

Abstract
p21-activated kinase-1 (Pak1) is frequently upregulated in human breast cancer and is required for transformation of mammary epithelial cells by ErbB2. Here we show that loss of Pak1, but not the closely related Pak2, leads to diminished expression of β-catenin and its target genes. In MMTV-ErbB2 transgenic mice, loss of Pak1 prolonged survival, and mammary tissues of such mice showed loss of β-catenin. Expression of a β-catenin mutant bearing a phospho-mimetic mutation at Ser 675, a specific Pak1 phosphorylation site, restored transformation to ErbB2-positive, Pak1-deficient mammary epithelial cells. Mice bearing xenografts of ErbB2-positive breast cancer cells showed tumor regression when treated with small molecule inhibitors of Pak or β-catenin, and combined inhibition by both agents was synergistic. These data delineate a signaling pathway from ErbB2 to Pak to β-catenin that is required for efficient transformation of mammary epithelial cells, and suggest new therapeutic strategies in ErbB2-positive breast cancer.
Keywords: transformation, small GTPase, protein kinase, ErbB2, signal transduction, breast cancer
Introduction
The ErbB2 oncogene is amplified in approximately 30% of human breast cancers and represents a clinically useful therapeutic target (1). ErbB2 proteins frequently heterodimerize with ErbB1 or ErbB3, and these heterodimers activate a signaling program that drives cell proliferation, resistance to apoptosis, loss of polarity, and increased motility and invasiveness (2). Key signaling pathways that emanate from ErbB2 include, but are not limited to, the phosphoinositol-3 kinase (PI3K)/Akt/mTOR, Ras/ERK, and Src/Fak networks (3).
p21-activated kinases (Paks) are Cdc42/Rac activated serine-threonine protein kinases that regulate the PI3K/Akt, Ras/ERK, and Src/Fak signaling pathways (4, 5). Pak1 is required for activation of Akt, perhaps due to a scaffolding function that links Pdk1 to Akt (6, 7). In Ras/ERK signaling, Pak1 phosphorylates c-Raf at S338 and Mek1 at S298, sites that are required for full activation of these proteins in some cell types (8, 9). Pak1 also acts downstream in the Src/Fak pathway, as an effector for the small GTPase Rac1 (10, 11) Loss of Pak1, induced by siRNA, expression of dominant negative alleles, gene-disruption, or small molecule inhibitors, has been shown to block transformation in vitro by oncogenic forms of Kras, ErbB2, and KSHV (9, 12–16). In addition, Pak1 is frequently overexpressed in human breast, ovary, bladder, uterine, and brain cancer, due to amplification of the PAK1 gene in an 11q13 amplicon (9), and has oncogenic properties when expressed in mouse breast epithelial cells and tissues (17, 18). However, the role of Pak1 in tumorigenesis in vivo, and the particular signaling pathways affected, are not defined. In addition, the apparent special role of Pak1, versus the closely related and broadly expressed Pak2, is not understood.
In this work, we examined the distinct roles of Pak1 and Pak2 in cellular and animal models of ErbB2-driven breast cancer. We found that inhibition of Pak1, but not Pak2, impedes transformation by ErbB2 in a 3D cell culture system, but that loss of either Pak1 or Pak2 causes loss of both ERK and Akt activation. A phospho-proteomic screen revealed that Pak1-deficient, but not Pak2-deficient, ErbB2 cells showed almost total loss of β-catenin expression, and that exogenous expression of a stabilized β-catenin or a mutant form bearing a phosphomimic residue at a known site of Pak1 phosphorylation (S675E), restored the ability of ErbB2 to transform Pak1-deficient cells. Lastly, we showed that small molecule inhibitors of Pak or β-catenin blocked transformation by ErbB2 in 3D culture and tumorigenesis by ErbB2 in mouse xenografts. Combined use of anti-Pak and anti-β-catenin agents was synergistic. These findings establish Pak1 as a new target in ErbB2-driven breast cancer and define a new mechanism of action primarily through the β-catenin, but not the ERK or Akt, signaling pathways.
Materials and Methods
Generation of transgenic mice and tumor measurement
All animal experiments were approved by the Fox Chase Cancer Center Institutional Animal Care and Use Committee (IACUC) and carried out according to NIH-approved protocols in compliance with the guide for the Care and Use of Laboratory Animals. Detailed methods are contained in SI Materials and Methods.
Cell lines and 3D cell culture
Tumors from MMTV-Neu:Pak1+/+ and MMTV-Neu:Pak1−/− animals were dissected, rinsed with PBS and minced, then treated with 0.2% collagenase for 2 hours at 37°C. Cells were washed several times with serum free DMEM, and finally with low calcium medium supplemented with 5% horse serum. Tissues kept in low calcium medium were transferred to T-25 flasks coated with 0.1% gelatin and incubated in a 37°C incubator overnight. The next day, supernatants containing floating cells were transferred and seeded into new flasks, and maintained with regular media changes until confluency, to prevent fibroblast growth. Fibroblast-free cell populations were derived typically after 6–8 weeks. In vitro proliferation was measured by seeding approximately 1 × 105 cells on 0.1% gelatin-coated T25 flasks. At specific time points, cells were trypsinized and counted using Trypan blue exclusion analysis. All analyses used cells passaged <6 times. 10A.ErbB2 cells (MCF-10A cells expressing a chimeric form of ErbB2) (19) were maintained in DMEM/F12 (Gibco BRL) supplemented with 5% donor horse serum, 20 ng/ml EGF (Harlan Bioproducts), 10 μg/ml insulin (Sigma), 1 ng/ml cholera toxin (Sigma), 100 μg/ml hydrocortisone (Sigma), 50 U/ml penicillin and 50 μg/ml streptomycin. For 3D cultures, ~5,000 cells were plated atop rBM in 8-well slide chambers as described (19). To activate chimeric ErbB proteins, 1 μM AP1510 was added to the growth medium. MCF-7, MDA-MB-231, BT-474 and SK-BR3 were obtained from American Type Culture Collection, MCF-7 and MDA-MB-231cells were grown in DMEM supplemented with 10% fetal bovine serum, BT-474 cells were grown in RPMI supplemented with 10% fetal bovine serum and SK-BR3 were grown in McCoy’s 5A supplemented with 10% fetal bovine serum. BT-474R cells were a kind gift from Dr. Jose Baselga (Massachusetts General Hospital).
