

Abstract
Preeclampsia is a major obstetric problem defined by new-onset hypertension and proteinuria associated with compromised placental perfusion. Although activation of the complement system is increased in preeclampsia compared to normal pregnancy, it remains unclear whether excess complement activation is a cause or consequence of placental ischemia. Therefore, we hypothesized that complement activation is critical for placental ischemia-induced hypertension. We employed the reduced utero-placental perfusion pressure (RUPP) model of placental ischemia in the rat to induce hypertension in the third trimester and evaluated the effect of inhibiting complement activation with a soluble recombinant form of an endogenous complement regulator, human complement receptor 1 (sCR1; CDX-1135). On day 14 of a 21-day gestation, rats received either RUPP or Sham surgery and 15 mg/kg/day sCR1 or saline intravenously on days 14–18. Circulating complement component 3 decreased and complement activation product C3a increased in RUPP vs Sham (p<0.05), indicating complement activation had occurred. Mean arterial pressure (MAP) measured on day 19 increased in RUPP vs Sham rats (109.8±2.8 mmHg vs 93.6±1.6 mmHg). Treatment with sCR1 significantly reduced elevated MAP in RUPP rats (98.4±3.6 mmHg, p<0.05) and reduced C3a production. Vascular endothelial growth factor (VEGF) decreased in RUPP compared to Sham rats, and the decrease in VEGF was not affected by sCR1 treatment. Thus, these studies have identified a mechanistic link between complement activation and the pregnancy complication of hypertension apart from free plasma VEGF and have identified complement inhibition as a potential treatment strategy for placental ischemia-induced hypertension in preeclampsia.
Keywords: pregnancy, hypertension, preeclampsia, complement, C3a, placental ischemia, vascular endothelial growth factor
1. Introduction
The pregnancy-specific condition preeclampsia is a leading cause of maternal and fetal morbidity and mortality (Steegers et al., 2010). Preeclampsia is hallmarked by new-onset hypertension (systolic blood pressure of >140 mmHg or a diastolic blood pressure of >90 mmHg) and proteinuria (>300 mg/L) over a period of 24 hours. The initiating event of preeclampsia is unknown. Inadequate trophoblast invasion is postulated to impair maternal spiral artery remodeling in the placenta that is necessary to accommodate increased blood flow required for pregnancy. This impaired spiral artery remodeling results in placental ischemia, accompanied by imbalances in angiogenic factors including vascular endothelial growth factor (VEGF) and soluble fms-like tyrosine kinase-1 (sFlt-1) (Baker et al., 1995; Maynard et al., 2008), oxidative stress (Hubel, 1999; Mistry et al., 2013), endothelial dysfunction (Gilbert et al., 2008) and inflammation with excessive complement activation (Lynch and Salmon, 2010). The impaired placental perfusion leads to hypertension, proteinuria and intrauterine growth restriction of the fetus (Steegers et al., 2010). Although many factors have been identified as contributors to this condition, delivery of the placenta remains the only definitive treatment and new treatment strategies are needed.
In pregnancy, maternal mechanisms are evoked to prevent an immune response against the fetus and involve both innate and adaptive immunity (Denny et al., 2013; Lynch and Salmon, 2010), including the complement system. The complement system is an enzymatic amplification system composed of endogenous plasma proteins that normally operates at a low steady state. The components are sequentially activated by any of three pathways (classical, mannose-binding lectin, and alternative) to function in host defense and inflammation and lead to pathogen opsonization and/or lysis. Central to all three pathways is formation of a C3 convertase that cleaves complement component 3 (C3) into C3a, a fluid phase inflammatory product, and C3b, a larger fragment that covalently binds to surfaces and is an essential part of the enzyme C5 convertase. Cleavage of C5 into C5a and C5b by C5 convertase can lead to formation of membrane attack complex, C5b-9, and lysis of the target. In uncomplicated pregnancies, a heightened inflammatory state is evident with the cleavage product C3a normally increasing in circulation as gestation progresses (Lynch and Salmon, 2010) and the acute phase protein C3 increasing (Baines et al., 1974).
Regulation of this complement activation is important for a successful pregnancy as demonstrated by the inability of offspring to survive if mice lack the normal membrane-bound complement regulator Crry (Xu et al., 2000). In addition, complement component C1q is necessary for normal placental development since C1q-deficient mice exhibit abnormal trophoblast migration and remodeling of spiral arteries and do not develop a normal placenta (Singh et al., 2011). In the rat, C3 synthesized in the uterus is necessary for normal fetal development and has been identified as an early post-implantation embryotrophic growth factor (Usami et al., 2010).
Though complement is necessary for a normal pregnancy, excessive activation of complement and generation of active fragments may be an important contributor to adverse pregnancy outcomes such as spontaneous miscarriage and preeclampsia. In mouse models of spontaneous miscarriage and antiphospholipid syndrome, complement activation product C5a is important in fetal loss and growth restriction (Girardi et al., 2003; Girardi et al., 2006; Holers et al., 2002; Qing et al., 2011). In preeclamptic pregnancies, elevated concentrations of complement activation products Bb, C3a and C5a have been observed both early in pregnancy as well as near term (Denny et al., 2013; Derzsy et al., 2010; Lynch et al., 2011; Lynch and Salmon, 2010). It is unclear whether this excessive complement activation contributes to placental ischemia and/or placental ischemia causes complement activation and contributes to hypertension. Since complement system activation has also been implicated in pathogenesis of ischemia/reperfusion injury in many different organs (Gorsuch et al., 2012) and the complement cleavage products C3a and C5a are vasoactive (Proctor et al., 2009; Regal and Klos, 2000), we hypothesized that complement activation occurs as a result of placental ischemia and leads to pregnancy-induced hypertension. To test this hypothesis, we used the well-established reduced utero-placental perfusion pressure (RUPP) model of placental ischemia-induced hypertension in rat that mimics features of preeclampsia. In the RUPP model, placental ischemia is induced in the third trimester of pregnancy resulting in a reduction in blood flow to the uteroplacental units and hypertension in the mother. Our studies were designed to determine if complement system activation is increased due to placental ischemia in the RUPP model and to evaluate the effectiveness of complement inhibition using a soluble form of an endogenous complement regulator, soluble complement receptor 1 (sCR1), to attenuate hypertension seen in this model.
