obtained of random fields of view at 100X magnification. The images shown are representative of three independent experiments, each performed with triplicate fields of view. (B) Cell migration profiles were quantified by enumerating the migrated cells in a minimum of three fields. Results are presented as the number of migrated cells per field and the bars represent the mean ± SEM from three independent experiments. (C) Silencing of endogenous Gα13 was monitored by immunoblot analysis using antibodies to Gα13, probing the lysates from cells expressing control non-specific shRNA (sh-C) and cells in which the expression of Gα13-was silenced using Gα13-specific shRNA (sh13). The blot was stripped and re-probed with antibodies to GAPDH to monitor equal loading of protein.
DISCUSSION
Our inability to recognize and treat patients with pancreatic cancer stems from a poor understanding of the pathophysiological mechanisms of the disease. Numerous GPCR agonists, including LPA, along with their cognate receptors have been implicated in the oncogenic process that drives progression and metastasis of pancreatic cancer6,9-12. However, the specific role of LPA in cancer cell survival, proliferation, migration, or metastasis is far from clear. In this context, the results from our study delineate the role of LPA in pancreatic cancer cell proliferation versus migration. The finding that LPA promotes migration in all of the pancreatic cancer cells, but stimulates proliferation only in BxPC3 cells suggests that the dominant role of LPA is to stimulate cancer cell migration and thereby metastasis.
Although previous studies have identified a role for LPA in pancreatic cancer cell migration, the identity of the G protein involved in the process is largely unknown. It has been previously suggested that LPA dependent migration involves Gαi-dependent mechanism12. However, the observations that the MDAPanc-28 cell line, which does not express Gαi or expresses undetectable levels of Gαi, exhibited a potent migratory response when stimulated with LPA suggests that Gαi may not be uniquely associated with cell migration. It is interesting to note here that among all the α-subunits that can be activated by LPA, Gα13 is the only α-subunit that potently stimulates cellular migration in response to diverse stimuli19-24,41. In spite of such strong correlation, the role of Gα13 in LPA-mediated invasive migration cancer cells, including those of pancreatic cancer cells, has not been investigated thus far. This could be due to the relatively recent emerging view that LPA plays a role in the genesis and progression of many different cancers11,36,38,42 and equally recent findings that of Gα13 acts as a major hub for cellular migration stimulated by diverse pathways23,24,41. Thus, the results presented here, establish for the first time that Gα13, which has been previously defined as the gep oncogene, is involved in LPA-mediated migration of pancreatic cancer cells. Although LPA receptors have been shown to activate the G12 family of G proteins and the α-subunits of G12 and G13 are by far the most potent oncogenic α-subunits that have been characterized, the role of Gα13, a critical mediator of cellular migration, has not been systematically analyzed. Thus, it is highly significant that the results presented here using cells expressing the dominant negative mutant of Gα13 or silencing of Gα13-expression has identified, for the first time, that Gα13 is the α-subunit involved in LPA-mediated migration of pancreatic cancer cells. Consistent with the previous findings that the ablation of Gα13, but not Gα12, leads to the inhibition of cell migration20,23, our results clearly rule out a role for Gα12, a closely related α-subunit-, in LPA-stimulated migration of pancreatic cancer cells. Thus, the results presented here clearly establish that Gα13 is a critical signaling component in LPA as well as serum stimulated migration in pancreatic cancer cells (Figure 5, 6, & 8). It is interesting to note here that the silencing of Gα12 promotes LPA- as well as serum-stimulated cell migration (Figure 7). Surprisingly, this is similar to the results we have observed in ovarian cancer cells in Gα12 was silenced19. Earlier studies have speculated that LPA1-receptor signaling is involved in the stimulation of pancreatic cancer cell migration whereas LPA2-receptor is involved in the inhibition of pancreatic cancer cell migration9,10. This is consistent with the observation that PaCa-2 cells, which express lower levels of LPA1, exhibit relatively weaker migration (Figure 3D). Based on these findings, it can be concluded that Gα12 is involved in transmitting the signals from LPA2-receptor there by inhibiting cell migration while silencing Gα12 relives such inhibition. Extending this further, one can speculate that Gα13 is more involved in signaling by LPA1-receptor. Further analyses are likely to provide evidence to this important corollary.