Tissue preparation, histology, immunohistochemistry, and immunoblotting
All tumor samples and control tissues were fixed overnight in 4% paraformaldehyde, dehydrated and embedded in paraffin. Hematoxylin and eosin (H&E) stained sections were used for diagnostic purposes and unstained sections for immunohistochemical (IHC) studies. Protein concentration was determined, and equal amounts of total proteins were separated on SDS-PAGE. A detailed list of antibodies used is contained in SI Materials and Methods.
Tumor Xenografts
All studies described in this article were done under approved protocols following Fox Chase Cancer Center Institutional Animal Care and Use Committee guidelines. Detailed methods are contained in SI Materials and Methods.
Statistical analysis
Statistical analysis was performed using the unpaired Student’s t test except for survival curves where the log P rank test was used. Values of p < 0.05 were considered significant.
Results
Pak1, but not Pak2, knockdown inhibits proliferation and rescues apoptosis in 10A.ErbB2 cells
To investigate the roles of Pak1 and Pak2 in ErbB2 signaling, we employed doxycycline-inducible shRNAs (15). Pak1 and Pak2 are both expressed in MCF10A cells and in ErbB2-positive or ErbB2-negative mammary epithelial cells such as BT-474, SK-BR3, MCF-7 and MDA-MB-231 (Fig. S1A). MCF10A-ErbB2 cells (henceforth termed 10A.ErbB2), which activate ErbB2 in response to AP1510 (19), were stably transduced with a doxycycline-inducible shRNA specific to Pak1 or Pak2, respectively, and the cells were assessed for proliferation and apoptosis. Upon doxycycline induction, the Pak1 and Pak2 shRNA-infected cells showed specific loss of their intended target (Fig. 1A). Loss of Pak1, but not Pak2, impeded the proliferation (Fig. 1B) and survival (Fig. 1C) of 10A.ErbB2 cells, similar results were observed in BT-474, SK-BR3, MCF-7 and MDA-MB-231 (Fig. S1B, S1C and S1D). These data suggest that only Pak1 is required for ErbB2 effects on two of the cardinal features of transformation: cell division and apoptosis.
Figure 1. Pak1, but not Pak2, is required for ErbB2-mediated transformation of MCF-10A cells.

(A) Western analysis shows specific loss of Pak1 and Pak2 in shRNA infected cells. (B) Proliferation of Pak1 and Pak2 10A.ErbB2 deficient cells upon ErbB2 stimulation. Cells were seeded, harvested and counted at 0, 24, 48, 72 and 96 h. The data are representative of three independent experiments. Points, mean; bars, SD. (C) Apoptosis of Pak1 and Pak2 10A.ErbB2 deficient cells. Apoptosis was measured calculating the percent of positive Annexin V-PE cells by flow cytometry. The data are representative of three independent experiments. Bars, SD. (D) Pak1 and Pak2 in shRNA-infected 10A.ErbB2 cells were plated atop reconstituted basement membrane. Cells treated with doxycycline and stimulated with vehicle or 1 μM AP1510 on day 3 and fixed on day 12 were stained with Oregon green-phalloidin, Ki-67, or anti-cleaved caspase-3. Percentage of unilamellar acini, Ki-67-positive, and anti-cleaved caspase-3-positive acini were scored based on assessment of 50 to 60 acini per well. Bar, 50 μm.
Pak1 knockdown restores normal acinar morphology in 3D cell culture
To assess the effects of Pak1 and Pak2 on acinar development and architecture, we plated the shRNA expressing 10A.ErbB2 cells in a 3D Matrigel matrix. Under such conditions, induction of ErbB2 induces a striking phenotype characterized by formation of multi-lobular acini (Fig. 1Da, Db), loss of central apoptosis (Fig. 1Dc, Dd), and increase in proliferation (Fig. 1De, Df). Expression of Pak1-directed shRNA had little effect on non-induced 10A.ErbB2 cells (Fig. 1Dg, Di, Dk), but strongly suppressed the transformed phenotype of induced cells (Fig. 1Dh, Dj, Dl). In contrast, Pak2-directed shRNA had little effect on acinar structure irrespective of ErbB2 expression (Fig. 1Dm – Dr).
As Pak1 and Pak2 have similar catalytic properties, we sought to uncover differences in their behavior in breast epithelial cells. We and others had earlier established that Pak1 can be driven into the nucleus upon EGF receptor activation in mammary epithelial cells (20, 21); we therefore asked if Pak2 responded in a similar manner. 10A.ErbB2 cells, stably transduced with GFP-Pak1 or GFP-Pak2, were stimulated with EGF or AP1510 for ten minutes and the distribution of Pak1 and Pak2 determined by immunofluorescence (Fig. S2A, S2B, S2C) and by subcellular fractionation (Fig. S2D). As expected, a substantial fraction of Pak1 redistributed from the cytoplasm to the nucleus upon EGF or AP1510 stimulation. In contrast, Pak2 was never detected in the nucleus.
Molecular pathways affected by Pak1 and Pak2
Paks have been implicated in regulating ERK signaling downstream of Ras, via phosphorylation of Mek1 and c-Raf (5, 12, 22, 23) and Pak1 has also been implicated in activating Akt via a scaffolding interaction with PDK1 (6, 13). Given that loss of Pak1, but not Pak2, impeded ErbB2 oncogenic signaling, we asked if these two Pak isoforms affected different signaling pathways. We examined the activation of ERK and Akt in 10A.ErbB2 cells bearing Pak1 or Pak2-specific shRNA. Interestingly, reduction of either Pak1 or Pak2 by shRNA had a profound negative effect on both ERK and Akt activation (Fig. 2A). In contrast, expression of a constitutively active form of Pak1 in a panel of breast cancer cell lines had only a modest effect in ERK and Akt activation (Fig. S3A). This result was expected, as Pak1 is generally thought to be necessary, but not sufficient, for activation of these pathways (4).
Figure 2. Effects of Pak depletion on proliferation and survival signaling pathways in MCF-10A cells.