2. Materials and Methods
2.1 Reduced utero-placental perfusion pressure (RUPP) procedure and sCR1 administration
The reduced utero-placental perfusion pressure (RUPP) procedure was employed to achieve chronic placental ischemia in the pregnant rat as described previously (Alexander et al., 2001; Crews et al., 2000; Gilbert et al., 2007a; Granger et al., 2006). In brief, surgical procedures were performed with timed pregnant Sprague Dawley dams (Crl:CD IGS, Charles River Laboratories, Portage, MI) under isoflurane anesthesia. All animal experiments were submitted to and approved by the University of Minnesota Institutional Animal Care and Use Committee and conformed to National Institutes of Health guidelines. On day 14 of pregnancy, the jugular vein was cannulated and the cannula exteriorized to the back of the neck. Heparin was not used in cannulas due to its complement inhibitory properties, and a 25% dextrose lock solution was used to maintain cannula patency. For RUPP surgery under isoflurane anesthesia, a vertical midline incision was made, the lower abdominal aorta isolated and a sterile silver clip (0.203 mm ID) placed around the aorta above the iliac bifurcation. This procedure reduces uterine perfusion pressure in the gravid rat by ~40%. Since compensation of blood flow to the placenta occurs in pregnant rats through an adaptive increase in ovarian blood flow, both right and left uterine arcades are clipped at the ovarian end, right before the first segmental artery using a silver clip (0.100 mm ID). All control dams underwent a sham operation, differing only in the absence of clips. sCR1 or saline vehicle (15 mg/kg/day in a volume of 2.6 ml/kg) was administered intravenously every 24 hours beginning 15–30 min prior to RUPP or Sham surgery. Selected experiments used 30 mg/kg/day sCR1. sCR1 (CDX-1135; Celldex Therapeutics, Needham, MA) was prepared under endotoxin-free conditions, dialyzed against nonpyrogenic normal saline, and aliquoted and stored at −80°C. Rats were randomly assigned to one of four experimental groups based on surgical procedure and drug treatment: 1) RUPP surgery receiving a normal saline vehicle (RUPP Saline); 2) RUPP surgery receiving sCR1 (RUPP sCR1); 3) sham surgery receiving a normal saline vehicle (Sham Saline); 4) sham surgery receiving sCR1 (Sham sCR1).
2.2 Measurement of mean arterial pressure and tissue collection
An intra-arterial carotid catheter was placed on gestation day 18 under isoflurane anesthesia for measurement of mean arterial pressure (MAP) in unanesthetized restrained animals on day 19 of gestation. Serum, plasma, and carotid arteries were harvested on day 19 as described previously (Alexander et al., 2001; Gilbert et al., 2007a; Gilbert et al., 2007b; Gilbert et al., 2010). The uterus was exteriorized, the total number of viable and resorbed pups counted and the pups and placentae of the right horn weighed to determine placental efficiency (fetal weight/placental weight). From the left horn, select utero-placental units from the ovarian, middle, and cervical uterine regions were fixed in 10% neutral buffered formalin for histological analysis.
2.3 Complement measurements
C3a
Serum concentrations of C3a were measured by Western immunoblot as previously described for guinea pig C3a (Regal and Klos, 2000). The primary antibody used for immunodetection was IgG fraction of rabbit polyclonal antibody to the 9 carboxy-terminal amino acids of rat C3a (Research Genetics, Inc., Huntsville, AL). The blot was probed with a 1:2,500 dilution of primary antibody followed by 80 ng/mL of goat anti-rabbit IgG coupled to horseradish peroxidase (Pierce, Rockford, IL). Images were acquired as described previously (Gilbert et al., 2012) using incubation with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Rockford, IL) for 5 minutes, image capture with FluoroChem camera (AlphaInnotech, San Leadro, CA), and pixel density quantification with Un-Scan-It Gel Analysis Software (Silk Scientific, Orem, UT). Dilutions of a standard pool of rat serum complement-activated by yeast were used to construct a standard curve on each gel and a regression equation was used to calculate the relative amount of C3a in the unknown samples. Relative amounts of C3a in each sample were expressed as C3a units/μl based on signal intensity of 1 μl of standard pool of rat serum activated by yeast.
C3 ELISA
C3 was determined as previously described for mouse C3 (Taktak and Stenning, 1992) with modifications for rat using goat anti-rat C3 IgG fraction (MP Biomedicals, LLC. Solon, OH) as the capture antibody and a peroxidase-conjugated goat IgG fraction to rat complement C3 antibody for detection (1:8,000, MP Biomedicals, LLC. Solon, OH). The concentration of C3 in each sample was expressed relative to a rat serum standard with one C3 unit/μl representing the optical density of a 1:4,000 dilution of that standard.
Total complement hemolytic activity
The inverse dilution of serum that causes 50% hemolysis of sensitized sheep erythrocytes (CH50) was determined as an indicator of total complement pathway function as previously described (Larsen et al., 2001).