It should be noted here, that LPA-LPAR signaling could recruit Gαi as well as Gαq in addition to Gα12 and Gα13 for intracellular signaling. It is possible that the oncogenic signaling by LPA-LPAR signaling involves the recruitment of multiple α-subunits to coordinate the complex array of signaling underlying cancer cell dissemination, migration, and metastasis. Now that the cellular model defining LPA signaling to Gα13 has been established (Figure 5, 6 and 8), further studies using this paradigm should identify the critical components and their role in the invasive migration of pancreatic cancer cells. Another significant corollary to our finding is the observation that the silencing or inhibition of Gα13 readily attenuates LPA- as well as serum-mediated invasive migration of pancreatic cancer cells. Since GPCRs have proven to be “druggable targets” for diverse diseases, the LPA-LPAR-Gα13 signaling pathway unraveled here can be targeted for the development of novel therapeutics for pancreatic cancer.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (CA 125752, CA 116984), The Pennsylvania Department of Health, and The World Class University project funded by Ministry of Education, Science and Technology Development, S. Korea [No.R32-2008-000-10098-0].
ABBREVIATIONS
- FCS
Fetal Calf Serum
- G protein
Guanine nucleotide-binding heterotrimeric protein
- GPCR
G protein-coupled receptor
- HA
Hemagglutinin
- LPA
Lysophosphatidic Acid
- LPAR
Lysophosphatidic Acid Receptor
- LPA1
Lysophosphatidic Acid Receptor-1
- LPA2
Lysophosphatidic Acid Receptor-2
- LPA3
Lysophosphatidic Acid Receptor-3
- NBCS
Newborn Calf Serum
- shRNA
Short Hairpin Ribonucleic Acid
Footnotes
Disclosure: The authors have no conflicts of interest or funding to disclose.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Siegel R, Desantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA-Cancer J Clin. 2012;62(4):220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 2.Farrow B, Albo D, Berger DH. The role of the tumor microenvironment in the progression of pancreatic cancer. J Surg Res. 2008;149(2):319–328. doi: 10.1016/j.jss.2007.12.757. [DOI] [PubMed] [Google Scholar]
- 3.Chang DZ, Ma Y, Ji B, et al. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2011;17(22):7015–7023. doi: 10.1158/1078-0432.CCR-11-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tjomsland V, Niklasson L, Sandstrom P, et al. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin Dev Immunol. 2011;2011:212810. doi: 10.1155/2011/212810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L, Friess H, Zhu Z, et al. Neurotensin receptor-1 mRNA analysis in normal pancreas and pancreatic disease. Clin Cancer Res. 2000;6(2):566–571. [PubMed] [Google Scholar]
- 6.Ryder NM, Guha S, Hines OJ, et al. G protein-coupled receptor signaling in human ductal pancreatic cancer cells: neurotensin responsiveness and mitogenic stimulation. J Cell Physiol. 2001;186(1):53–64. doi: 10.1002/1097-4652(200101)186:1<53::AID-JCP1004>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 7.Smith JP, Shih A, Wu Y, et al. Gastrin regulates growth of human pancreatic cancer in a tonic and autocrine fashion. Am J Physiol. 1996;270(5 Pt 2):R1078–1084. doi: 10.1152/ajpregu.1996.270.5.R1078. [DOI] [PubMed] [Google Scholar]
- 8.Kaufmann R, Schafberg H, Rudroff C, et al. Cholecystokinin B-type receptor signaling is involved in human pancreatic cancer cell growth. Neuropeptides. 1997;31(6):573–583. doi: 10.1016/s0143-4179(97)90003-2. [DOI] [PubMed] [Google Scholar]
- 9.Bian D, Mahanivong C, Yu J, et al. The G12/13-RhoA signaling pathway contributes to efficient lysophosphatidic acid-stimulated cell migration. Oncogene. 2006;25(15):2234–2244. doi: 10.1038/sj.onc.1209261. [DOI] [PubMed] [Google Scholar]