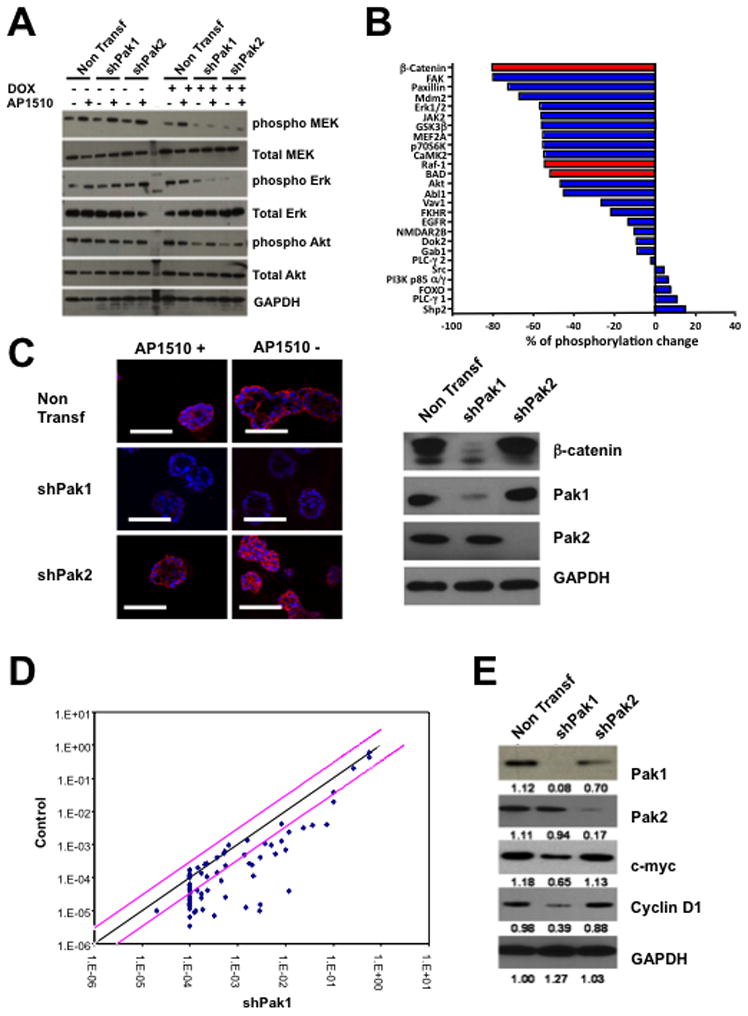
10A.ErbB2 cells stably expressing inducible Pak1 or Pak2 shRNAs were treated with doxycycline and stimulated with vehicle or 1μM AP1510. (A) The activities of Mek, ERK, and Akt was assessed by immunoblot using total and phosphospecific antibodies. (B) The activities of a panel of signaling proteins was assessed using a phospho-antibody array. Results are presented as changes in phosphorylation between control and Pak1-depleted cells (C) β-catenin levels in control and Pak1-depleted 10A.ErbB2 cells. (D) Pak1 depletion in 10A.ErbB2 cells negatively affects the transcription of β-catenin target genes. (E) Effects of Pak1 depletion on Cyclin D and c-Myc expression, as assessed by immunoblot.
As ERK and Akt activation was lost in cells depleted of either Pak1 or Pak2, but only Pak1 depletion affected transformation by ErbB2, we used a phospho-antibody array to examine additional signaling pathways that might be differentially regulated by these two Pak isoforms. An array containing several dozen ErbB2-relevant phospho-protein specific antibodies was probed with lysates from control and Pak1 knockdown 10A.ErbB2 cells. This experiment showed that phosphorylation of a number of known Pak1 direct and indirect substrates, including ERK, c-Raf, BAD, and Akt, were strongly reduced in Pak1 knock-down cells (Fig. 2B). The protein showing the greatest loss of phosphorylation was β-catenin, a protein that has recently been shown to be stabilized by Pak1-mediated phosphorylation (24, 25). Immunofluorescence and immunoblot analyses showed a severe reduction in total β-catenin (Fig. 2C, 2D). Similar effects were noted in SK-BR-3 and BT-474 cells, in which expression of a dominant negative form of Pak1, or treatment with the Pak inhibitor PF-3758309 (26) induced loss of β-catenin expression (Fig. S3B). This loss of β-catenin was accompanied by reduction in almost all known β-catenin target genes (Fig. 2E), including c-Myc and cyclin D (Fig. 2E). Similar effects on β-catenin, c-Myc, and cyclin D expression were noted in 10A.ErbB2 cells treated with the Pak inhibitor PF-3758309 (Fig. S3C).
As relatively few studies have measured the effect of ErbB2 activation on Wnt signaling (27–29), we asked if crossing-linking ErbB2 promoted relocalization of β-catenin from the plasma membrane to the nucleus in human mammary epithelial cells. As a positive control, we treated 10A.ErbB2 cells with Wnt3a, which resulted in relocalization of ~50% of the β-catenin to the nucleus within 1–2 hr of treatment (Fig. S4A). Similarly, a substantial (~40%) of β-catenin relocalized to the nucleus in response to ErbB2 activation. These results suggest that, in this cell type, ErbB2 activation activates Wnt signaling.
Loss of Pak1 impedes ErbB2-mediated carcinogenesis and is associated with reduced levels of β-catenin in vivo
To determine if the observed effects of Pak1 in ErbB2 signaling were relevant in vivo, we crossed MMTV-Neu mice with WT and Pak1−/− mice and followed the natural history of Neu:Pak1+/+ and Neu:Pak1−/− female mice over the course of two years. Pak1 deletion is well tolerated in mice, with no effects on general health, longevity, or fertility (30). Consistent with prior reports (31), half the MMTV-Neu mice developed palpable breast tumors by 9 months of age (Fig. 3A). In contrast, the MMTV- Neu/Pak1−/− mice showed a much longer latency to tumor formation and tumor growth, with half the mice showing detectable disease by 16 months. This result shows that Pak1 negatively affects the progression of ErbB2/Neu-initiated breast cancer in this mouse model.
Figure 3. Pak1 deficiency delays tumorigenesis and impacts proliferation, survival, migration and invasion of ErbB2/neu-expressing tumor cells.