2.4 Carotid artery myography
Uncannulated carotids were excised during necropsy, cleaned of adipose tissue, and a 1–2 mm segment cut from each carotid for myography. Carotid segments were placed in Krebs-Henseleit buffer (Regal et al., 1980) in DMT system baths (Model 610M, Danish Myo Technology, Aarhus, Denmark), normalized to transmural pressure of 100 mmHg, and equilibrated for 60 minutes with 3–4 washes of Krebs-Henseleit buffer as previously described (Gilbert et al., 2010). Segments were pre-contracted with thromboxane mimetic U46619 (5.7×10−7 M), followed by addition of half-log increments of acetylcholine (1.38×10−8 M to 4.14×10−4 M) to assess endothelial-dependent relaxation. After washing, vessels were again contracted with U46619 and relaxed in response to half-log doses of sodium nitroprusside (1.0×10−8 M to 1.12×10−3 M) to assess endothelial-independent relaxation. Finally, after washing, a cumulative concentration response curve to U46619 verified vessel reactivity at the end of the experiment.
2.5 Plasma VEGF and sFlt-1 assays
Circulating free VEGF and sFlt-1 concentrations in EDTA plasma collected on day 19 of gestation were measured using commercially available kits for Mouse VEGF and Mouse sVEGF R1 from R&D Systems (Quantikine, Minneapolis, MN) according to manufacturer’s directions and as described previously (Gilbert et al., 2007b; Gilbert et al., 2010).
2.6 Statistical analyses
Data are presented as mean or geometric mean ± SE, and differences were considered significant when p<0.05. Data were analyzed using two-way ANOVA with three individual contrasts considered most relevant for comparison of means: Sham Saline vs RUPP Saline, RUPP Saline vs RUPP sCR1, and Sham Saline vs Sham sCR1. C3a, CH50 and VEGF values were logged to meet model assumptions.
3. Results
3.1 sCR1 significantly inhibits placental ischemia-induced increase in MAP
MAP increased in RUPP Saline compared to Sham Saline dams (Figure 1). 15 mg/kg/day sCR1 treatment on days 14–18 markedly attenuated the elevated MAP in the RUPP group with no difference between the two Sham groups. Selected experiments were conducted using 30 mg/kg/day sCR1 on days 14–18 with a similar significant inhibition of MAP (p<0.05 vs RUPP Saline; RUPP sCR1 at 30 mg/kg MAP 100.3±4.6 mm Hg).
Figure 1.

sCR1 significantly inhibits placental ischemia-induced increase in mean arterial pressure (MAP). The increase in MAP in RUPP Saline (n=22) compared to Sham Saline rats (n=19) was significantly inhibited by daily iv administration of 15 mg/kg sCR1 (RUPP sCR1, n=9). MAP did not differ between Sham sCR1 (n=6) and Sham Saline groups. Values represent mean ± SE of MAP measured day 19 of gestation (term=21 days). *p<0.05 for indicated comparisons.
3.2 sCR1 inhibits placental ischemia-induced complement activation
With excessive complement activation, the cleavage of C3 can outpace new synthesis of protein leading to reduced C3. As seen in Figure 2, the RUPP procedure results in significantly decreased C3 in the circulation, suggesting excessive complement activation occurred and C3 substrate was being consumed. To more directly assess complement activation and the effect of sCR1, C3a in circulation was determined. Placental ischemia induced a significant increase in C3a when compared to controls (Figure 3). However, sCR1 effectively inhibited this complement activation as evidenced by decreased C3a concentrations in RUPP sCR1 animals. As previously reported (Balta et al., 2011) and confirmed in our studies, RUPP surgery with clip placement in a non-pregnant female rat did not increase blood pressure. In addition, C3a did not significantly change over a 5 day period in a non-pregnant female with RUPP surgery and clip placement (C3a change of 0.05±0.07 units/μl).
Figure 2.

Complement component C3 is reduced in serum obtained day 19 of gestation in RUPP Saline (n=10) compared to Sham Saline rats (n=12). Units of C3 are relative to a rat serum standard as described in Materials and Methods. *p<0.05 vs Sham.
Figure 3.

sCR1 significantly inhibits placental ischemia-induced increase in C3a. C3a concentrations increased in RUPP Saline (n=13) compared to Sham Saline dams (n=12). Treatment with 15 mg/kg/day sCR1 decreased C3a serum concentrations in both RUPP (n=5) and Sham (n=6) groups compared to RUPP Saline. Values represent geometric mean ± SE of C3a units/μl in serum obtained day 19 of gestation. Units of C3a are relative to a standard pool of yeast activated rat serum as described in Materials and Methods. *p<0.05 for indicated comparisons, #p<0.05 comparing Sham Saline to Sham sCR1.
The efficacy of sCR1 in inhibiting the complement system in vivo was also evaluated by measuring the ability of serum collected on gestation day 19 to lyse antibody-coated sheep erythrocytes via a total hemolytic complement assay. Total hemolytic complement activity was not different in RUPP Saline and Sham Saline groups, but treatment with sCR1 significantly decreased the CH50 in serum from both RUPP and Sham animals (Figure 4). In fact, CH50 was significantly less in Sham sCR1 animals compared to RUPP sCR1 animals suggesting that it was a more effective inhibitor in control animals.
Figure 4.

Treatment with 15 mg/kg/day sCR1 for 5 days significantly reduced CH50 in serum of RUPP and Sham animals. The CH50 in RUPP Saline (n=12) and Sham Saline (n=12) groups were not different. Treatment with sCR1 significantly decreased CH50 in serum collected on day 19 from RUPP sCR1 (n=5) and Sham sCR1 (n=6) compared to controls. Values represent geometric mean ± SE of CH50 in serum collected day 19 of gestation. *p<0.05 for indicated comparisons, #p<0.05 comparing Sham Saline to Sham sCR1. Sham sCR1 vs RUPP sCR1 was also tested for this outcome and was statistically different, p<0.05.