- 10.Yamada T, Sato K, Komachi M, et al. Lysophosphatidic acid (LPA) in malignant ascites stimulates motility of human pancreatic cancer cells through LPA1. J Biol Chem. 2004;279(8):6595–6605. doi: 10.1074/jbc.M308133200. [DOI] [PubMed] [Google Scholar]
- 11.Komachi M, Tomura H, Malchinkhuu E, et al. LPA1 receptors mediate stimulation, whereas LPA2 receptors mediate inhibition, of migration of pancreatic cancer cells in response to lysophosphatidic acid and malignant ascites. Carcinogenesis. 2009;30(3):457–465. doi: 10.1093/carcin/bgp011. [DOI] [PubMed] [Google Scholar]
- 12.Lv GM, Li P, Wang WD, Wang ShK, Chen JF, Gong YL. Lysophosphatidic acid (LPA) and endothelial differentiation gene (Edg) receptors in human pancreatic cancer. J Surg Oncol. 2011;104(6):685–691. doi: 10.1002/jso.22016. [DOI] [PubMed] [Google Scholar]
- 13.Stahle M, Veit C, Bachfischer U, et al. Mechanisms in LPA-induced tumor cell migration: critical role of phosphorylated ERK. J Cell Sci. 2003;116(Pt 18):3835–3846. doi: 10.1242/jcs.00679. [DOI] [PubMed] [Google Scholar]
- 14.Choi JW, Lim S, Oh YS, et al. Subtype-specific role of phospholipase C-β in bradykinin and LPA signaling through differential binding of different PDZ scaffold proteins. Cell Signal. 2010;22(7):1153–1161. doi: 10.1016/j.cellsig.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3(8):582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 16.Radeff-Huang J, Seasholtz TM, Matteo RG, et al. G protein mediated signaling pathways in lysophospholipid induced cell proliferation and survival. J Cell Biochem. 2004;92(5):949–966. doi: 10.1002/jcb.20094. [DOI] [PubMed] [Google Scholar]
- 17.Radhika V, Dhanasekaran N. Transforming G proteins. Oncogene. 2001;20(13):1607–1614. doi: 10.1038/sj.onc.1204274. [DOI] [PubMed] [Google Scholar]
- 18.Radhakrishnan R, Ha JH, Dhanasekaran DN. Mitogenic Signaling by the gep Oncogene Involves the Upregulation of S-Phase Kinase-Associated Protein 2. Genes Cancer. 2010;1(10):1033–1043. doi: 10.1177/1947601910390516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kashef K, Radhakrishnan R, Lee CM, et al. Neoplastic transformation induced by the gep oncogenes involves the scaffold protein JNK interacting leucine zipper protein. Neoplasia. 2011;13(4):358–364. doi: 10.1593/neo.101622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldsmith ZG, Ha JH, Jayaraman M, et al. Lysophosphatidic Acid Stimulates the Proliferation of Ovarian Cancer Cells via the gep Proto-Oncogene Gα12. Genes Cancer. 2011;2(5):563–575. doi: 10.1177/1947601911419362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Offermanns S, Mancino V, Revel JP, et al. Vascular system defects and impaired cell chemokinesis as a result of Gα13 deficiency. Science. 1997;275(5299):533–536. doi: 10.1126/science.275.5299.533. [DOI] [PubMed] [Google Scholar]
- 22.Gu JL, Muller S, Mancino V, et al. Interaction of Gα12 with Gα13 and Gαq signaling pathways. Proc Natl Acad Sci U S A. 2002 Jul 9;99(14):9352–9357. doi: 10.1073/pnas.102291599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radhika V, Onesime D, Ha JH, et al. Gα13 stimulates cell migration through cortactin-interacting protein Hax-1. J Biol Chem. 2004;279(47):49406–49413. doi: 10.1074/jbc.M408836200. [DOI] [PubMed] [Google Scholar]
- 24.Shan D, Chen L, Wang D, et al. The G protein Gα13 is required for growth factor-induced cell migration. Dev Cell. 2006;10(6):707–718. doi: 10.1016/j.devcel.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 25.Gantulga D, Tuvshintugs B, Endo Y, et al. The scaffold protein c-Jun NH2-terminal kinase-associated leucine zipper protein regulates cell migration through interaction with the G protein Gα13. J Biochem. 2008;144(6):693–700. doi: 10.1093/jb/mvn123. [DOI] [PubMed] [Google Scholar]