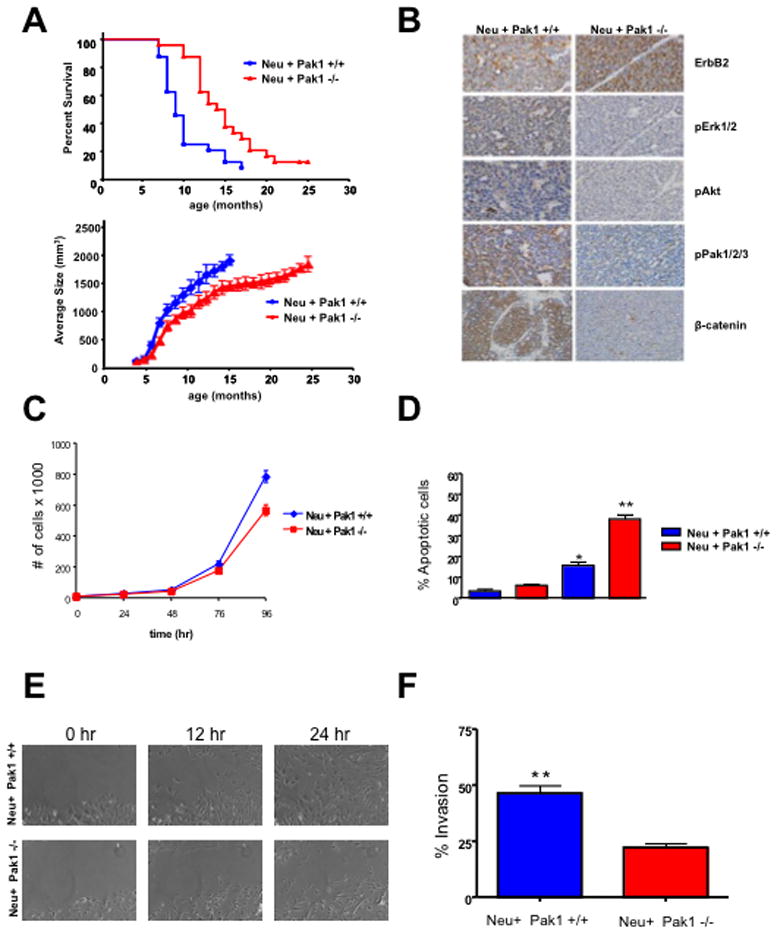
(A), Kaplan-Meier curve (upper panel) indicate significant increase in latency of tumor formation in MMTV-Neu:Pak1−/− (n = 22) versus MMTV-Neu:Pak1+/+ (n = 21) mice (P = 0.0015). Reduced tumor burden (lower panel) in MMTV-Neu:Pak1−/− mice (p<0.0001) (B) Representative example of MMTV-Neu:Pak1+/+ and MMTV-Neu:Pak1−/− breast cancer specimens stained for ErbB2, phospho-ERK, phospho-Akt, β-catenin, and phospho-Pak. (C) Proliferation of MMTV-Neu:Pak1+/+ and MMTV-Neu:Pak1−/− tumor derived cells. Cells were seeded, harvested and counted at 0, 24, 48, 72 and 96 h. The data are representative of three independent experiments. Points, mean; bars, SD. (D) Apoptosis of MMTV-Neu:Pak1+/+ and MMTV-Neu:Pak1−/− tumor derived cells. Apoptosis was measured calculating the percent of positive Annexin V-PE cells by flow cytometry. The data are representative of three independent experiments. Bars, SD. (E) in vitro scratch assay using MMTV-Neu:Pak1+/+ and MMTV-Neu:Pak1−/− tumor derived cells. (F) Matrigel invasion assay using MMTV-Neu:Pak1+/+ and MMTV-Neu:Pak1−/− tumor derived cells. The data are representative of three independent experiments. Bars, SD.
Immunohistochemical staining of tumor tissue revealed strong activity for ErbB2, ERK, Akt, β-catenin, and Pak in Neu:Pak1+/+ mice, and almost absent staining for active ERK, Akt, β-catenin, and Pak in Neu:Pak1−/− mice (Fig. 3B). These results show that, as in mammary epithelial cell lines (Fig. 2 and Fig. S3), Pak1 is required for the activation of ERK, Akt, and β-catenin downstream of ErbB2 in vivo. These results also suggest that other Pak isoforms (e.g., Pak2 and Pak3) are not redundant for Pak1 in ErbB2 signaling in mammary epithelial cells.
We derived epithelial cell lines from mammary tumors from Neu/Pak1+/+ and Neu:Pak1−/−mice and assessed their growth and signaling properties. Neu/Pak1+/+ cells grew faster than Neu:Pak1−/− cells (Fig. 3C), showed greater viability following treatment with actinomycin D (Fig. 3D), had greater motility (Fig. 3E, Supplemental movies 1 and 2), and were more invasive (Fig. 3F). Moreover, Neu:Pak1−/−cells displayed a defective cell cycle and a uni-lobular morphology when plated in a 3D Matrigel matrix and compared to Neu:Pak1+/+ and other breast cancer cell lines (Figure S5 and S6). Thus, many of the hallmark features of transformation were impeded in mouse-derived ErbB2 mammary epithelial cells lacking Pak1. As in 10A.ErbB2 cells, basal and EGF-stimulated levels of phospho-ERK, phospho-Akt, and total β-catenin were decreased in mammary epithelial cells derived from Neu:Pak1−/− mice (Fig. S7). Phosphoylation of β-catenin at a destabilizing site (S33) was augmented in Pak1−/− cells, whereas phosphorylation at a stabilizing, Pak1-catalyzed site (S675), was diminished, consistent with the overall reduction in β-catenin expression noted in these cells. Phosphorylation of glycogen synthase kinase 3β at an inhibitory site (S9) was also decreased in Pak1−/− cells, as might be expected in cells with reduced Akt activity. These data suggest that Pak1 is required for β-catenin stabilization in mammary epithelial cells derived from Neu mice.
Role of β-catenin in ErbB2-mediated signaling
Since Pak1 was required for β-catenin expression in mammary epithelial cells as well as for ErbB2-mediated oncogenesis, we asked if activators or inhibitors of Wnt signaling affected ErbB2 signaling. We first tested the effects of over-expressing wild-type or a stabilized form of β-catenin (S33Y), as well as a phosphomimetic mutant at the putative Pak1 phosphorylation site (S675E) in 10A.ErbB2 cells (24, 32). All these forms of β-catenin induced multi-lobular acini even in the absence of ErbB2 stimulation (Fig. 4A–C). Conversely, expression of β-catenin S675A, which lacks the Pak1 phosphorylation site, did not on its own induce this multi-lobular phenotype and blocked the ability of ErbB2 to confer this phenotype. Finally, treatment of cells with a small molecule inhibitor of Pak or β-catenin blocked formation of aberrant acini in ErbB2-stimulated cells, but the Pak inhibitor did not affect β-catenin S675E mediated transformation (Fig. 4E–H).
Figure 4. Downregulation of β-catenin blocks the effects of ErbB2 activation on acinar morphology.