Placentae of RUPP and Sham animals were examined in all treatment groups by immunohistochemistry using a polyclonal anti-rat C3 antibody to determine if C3 deposition was evident. No difference in intensity or location of immunoreactive C3 was detected between the two groups (data not shown).
3.3 sCR1 does not affect the VEGF decrease observed in placental ischemia
Hypertension following placental ischemia is associated with a decreased free plasma VEGF and increased sFlt-1, and infusion of VEGF attenuates placental ischemia-induced hypertension (Gilbert et al., 2010). No increase in circulating sFlt-1 was observed in RUPP vs Sham (118.4±19.4 pg/mL vs 114.9±20.4 pg/mL) with measured values near the detection limit of the assay. VEGF significantly decreased with placental ischemia (Figure 5) and no restoration of VEGF was evident in RUPP animals with sCR1 treatment. The sFlt-1/VEGF ratio did not differ amongst treatment groups (data not shown).
Figure 5.

sCR1 treatment did not reverse RUPP-induced decrease in free VEGF concentrations in plasma. RUPP Saline animals (n= 21) had a decrease in VEGF compared to Sham Saline animals (n= 19). VEGF concentrations did not change in RUPP sCR1 animals (n=9) compared to RUPP Saline animals. Concentration of VEGF did not differ in Sham Saline and Sham sCR1 (n=6) animals. Values represent geometric mean ± SE of free VEGF measured by ELISA in plasma obtained from animals day 19 of gestation. *p<0.05 for indicated comparisons.
3.4 sCR1 alters endothelial-independent arterial relaxation
The contractile response to submaximal concentration of thromboxane mimetic U46619 did not differ between RUPP and Sham animals. The RUPP procedure did not significantly alter endothelial-dependent acetylcholine-induced relaxation of the carotid artery (Figure 6A). Histological analysis of select vessels clearly showed intact endothelium. Dilation in response to the endothelial-independent vasodilator sodium nitroprusside (SNP) was also not altered in RUPP compared to Sham (data not shown). In carotid arteries from either RUPP or Sham animals treated with sCR1, the vasodilatory response to SNP was enhanced (Figure 6B) with no effect on acetylcholine-induced relaxation (data not shown). sCR1 itself did not relax an isolated carotid artery from a pregnant animal or cause relaxation of an artery pre-contracted with U46619 (data not shown). In addition, presence of sCR1 in the myography bath did not significantly alter vasodilation to either acetylcholine or SNP in carotid artery from a normal pregnant rat (data not shown).
Figure 6.
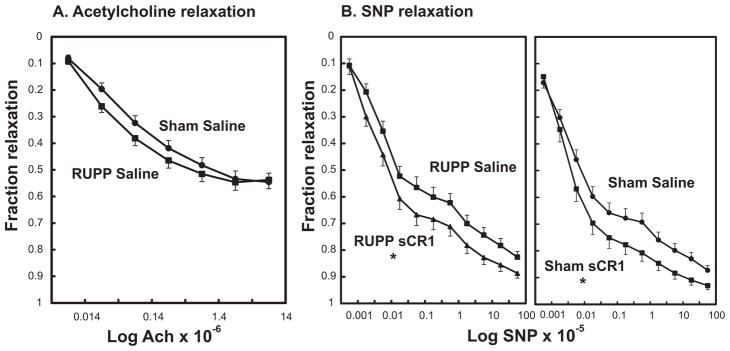
Effect of placental ischemia and sCR1 on relaxation responses of carotid artery in vitro. Values represent mean ± SE of the fraction relaxation of arteries pre-contracted with thromboxane mimetic U46619. A. Carotid arteries isolated from RUPP Saline (n=16) or Sham Saline (n=13) animals relaxed to increasing concentrations of acetylcholine with no significant treatment effect. B. sCR1 enhances endothelial-independent relaxation in carotid arteries isolated from RUPP and Sham animals. Carotid arteries isolated from animals either treated with saline or sCR1 were pre-contracted with U46619 and the fraction relaxation to increasing concentrations of sodium nitroprusside (SNP) determined. *Repeated measures ANOVA demonstrated a significant sCR1 effect in carotid arteries from RUPP and Sham animals. Values represent geometric mean ± SE of 6–16 animals is each group.
3.5 sCR1 does not affect the RUPP-induced increase in fetal resorptions
Fetal resorptions were significantly increased in RUPP Saline and RUPP sCR1 groups compared to Sham Saline or Sham sCR1 groups with no significant sCR1 treatment effect (Figure 7). Neither average placental weights nor placental efficiencies (fetal weight/placental weight) were significantly different amongst any of the treatment groups (data not shown).
Figure 7.

sCR1 treatment did not affect RUPP-induced fetal resorptions. RUPP Saline animals (n= 22) had an increase in resorptions compared to Sham Saline animals (n= 19). The fraction of fetal resorptions did not change in RUPP sCR1 animals (n=9) compared to RUPP Saline animals. Values for Sham Saline did not differ compared to Sham sCR1 (n=6). Values represent mean ± SE of the fraction of resorbed fetuses on day 19 of gestation. *p<0.05 for indicated comparisons.
4. Discussion
Our studies are the first to demonstrate that inhibition of complement activation attenuates development of high blood pressure following placental ischemia in pregnancy, indicating that complement activation is a critical event leading to placental ischemia-induced hypertension. Using the RUPP model, we established that complement activation occurs in concert with placental ischemia-induced hypertension and that sCR1 effectively inhibits complement activation to attenuate this hypertension. Previous studies had indicated an important role for angiogenic imbalance in mediating the blood pressure increase (Gilbert et al., 2010), but inhibition of complement activation attenuated placental ischemia-induced hypertension regardless of concurrent decreases in free plasma VEGF. These studies provide evidence for an important pathway leading to hypertension distinct from angiogenic imbalance and highlight the potential utility of inhibiting complement activation to manage hypertension in pregnancy.