- 26.Gilchrist A, Bunemann M, Li A, et al. A dominant-negative strategy for studying roles of G proteins in vivo. J Biol Chem. 1999;274(10):6610–6616. doi: 10.1074/jbc.274.10.6610. [DOI] [PubMed] [Google Scholar]
- 27.Gilchrist A, Vanhauwe JF, Li A, et al. Gα minigenes expressing C-terminal peptides serve as specific inhibitors of thrombin mediated endothelial activation. J Biol Chem. 2001;276(28):25672–25679. doi: 10.1074/jbc.M100914200. [DOI] [PubMed] [Google Scholar]
- 28.Gilchrist A, Li A, Hamm HE. Design and use of C-terminal minigene vectors for studying role of heterotrimeric G proteins. Methods Enzymol. 2002;344:58–69. doi: 10.1016/s0076-6879(02)44705-2. [DOI] [PubMed] [Google Scholar]
- 29.Dermott JM, Dhanasekaran N. Determining cellular role of Gα12. Methods Enzymol. 2002;344:298–309. doi: 10.1016/s0076-6879(02)44722-2. [DOI] [PubMed] [Google Scholar]
- 30.Sawada K, Morishige K, Tahara M, et al. Lysophosphatidic acid induces focal adhesion assembly through Rho/Rho-associated kinase pathway in human ovarian cancer cells. Gynecol Oncol. 2002;87(3):252–259. doi: 10.1006/gyno.2002.6831. [DOI] [PubMed] [Google Scholar]
- 31.Mills GB, Eder A, Fang X, et al. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer Treat Res. 2002;107:259–283. doi: 10.1007/978-1-4757-3587-1_12. [DOI] [PubMed] [Google Scholar]
- 32.Bian D, Su S, Mahanivong C, et al. Lysophosphatidic Acid Stimulates Ovarian Cancer Cell Migration via a Ras-MEK Kinase 1 Pathway. Cancer Res. 2004;64(12):4209–4217. doi: 10.1158/0008-5472.CAN-04-0060. [DOI] [PubMed] [Google Scholar]
- 33.Daaka Y. Mitogenic action of LPA in prostate. Biochim Biophys Acta. 2002;1582(1-3):265–269. doi: 10.1016/s1388-1981(02)00180-4. [DOI] [PubMed] [Google Scholar]
- 34.Shida D, Kitayama J, Yamaguchi H, et al. Dual mode regulation of migration by lysophosphatidic acid in human gastric cancer cells. Exp Cell Res. 2004;301(2):168–178. doi: 10.1016/j.yexcr.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Shida D, Watanabe T, Aoki J, et al. Aberrant expression of lysophosphatidic acid (LPA) receptors in human colorectal cancer. Lab Invest. 2004;84(10):1352–1362. doi: 10.1038/labinvest.3700146. [DOI] [PubMed] [Google Scholar]
- 36.Panupinthu N, Lee HY, Mills GB. Lysophosphatidic acid production and action: critical new players in breast cancer initiation and progression. Br J Cancer. 2010;102(6):941–946. doi: 10.1038/sj.bjc.6605588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park SY, Jeong KJ, Panupinthu N, et al. Lysophosphatidic acid augments humanhepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene. 2011;30(11):1351–1359. doi: 10.1038/onc.2010.517. [DOI] [PubMed] [Google Scholar]
- 38.Lee SJ, Yun CC. Colorectal cancer cells - Proliferation, survival and invasion by lysophosphatidic acid. Int J Biochem Cell Biol. 2010;42(12):1907–1910. doi: 10.1016/j.biocel.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitayama J, Shida D, Sako A, et al. Over-expression of lysophosphatidic acid receptor-2 in human invasive ductal carcinoma. Breast Cancer Res. 2004;6(6):R640–646. doi: 10.1186/bcr935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhanasekaran N, Dermott JM. Signaling by the G12 class of G proteins. Cell Signal. 1996;8(4):235–245. doi: 10.1016/0898-6568(96)00048-4. [DOI] [PubMed] [Google Scholar]
- 41.Dhanasekaran DN. Transducing the signals: a G protein takes a new identity. Sci STKE. 2006;2006(347):pe31. doi: 10.1126/stke.3472006pe31. [DOI] [PubMed] [Google Scholar]
- 42.Houben AJ, Moolenaar WH. Autotaxin and LPA receptor signaling in cancer. Cancer Metastasis Rev. 2011;30(3-4):557–565. doi: 10.1007/s10555-011-9319-7. [DOI] [PubMed] [Google Scholar]