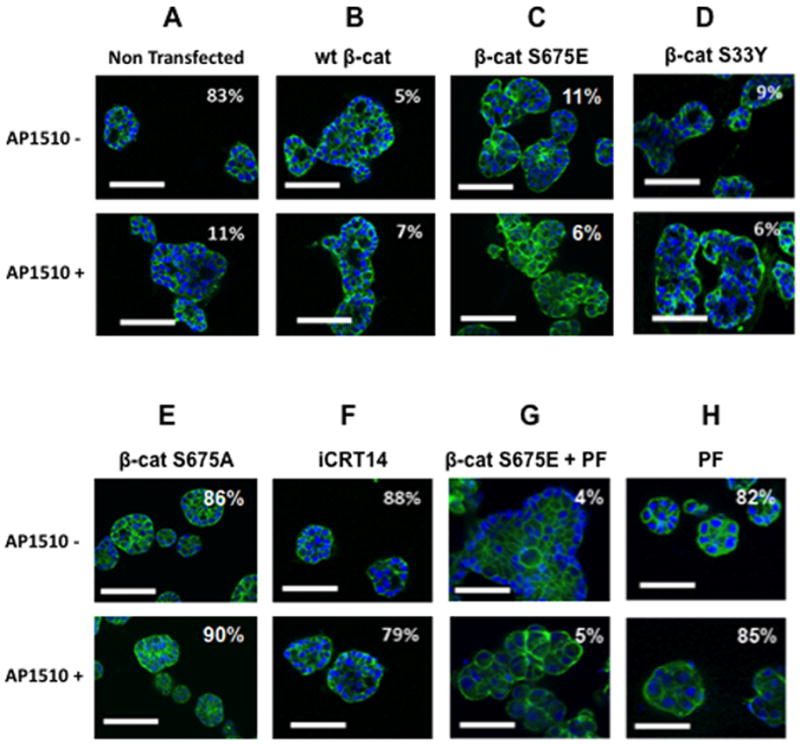
10A.ErbB2 cells transfected with expression vectors encoding the indicated forms of β-catenin, or treated with 25 μM iCRT14 or 100 nM PF-3758309 (PF), were plated atop reconstituted basement membrane. Cells were stimulated with vehicle or 1 μM AP1510 on day 3, and fixed on day 12 were stained with Oregon green-phalloidin, Ki-67, or anti-cleaved caspase-3. Percentage of unilamellar acini, Ki-67-positive, and anti-cleaved caspase-3-positive acini were scored based on assessment of 50 to 60 acini per well. Bar, 50 μm.
In-vitro synergy between Pak and β-catenin inhibitors
As Pak1 and β-catenin appear to act in a linked pathway downstream of ErbB2 (Fig. 5A), we next tested the effects of small molecule inhibitors of Pak1 and β-catenin, alone and together, on the survival of ErbB2-driven breast cancer cells. These compounds included PF-3758309, which potently suppresses both group A and B Paks (26), iCRT14 and iCRT3, which inhibit β-catenin interaction with T-cell factor (TCF)-4 (33), and JW55, a tankyrase inhibitor that suppresses the activity of the PARP domain of TNKS1/2, leading to the stabilization of AXIN2 followed by increased degradation of β-catenin (34). BT-474 and SK-BR3 cells were treated with varying concentrations of these inhibitors and the effect on cell survival was assessed following 3 days of treatment. Each compound alone, except JW55 (IC50 values of 7.3 μM and 6.6 μM in BT-474 and SK-BR3 respectively), had a nearly identical effect on cell survival. The IC50 values for iCRT14 in BT-474 and SK-BR3 cells were 71 nM and 107 nM respectively; the IC50 values for iCRT 3 were 52.3 nM and 82 nM respectively; whereas the IC50 values for PF-3758309 were 62.5 nM and 85.7 nM respectively (Figure 5B). When the compounds were co-administered, however, a marked synergistic effect was noted [combination index (CI) <0.5] (Figure 5B and Table 1). Co-administration of PF-3758309 and iCRT14 yielded CI values of 13.3 nM and 20.4 nM respectively, and co-administration of PF-3758309 and iCRT3 yielded CI values of 19.3 nM and 31 nM respectively, indicating a high degree of synergy. Interestingly, synergistic effects were not seen in cells treated with the tankyrase inhibitor JW55 plus PF-3758309, most likely because β-catenin, once stabilized by phosphorylation at S675, is not readily susceptible to destruction by cleavage. However, the combination of Pak and either of the iCRT β-catenin-targeting agents, which block interactions of β-catenin with TCF, did not merely produce cytostasis, but also resulted in cell death, increasing the frequency of apoptosis by nearly a factor of two (Figure 5C).
Figure 5. Synergistic interactions between Pak and β-catenin inhibitors.
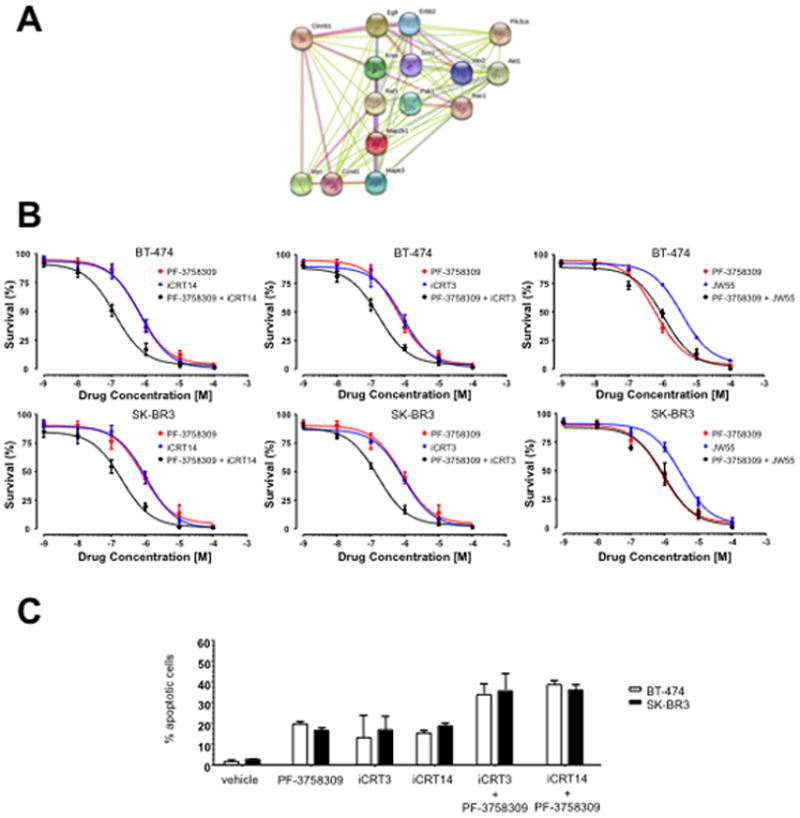
(A) Network of Pak1 signaling. (Graphical representation of some of the molecular pathways affected by Pak1 loss in 10A.ErbB2 cells). Pink lines represent connections based on experimental evidence; green and blue lines represent connections based on databases and text mining respectively. (B) Effect of Pak and β-catenin inhibitors on survival of BT-474 and SK-BR3 cells. Cells were treated with the indicated amounts of PF-3758309 and/or iCRT14, iCRT3 or JW55 for 4 days, cell viability was determined by trypan blue exclusion. (C) Combination of PF-3758309 and/or iCRT14 or iCRT3 treatment increases apoptosis of BT-474 and SK-BR3 cells. Cells were treated with the indicated amounts of PF-3758309 and/or iCRT14 or iCRT3 for 4 days, collected and apoptosis was measured calculating the percent of positive Annexin V-PE cells by flow cytometry. The data are representative of three independent experiments. Bars, SD.