Numerous mediators have been implicated in the pathogenesis of pregnancy-induced hypertension including TNFα, sFlt-1, endothelin, oxidative stress, CD4+ T cells and agonistic autoantibody to angiotensin II type 1 receptor (Li et al., 2012). Many animal models of preeclampsia have been described using a single mediator as a discrete initiating event leading to symptoms of preeclampsia. However, preeclampsia is a heterogeneous disorder with varying degrees of angiogenic imbalance and autoantibody production, so we chose to use a model that employs placental ischemia as the initiating event for the wide range of characteristics that mimic preeclampsia to determine the role of complement system activation. Placental ischemia is a consistent finding in preeclampsia and does not presume the mechanistic pathways leading to the hypertension. In addition, placental ischemia as the initiator of hypertension allows concurrent inflammatory and angiogenic pathways to operate in situ resulting in hypertension. The RUPP procedure is an established and effective means of inducing hypertension following placental ischemia in animal models and closely mimics many characteristics of preeclampsia (Li et al., 2012) without inducing symptoms of severe preeclampsia (Balta et al., 2011).
During pregnancy, complement activation has been implicated in pathogenesis of hypertension in two different studies in mice; abnormal placental development (C1q deficiency) or infusion of autoantibody to angiotensin II type 1 receptor. The C1q-deficient model investigates events leading to placental ischemia as well as resulting from placental ischemia. Mice deficient in C1q exhibit abnormal placental development during pregnancy resulting in preeclamptic symptoms including high blood pressure. Thus C1q early in pregnancy is essential for normal placentation and to maintain normal blood pressure throughout pregnancy (Singh et al., 2011). In third trimester pregnant mice with the placenta already formed, adoptive transfer of human autoantibodies to the angiotensin II type I receptor (AT1-AA) results in symptoms resembling preeclampsia including increased blood pressure. A C3a antagonist prevents the AT1-AA-induced hypertension indicating that excessive complement activation during pregnancy mediates hypertension initiated by the immune complex formation of AT1-AA with its receptor (Wang et al., 2012; Zhou et al., 2008). Whether complement activation following placental ischemia involves AT1-AA is a subject of future investigations.
Inhibition via sCR1 targets complement activation products C3b and C4b to promote their degradation and accelerate decay of convertase enzymes in the complement cascade. sCR1 has been extensively used to assess the importance of complement system activation in numerous rodent models of autoimmune and inflammatory diseases (Goodfellow et al., 1997; Piddlesden et al., 1994) and was clearly effective in inhibiting both C3a production and resultant hypertension in the RUPP model. The sCR1 reduction in CH50 at day 19 was significant but not as marked as some previously published studies. sCR1 effectiveness in inhibiting total hemolytic complement activity may be limited by the 5 day treatment course in our study, consistent with the observations made by Pratt et al (Pratt et al., 1997) who demonstrated rat anti-human sCR1 antibody production after several days that limits effectiveness. sCR1 is also known to bind C1q (Klickstein et al., 1997) and MBL (Ghiran et al., 2000) and thus may affect processes beyond inhibition of the complement cascade at the level of the C3 and C5 convertases. In our rat studies, sCR1 treatment does not begin until day 14 of gestation so any possible effects on C1q would be restricted to those occurring after placentae are established. Our studies address the role of complement activation downstream of placental ischemia as opposed to the previously reported role for complement upstream of placental ischemia in a C1q-deficient mouse (Singh et al., 2011).
Clearly, inhibition of complement activation has the potential to increase susceptibility to infection and limits its usefulness for long term therapy. Any human use of sCR1 or other complement inhibitors would employ appropriate immunizations and close monitoring similar to that used with the anti-C5 antibody eculizumab for treatment of paroxysmal nocturnal hemoglobinuria (Kelly et al., 2010). A recent case report using eculizumab in a woman with preeclampsia/HELLP syndrome successfully normalized lab values and prolonged pregnancy by 17 days suggesting that therapeutic manipulation of the complement system during pregnancy may be feasible (Burwick and Feinberg, 2013).
Data from previous investigations show significant changes in circulating pro- and anti-angiogenic factors following the RUPP procedure; specifically, RUPP rats exhibit decreased VEGF and increased sFlt-1 compared to Sham (Gilbert et al., 2007b; Gilbert et al., 2010). A decrease in free plasma VEGF was apparent in our studies, but was not restored with sCR1 treatment, indicating that complement activation mediates hypertension regardless of changes in free plasma VEGF. This is in contrast to studies of fetal rejection and growth restriction in the mouse where inhibition of C5 resulted in an increase in plasma free VEGF and a decrease in sFlt-1 (Girardi et al., 2006). In our study, inhibiting complement activation did not restore VEGF but attenuated hypertension introducing the possibility that low VEGF may result in increased complement activation. A recent report (Keir, 2012) suggests that decreased VEGF in the kidney might allow excessive complement activation to occur due to a decrease in complement regulators.
Unlike other studies in the RUPP model, heparin was not used in our experimental protocol due to its demonstrated ability to inhibit complement activation (Girardi et al., 2004). It is known that heparin treatment in coronary angiography results in elevated sFlt-1 and that sFlt-1 increases in the circulation of mice treated with heparin (Searle et al., 2011). In a recent abstract, heparin is also reported to displace sFlt-1 from rat placenta and increase circulating sFlt-1 (George, 2012). Thus, it is possible that the lack of increased circulating sFlt-1 in our study may be due to its continued sequestration in the placenta in absence of added heparin in the experimental protocol. Regardless, hypertension following placental ischemia was evident in the absence of changes in circulating sFlt-1.