Table 1. Synergistic effects of Pak plusβ-catenin inhibitors.
Summary results of drug interactions calculated as Chou-Talalay CI based on CellTiter-Blue viability determinations. CI values <1 indicate synergy, and <0.5 (in bold) strong synergy, between the two agents in producing cytotoxic effect
| Cell line | Inhibitors | Molar Ratio | CI (average ± std. dev) | |||
|---|---|---|---|---|---|---|
| ED50 | ED75 | ED95 | ||||
| BT-474 | PF-3758309 | iCRT14 | 1:1 | 0.368 ± 0.025 | 0.416 ± 0.026 | 0.512 ± 0.033 |
| PF-3758309 | iCRT3 | 1:1 | 0.396 ± 0.037 | 0.429 ± 0.032 | 0.543 ± 0.042 | |
| PF-3758309 | JW55 | 1:1 | 0.982 ± 0.015 | 1.002 ± 0.013 | 1.021 ± 0.098 | |
| SK-BR3 | PF-3758309 | iCRT14 | 1:1 | 0.314 ± 0.002 | 0.330 ± 0.114 | 0.417 ± 0.144 |
| PF-3758309 | iCRT3 | 1:1 | 0.402± 0.096 | 0.472± 0.103 | 0.582 ± 0.025 | |
| PF-3758309 | JW55 | 1:1 | 0.928 ± 0.016 | 0.982 ± 0.031 | 1.052 ± 0.028 | |
Inhibition of tumor growth by small molecule inhibitors of Pak and β-catenin
We next tested the effects of these signaling inhibitors on the growth of BT-474 xenografts. BT-474 cells were xenografted to SCID mice and tumors were allowed to form for seven days. The mice were then treated with vehicle, Pak inhibitor PF-3758309, β-catenin inhibitor iCRT14, or PF-3758309 plus iCRT14, for fifteen days. Tumor volumes were assessed every five days at which time the animals were sacrificed. Treatment with iCRT14 had a marked negative effect on tumor growth, yielding tumors of about one-half the volume, and one-fourth the weight, of tumors in untreated animals (Figure 6A, 6B, and 6C). PF-3758309 had an even more dramatic effect, essentially abrogating tumor growth. Interestingly, animals treated with the combined Pak and β-catenin inhibitors showed no tumor growth but also displayed extensive areas of necrosis in tumor remnants (Fig. 6C and 6E).
Figure 6. Inhibition of Pak or β-catenin impedes the tumorigenicity of ErbB2 positive breast cancer cells.
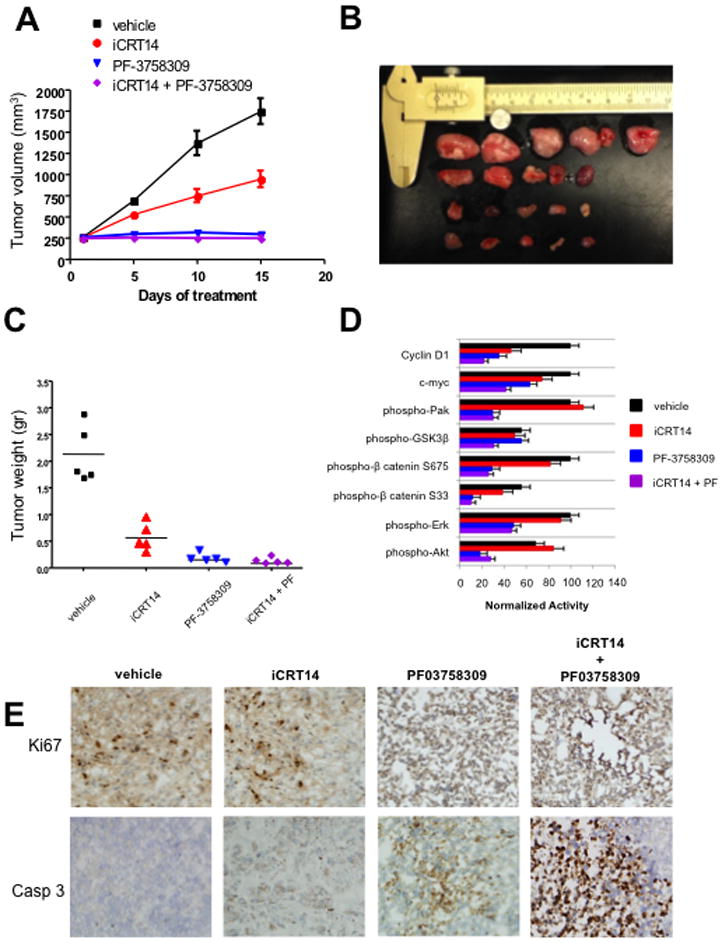
BT-474 cells were injected into the flanks of C.B17/IcrSCID mice. Ten days post-innoculation, the animals were treated with vehicle or inhibitors for 15 days. (A and B) Volumetric changes in tumor size between untreated mice (vehicle) and mice treated with inhibitors. (C) Average tumor size of untreated mice (vehicle) and mice treated with inhibitors. (D) Quantification of relative difference in activation of the indicated proteins between the different drug treatments. Each bar represents percent of inhibition relative to the controls. Data derived from immunoblots from 5 mice per antibody. Bars, SD. *p<0.05. (E) Representative example of tumor sections between untreated mice and mice treated with Pak inhibitor, β-catenin inhibitor and a combination of Pak and β-catenin inhibitors stained for cleaved caspase-3 and Ki67.
We then assessed signaling activity in these tumor tissues, using immunoblots (Fig. 6D). Treatment with the Pak inhibitor reduced Pak activity as expected, whereas treatment with the β-catenin inhibitor did not. Conversely, tumor tissues from iCRT14-treated mice showed loss of β-catenin targets (Myc and Cyclin D), but little change in pPak1, pERK, or pAkt. Combined treatment with PF-3758309 and iCRT14 reduced signaling activity in the ERK, Akt, and β-catenin pathways. Analysis of markers of cell proliferation and apoptosis revealed that PF-3758309 treatment prevented cell-cycle progression and induced apoptosis (Fig. 6E), whereas iCRT14 had a little effect on cell cycle progression but increased apoptosis. When co-administered, the inhibitors blocked cell-cycle progression and caused extensive apoptosis and necrosis (Fig. 6E).
Sensitization of trastuzumab resistant breast cancer cells by Pak and β-catenin inhibitors
As Pak1 and β-catenin appear to act in a linked pathway downstream of ErbB2, we assessed whether blocking these proteins would augment growth inhibition by an anti-ErbB2 agent. For these experiments, we chose the cell line BT-474R, known to be resistant to trastuzumab in vitro (35). As with parental BT-474 and SK-BR3 cells, we observed a synergistic inhibition of survival when BT-474R cells were treated with Pak plus β-catenin inhibitors (Fig. 7A). As expected, addition of trastuzumab alone had little effect on these resistant cells (Fig. 7B). Addition of either the Pak or the β-catenin inhibitor slightly increased apoptosis in BT-474R cells; however, in combination with trastuzumab, these inhibitors increased the number of apoptotic cells to >30% and ~40%, respectively. When combined, the Pak and β-catenin inhibitors increased apoptosis in BT-474R cells to ~15%; this effect was greatly accentuated by the addition of trastuzumab (Fig. 7B). These results suggest that inhibiting downstream components of ErbB2 signaling such as the Pak/β-catenin signaling axis, can re-sensitize trastuzumab-resistant cells to ErbB2-targeted therapeutic antibodies.
Figure 7. Sensitization of BT-474R cells to Trastuzumab.
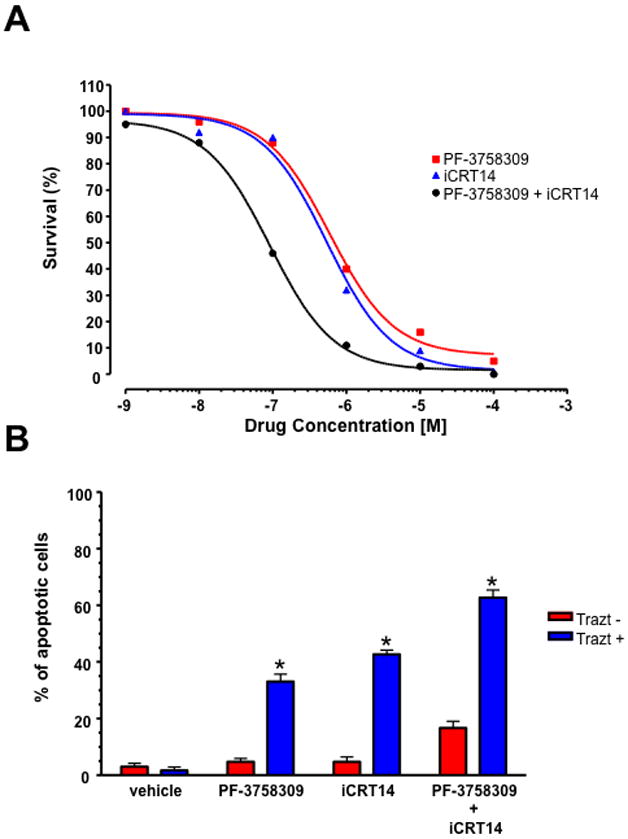
Effect of Pak and β-catenin inhibitors on survival of BT-474R cells. (A) Cells were treated with the indicated amounts of PF-3758309 and/or iCRT14 for 4 days and the viability was determined by trypan blue exclusion. (B) Combination of PF-3758309 and/or iCRT14 treatment sensitizes BT-474R cells to trastuzumab. Cells were treated with the indicated amounts of PF-3758309 and/or iCRT14 in the presence or absence of trastuzumab for 4 days, collected and apoptosis was measured calculating the percent of positive Annexin V-PE cells by flow cytometry. The data are representative of three independent experiments. Bars, SD. *p<0.05.
Discussion
Pak1 has previously been implicated in breast cancer for the following reasons: the PAK1 gene is frequently amplified in human breast cancer; PAK1 amplification is associated with resistance to tamoxifen; transgenic expression of an activated PAK1 allele induces transformation of mammary epithelial cells in culture and induces breast cancer in mice; and expression of dominant negative alleles, shRNAs, or treatment with Pak inhibitors, impede the growth and/or normalize the morphology of various breast cancer cell lines in tissue culture (13, 15–18). However, none of these studies examined the particular roles of the two most highly expressed group A Pak alleles, Pak1 and Pak2, nor did they employ clinically relevant inhibitors, nor establish key oncogenic signaling pathways activated by Pak in mouse breast cancer models. Here, we show that 1) blockade of Pak1, but not Pak2 expression or activity in vitro inhibits ErbB2 function; 2) loss of either Pak1 or Pak2 causes loss of both ERK and Akt activation, but loss of Pak1 uniquely causes destabilization of beta catenin; 5) loss of Pak1 in vivo delays oncogenesis and progression in MMTV-Neu mice; 6) restoring β-catenin expression by means of a stabilized β-catenin mutant or a Pak1 phospho-site mimic of β-catenin overcomes the effects of Pak inhibition; and 7) small molecule inhibitors of Pak or β-catenin block transformation by ErbB2 in cells and in mice. These results, coupled with recent work showing that Pak1 can phosphorylate β-catenin, establish an ErbB2-Pak-β-catenin linkage that is essential for transformation of mammary epithelial cells by ErbB2.