A difference in endothelial-dependent relaxation of aorta or carotid from RUPP rats vs Sham was not detected in our study suggesting that endothelial dysfunction of the larger blood vessels did not occur. This is in contrast to the spectrum of impaired endothelial-dependent relaxation of carotid and aorta reported in Sprague Dawley rats obtained from Harlan following RUPP surgery (Crews et al., 2000; Gilbert et al., 2010). In light of previous findings that inhibition of NO synthase increases blood pressure more in Sprague Dawley rats from Harlan compared to Charles River, our data support the possibility of differences in the relative contribution of endothelium-derived NO to carotid artery function in Charles River rats compared to Harlan (Buhimschi et al., 2001; Pollock and Rekito, 1998). In addition, more recent studies (Griffin et al., 2012) have also reported differences in nephropathy susceptibility and systemic and renal hemodynamic responses to the NO synthase inhibitor L-NAME in rats from the two suppliers. Therefore, differences in the effect of the RUPP procedure on endothelial-dependent relaxation may be due to strain differences in Sprague Dawley rats obtained from different distributors. Treatment with sCR1 in vivo resulted in greater carotid relaxation in both the RUPP and Sham groups in response to SNP, indicating complement may be acting independently of endothelium; however, sCR1 itself did not have direct effects on the vessel so further investigations are warranted.
Our data are first to demonstrate that inhibiting complement system activation may be a novel therapeutic strategy for managing hypertension following placental ischemia in preeclampsia. Using a well-characterized and highly relevant model of preeclampsia in the rat, we have demonstrated a mechanistic link between complement activation and hypertension in pregnancy. Our data suggest that placental ischemia, a consistent feature of preeclampsia, leads to complement activation and hypertension in the rat by a pathway distinct from VEGF. General complement inhibition may not be an optimal therapeutic strategy for preeclampsia, however, because of the potential increased risk of maternal and fetal infection. Certainly, further research is necessary to identify how complement is activated following placental ischemia and to determine the complement activation product(s) (e.g. C3a, C5a, C5b-9) responsible.
Highlights.
Complement activation is critical for hypertension caused by reduced placental blood flow
Inhibiting complement activation with sCR1 attenuates placental ischemia-induced hypertension
Role of complement in placental ischemia-induced hypertension does not depend on VEGF
Inhibiting complement activation may offer a therapeutic avenue in preeclampsia
Acknowledgments
The authors gratefully acknowledge Barbara Elmquist for technical assistance, Dr. Ronald Regal, Department of Mathematics and Statistics, University of Minnesota Duluth for statistical consultation, and Drs Ronald Taylor and Margaret Lindorfer, University of Virginia, for helpful review of the manuscript. This work was supported by NIH R15 HL109843 (JFR), R01 HL114096 and AHA 10SDG260040 (JSG) and student stipend support R25 GM086669 (AJB and SJL).
Abbreviations
- MAP
mean arterial pressure
- RUPP
reduced utero-placental perfusion pressure
- sCR1
soluble complement receptor 1
- sFlt-1
soluble fms-like tyrosine kinase-1
- SNP
sodium nitroprusside
- VEGF
vascular endothelial growth factor
Footnotes
Disclosures
Dr. Marsh is employed by Celldex Therapeutics, Inc. and Celldex Therapeutics, Inc. provided the sCR1 used in this study. The other authors have no conflicts to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Kathryn E Lillegard, Email: lille067@umn.edu.
Alex C Johnson, Email: joh06981@d.umn.edu.
Sarah J Lojovich, Email: sarah.lojovich@medtronic.com.
Ashley J Bauer, Email: bauer489@d.umn.edu.
Henry C Marsh, Email: hmarsh@celldextherapeutics.com.
Jeffrey S Gilbert, Email: jgilbe@uoregon.edu.
Jean F Regal, Email: jregal@d.umn.edu.
References
- Alexander BT, Kassab SE, Miller MT, Abram SR, Reckelhoff JF, Bennett WA, Granger JP. Reduced uterine perfusion pressure during pregnancy in the rat is associated with increases in arterial pressure and changes in renal nitric oxide. Hypertension. 2001;37:1191–5. doi: 10.1161/01.hyp.37.4.1191. [DOI] [PubMed] [Google Scholar]
- Baines MG, Millar KG, Mills P. Studies of complement levels in normal human pregnancy. Obstet Gynecol. 1974;43:806–10. [PubMed] [Google Scholar]
- Baker PN, Krasnow J, Roberts JM, Yeo KT. Elevated serum levels of vascular endothelial growth factor in patients with preeclampsia. Obstet Gynecol. 1995;86:815–21. doi: 10.1016/0029-7844(95)00259-T. [DOI] [PubMed] [Google Scholar]
- Balta O, Boztosun A, Deveci K, Gulturk S, Ekici F, Kaya A, Cetin A, Cetin M. Reduced uterine perfusion pressure model is not successful to mimic severe preeclampsia. Placenta. 2011;32:675–80. doi: 10.1016/j.placenta.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Buhimschi IA, Shi SQ, Saade GR, Garfield RE. Marked variation in responses to long-term nitric oxide inhibition during pregnancy in outbred rats from two different colonies. Am J Obstet Gynecol. 2001;184:686–93. doi: 10.1067/mob.2001.110448. [DOI] [PubMed] [Google Scholar]
- Burwick RM, Feinberg BB. Eculizumab for the treatment of preeclampsia/HELLP syndrome. Placenta. 2013;34:201–3. doi: 10.1016/j.placenta.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Crews JK, Herrington JN, Granger JP, Khalil RA. Decreased endothelium-dependent vascular relaxation during reduction of uterine perfusion pressure in pregnant rat. Hypertension. 2000;35:367–72. doi: 10.1161/01.hyp.35.1.367. [DOI] [PubMed] [Google Scholar]
- Denny KJ, Woodruff TM, Taylor SM, Callaway LK. Complement in pregnancy: a delicate balance. Am J Reprod Immunol. 2013;69:3–11. doi: 10.1111/aji.12000. [DOI] [PubMed] [Google Scholar]
- Derzsy Z, Prohaszka Z, Rigo J, Jr, Fust G, Molvarec A. Activation of the complement system in normal pregnancy and preeclampsia. Mol Immunol. 2010;47:1500–6. doi: 10.1016/j.molimm.2010.01.021. [DOI] [PubMed] [Google Scholar]
- George EM. Hypoxia-induced Heparanase Regulates sFlt-1 Release from Placental Chorionic Villi. Hypertension. 2012;60:A48. [Google Scholar]
- Ghiran I, Barbashov SF, Klickstein LB, Tas SW, Jensenius JC, Nicholson-Weller A. Complement receptor 1/CD35 is a receptor for mannan-binding lectin. J Exp Med. 2000;192:1797–808. doi: 10.1084/jem.192.12.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Dukes M, LaMarca B, Cockrell K, Babcock S, Granger J. Effects of reduced uterine perfusion pressure on blood pressure and metabolic factors in pregnant rats. Am J Hypertens. 2007a;20:686–91. doi: 10.1016/j.amjhyper.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Gilbert JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension. 2007b;50:1142–7. doi: 10.1161/HYPERTENSIONAHA.107.096594. [DOI] [PubMed] [Google Scholar]
- Gilbert JS, Bauer AJ, Gingery A, Banek CT, Chasson S. Circulating and utero-placental adaptations to chronic placental ischemia in the rat. Placenta. 2012;33:100–5. doi: 10.1016/j.placenta.2011.11.025. [DOI] [PubMed] [Google Scholar]
- Gilbert JS, Ryan MJ, LaMarca BB, Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension during preeclampsia: linking placental ischemia with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008;294:H541–50. doi: 10.1152/ajpheart.01113.2007. [DOI] [PubMed] [Google Scholar]
- Gilbert JS, Verzwyvelt J, Colson D, Arany M, Karumanchi SA, Granger JP. Recombinant vascular endothelial growth factor 121 infusion lowers blood pressure and improves renal function in rats with placentalischemia-induced hypertension. Hypertension. 2010;55:380–5. doi: 10.1161/HYPERTENSIONAHA.109.141937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi G, Berman J, Redecha P, Spruce L, Thurman JM, Kraus D, Hollmann TJ, Casali P, Caroll MC, Wetsel RA, Lambris JD, Holers VM, Salmon JE. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112:1644–54. doi: 10.1172/JCI18817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi G, Redecha P, Salmon JE. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nature medicine. 2004;10:1222–6. doi: 10.1038/nm1121. [DOI] [PubMed] [Google Scholar]
- Girardi G, Yarilin D, Thurman JM, Holers VM, Salmon JE. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med. 2006;203:2165–75. doi: 10.1084/jem.20061022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow RM, Williams AS, Levin JL, Williams BD, Morgan BP. Local therapy with soluble complement receptor 1 (sCR1) suppresses inflammation in rat mono-articular arthritis. Clinical and experimental immunology. 1997;110:45–52. doi: 10.1046/j.1365-2249.1997.5111408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia-reperfusion injuries. Immunobiology. 2012;217:1026–33. doi: 10.1016/j.imbio.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger JP, LaMarca BB, Cockrell K, Sedeek M, Balzi C, Chandler D, Bennett W. Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol Med. 2006;122:383–92. doi: 10.1385/1-59259-989-3:381. [DOI] [PubMed] [Google Scholar]
- Griffin K, Polichnowski A, Licea-Vargas H, Picken M, Long J, Williamson G, Bidani A. Large BP-dependent and -independent differences in susceptibility to nephropathy after nitric oxide inhibition in Sprague-Dawley rats from two major suppliers. American journal of physiology Renal physiology. 2012;302:F173–82. doi: 10.1152/ajprenal.00070.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holers VM, Girardi G, Mo L, Guthridge JM, Molina H, Pierangeli SS, Espinola R, Xiaowei LE, Mao D, Vialpando CG, Salmon JE. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. 2002;195:211–20. doi: 10.1084/jem.200116116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel CA. Oxidative stress in the pathogenesis of preeclampsia. Proc Soc Exp Biol Med. 1999;222:222–35. doi: 10.1177/153537029922200305. [DOI] [PubMed] [Google Scholar]
- Keir L, Welsh GI, Coward RJ, Satchell S, Saleem MA, Richards A. VEGF-A regulates glomerular endothelial cell expression of protective complement regulators involved in the pathogenesis of atypical haemolytic uraemic syndrome. Immunobiology. 2012;217:1215. [Google Scholar]
- Kelly R, Arnold L, Richards S, Hill A, Bomken C, Hanley J, Loughney A, Beauchamp J, Khursigara G, Rother RP, Chalmers E, Fyfe A, Fitzsimons E, Nakamura R, Gaya A, Risitano AM, Schubert J, Norfolk D, Simpson N, Hillmen P. The management of pregnancy in paroxysmal nocturnal haemoglobinuria on long term eculizumab. British journal of haematology. 2010;149:446–50. doi: 10.1111/j.1365-2141.2010.08099.x. [DOI] [PubMed] [Google Scholar]
- Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity. 1997;7:345–55. doi: 10.1016/s1074-7613(00)80356-8. [DOI] [PubMed] [Google Scholar]
- Larsen CP, Regal RR, Regal JF. Trimellitic anhydride-induced allergic response in the guinea pig lung involves antibody-dependent and -independent complement system activation. J Pharmacol Exp Ther. 2001;296:284–92. [PubMed] [Google Scholar]
- Li J, LaMarca B, Reckelhoff JF. A model of preeclampsia in rats: the reduced uterine perfusion pressure (RUPP) model. Am J Physiol Heart Circ Physiol. 2012;303:H1–8. doi: 10.1152/ajpheart.00117.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch AM, Gibbs RS, Murphy JR, Giclas PC, Salmon JE, Holers VM. Early elevations of the complement activation fragment C3a and adverse pregnancy outcomes. Obstet Gynecol. 2011;117:75–83. doi: 10.1097/AOG.0b013e3181fc3afa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch AM, Salmon JE. Dysregulated complement activation as a common pathway of injury in preeclampsia and other pregnancy complications. Placenta. 2010;31:561–7. doi: 10.1016/j.placenta.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard S, Epstein FH, Karumanchi SA. Preeclampsia and angiogenic imbalance. Annual review of medicine. 2008;59:61–78. doi: 10.1146/annurev.med.59.110106.214058. [DOI] [PubMed] [Google Scholar]
- Mistry HD, Kurlak LO, Broughton Pipkin F. The placental renin-angiotensin system and oxidative stress in pre-eclampsia. Placenta. 2013;34:182–6. doi: 10.1016/j.placenta.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Piddlesden SJ, Storch MK, Hibbs M, Freeman AM, Lassmann H, Morgan BP. Soluble recombinant complement receptor 1 inhibits inflammation and demyelination in antibody-mediated demyelinating experimental allergic encephalomyelitis. J Immunol. 1994;152:5477–84. [PubMed] [Google Scholar]
- Pollock DM, Rekito A. Hypertensive response to chronic NO synthase inhibition is different in Sprague-Dawley rats from two suppliers. The American journal of physiology. 1998;275:R1719–23. doi: 10.1152/ajpregu.1998.275.5.R1719. [DOI] [PubMed] [Google Scholar]
- Pratt JR, Harmer AW, Levin J, Sacks SH. Influence of complement on the allospecific antibody response to a primary vascularized organ graft. Eur J Immunol. 1997;27:2848–53. doi: 10.1002/eji.1830271116. [DOI] [PubMed] [Google Scholar]
- Proctor LM, Moore TA, Monk PN, Sanderson SD, Taylor SM, Woodruff TM. Complement factors C3a and C5a have distinct hemodynamic effects in the rat. International immunopharmacology. 2009;9:800–6. doi: 10.1016/j.intimp.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Qing X, Redecha PB, Burmeister MA, Tomlinson S, D’Agati VD, Davisson RL, Salmon JE. Targeted inhibition of complement activation prevents features of preeclampsia in mice. Kidney Int. 2011;79:331–9. doi: 10.1038/ki.2010.393. [DOI] [PubMed] [Google Scholar]
- Regal JF, Eastman AY, Pickering RJ. C5a induced tracheal contraction: a histamine independent mechanism. J Immunol. 1980;124:2876–8. [PubMed] [Google Scholar]
- Regal JF, Klos A. Minor role of the C3a receptor in systemic anaphylaxis in the guinea pig. Immunopharmacology. 2000;46:15–28. doi: 10.1016/s0162-3109(99)00152-6. [DOI] [PubMed] [Google Scholar]
- Searle J, Mockel M, Gwosc S, Datwyler SA, Qadri F, Albert GI, Holert F, Isbruch A, Klug L, Muller DN, Dechend R, Muller R, Vollert JO, Slagman A, Mueller C, Herse F. Heparin strongly induces soluble fms-like tyrosine kinase 1 release in vivo and in vitro--brief report. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:2972–4. doi: 10.1161/ATVBAHA.111.237784. [DOI] [PubMed] [Google Scholar]
- Singh J, Ahmed A, Girardi G. Role of complement component C1q in the onset of preeclampsia in mice. Hypertension. 2011;58:716–24. doi: 10.1161/HYPERTENSIONAHA.111.175919. [DOI] [PubMed] [Google Scholar]
- Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. 2010;376:631–44. doi: 10.1016/S0140-6736(10)60279-6. [DOI] [PubMed] [Google Scholar]
- Taktak YS, Stenning B. Solid phase enzyme immunoassays for the quantification of serum amyloid P (SAP) and complement component 3 (C3) proteins in acute-phase mouse sera. Hormone and metabolic research = Hormon-und Stoffwechselforschung = Hormones et metabolisme. 1992;24:371–4. doi: 10.1055/s-2007-1003338. [DOI] [PubMed] [Google Scholar]
- Usami M, Mitsunaga K, Miyajima A, Sunouchi M, Doi O. Complement component C3 functions as an embryotrophic factor in early postimplantation rat embryos. The International journal of developmental biology. 2010;54:1277–85. doi: 10.1387/ijdb.092993mu. [DOI] [PubMed] [Google Scholar]
- Wang W, Irani RA, Zhang Y, Ramin SM, Blackwell SC, Tao L, Kellems RE, Xia Y. Autoantibody-mediated complement C3a receptor activation contributes to the pathogenesis of preeclampsia. Hypertension. 2012;60:712–21. doi: 10.1161/HYPERTENSIONAHA.112.191817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Mao D, Holers VM, Palanca B, Cheng AM, Molina H. A critical role for murine complement regulator crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nature medicine. 2008;14:855–62. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]