Of the six Pak isoforms, Pak1 in particular has garnered much attention with respect to tumorigenesis, as amplification of the PAK1 gene, with concomitant overexpression of the Pak1 protein, is commonly observed in human cancers of the breast, ovary, and bladder (9). Unlike other oncogenic serine/threonine protein kinases such as BRAF, activating point mutations or deletions of PAK1 have not been found in human cancers, despite the ability of activated PAK1 mutants to transform cells in vitro and in vivo. PAK1 gene amplification appears to be particularly relevant in human breast cancer, as it has been shown that such amplification is associated with resistance to tamoxifen treatment and decreased survival (36, 37). In addition, cells bearing such amplifications have recently been shown to be “addicted” to Pak1 signaling, with elevated sensitivity to Pak1 depletion (15). In contrast, the role of other Paks such as Pak2 in breast cancer is not well established. Pak1 and Pak2 have very similar catalytic properties (38) and, in some cases, share redundant functions. Interestingly, we found that both Pak1 and Pak2 are required for activation of ERK and Akt in 10.ErbB2 cells (Fig. 2). It is possible that these proteins heterodimerize or, in the case of ERK activation, that each Pak isoform targets a different element in the pathway, e.g., one Pak isoform might phosphorylate c-Raf, and the other might phosphorylate Mek. In either event, effects on ERK and Akt signaling cannot explain our observation that loss of Pak1, but not Pak2, impedes ErbB2 oncogenic signaling. Instead, our results suggest that other pathways, such as the Wnt/β-catenin pathway, are more likely to mediate these effects, as only Pak1 depletion affects β-catenin levels and β-catenin target gene transcription in the cell types we examined. Why is Pak1 unique in this respect? Although the catalytic properties of Pak1 and Pak2 are indistinguishable in vitro, the two proteins localize differently (39, 40). For example, Pak1 is driven into the nucleus following EGF stimulation of MCF-7 and MDA-MB-435 human breast cancer cells (20) and in MCF10A cells ((21) and Fig. S2), whereas Pak2 is not (Fig. S2). In addition, the effects of gene deletion are quite different for Pak1 and Pak2: deletion of the former is associated with a mild phenotype whereas deletion of the latter results in early embryonic lethality (8). Why Pak1, but not Pak2, affects β-catenin levels in mammary epithelial cells is unknown, and it will be of interest to determine whether these different behaviors reside in different intrinsic substrate selection or differences in the non-catalytic, N-terminal regulatory domains of these kinases.
The role of the Wnt/β-catenin pathway in ErbB2 signaling in breast cancer is incompletely understood. In general, ErbB2 positivity in breast cancer is associated with a luminal phenotype, whereas Wnt activation is more commonly associated with a basal phenotype (41). However, multiple lines of evidence suggest important links between ErbB2 and β-catenin signaling. First, ErbB2 physically associates with β-catenin, and such complexes are associated with human infiltrating ductal breast and also with MMTV-c-Neu and MMTV-Wnt-1 mouse models of breast cancer (28). Second, ErbB2 phosphorylates β-catenin at Tyr 654, leading to dissociation of the E-cadherin-β-catenin membrane complex and increased signaling to Wnt target genes such as cyclin D1 (29). In addition, ErbB2 has been reported to transcriptionally activate expression of the Wnt pathway target gene Jab1 via an Akt/β-catenin pathway in breast cancer cells (42). These observations are supported by recent publications showing a positive correlation between HER2/neu expression and nucleocytoplasmic (i.e., non plasma membrane) β-catenin in node-positive carcinomas (P = 0.02) and in HER2/neu-induced mouse mammary tumors, with activation of Wnt pathway genes (27), as well as eradication of breast tumor initiating cells by pharmacological inhibitors of Wnt/β-catenin signaling in this mouse model (43).
Recently, it has been reported that Pak1 has a direct role in β-catenin stabilization in colon cancer cells, via phosphorylation of β-catenin at Ser 675 (24, 44) and Ser 663/675 (45). As we have previously shown that ErbB2 activates Pak1 (16), we here propose that Pak1 is a required signaling element linking ErbB2 activation to β-catenin in mammary epithelial cells, and that such activation is necessary for oncogenesis. This view is supported by the loss of β-catenin and target gene activation in Pak1-depleted mammary epithelial cells or Pak1-deleted mammary tissue, and the blockade of tumor growth by Pak or β-catenin inhibition seen in animals xenografted with ErbB2-positive tumor cells.
Small molecule Pak inhibitors have recently entered clinical trials and it should soon be possible to evaluate the potential of such agents in human cancer. We suggest that such agents may be useful in two general settings: cancers that have 11q13 amplification, such as a large fraction of breast, ovarian, thyroid, and bladder cancers, and cancers driven by oncogenes or loss of tumor suppressors for which Pak1 is thought to be an obligate signaling element, such as Kras, ErbB2, NF1, and NF2 (4, 9, 46). Given the results reported in this study, it will also be of interest to determine if such Pak inhibitors are also useful in the setting of activated Wnt signaling, and/or if such agents could be effectively combined with Wnt pathway inhibitors.
Supplementary Material
Acknowledgments
We thank Brion Murray (Pfizer) for PF-3758309, Jose Baselga (Massachusetts General Hospital) for the BT-474R cells, Igor Astsaturov (Fox Chase Cancer Center) for assistance with synergy calculations, Fang Zhu (Fox Chase Cancer Center Biostatistics Facility) for statistical analyses, Christy Ong, Kamran Ahmad and Daniel Feinberg for technical assistance, and Dr. Zhimin (James) Luo (MD Anderson Cancer Center) for providing the pcDNA3.1 Flag-β-catenin vector.
Financial Support: This work was supported by grants from the NIH to JC (R01 CA58836 and R01 CA098830) and to the Fox Chase Cancer Center (P30 CA006927)
Footnotes
No potential conflict of interests are disclosed
References
- 1.Carlsson J, Nordgren H, Sjostrom J, Wester K, Villman K, Bengtsson NO, et al. HER2 expression in breast cancer primary tumours and corresponding metastases. Original data and literature review. Br J Cancer. 2004;90:2344–8. doi: 10.1038/sj.bjc.6601881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moasser MM. The oncogene HER2; Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469–87. doi: 10.1038/sj.onc.1210477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcotte R, Muller WJ. Signal transduction in transgenic mouse models of human breast cancer--implications for human breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:323–35. doi: 10.1007/s10911-008-9087-3. [DOI] [PubMed] [Google Scholar]
- 4.Arias-Romero LE, Chernoff J. A tale of two Paks. Biol Cell. 2008;100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- 5.Chow HY, Jubb AM, Koch JN, Jaffer ZM, Stepanova D, Campbell DA, et al. p21-activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Research. 2012 doi: 10.1158/0008-5472.CAN-12-2246. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higuchi M, Onishi1 K, Kikuchii C, Gotoh Y. Scaffolding function of PAK in the PDK1–Akt pathway. Nat Cell Biol. 2008;10:1356–64. doi: 10.1038/ncb1795. [DOI] [PubMed] [Google Scholar]
- 7.Mao K, Kobayashi S, Jaffer ZM, Huang Y, Volden P, Chernoff J, et al. Regulation of Akt/PKB activity by P21-activated kinase in cardiomyocytes. J Mol Cell Cardiol. 2008;44:429–34. doi: 10.1016/j.yjmcc.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann C, Shepelev M, Chernoff J. The genetics of Pak. J Cell Sci. 2004;117:4343–54. doi: 10.1242/jcs.01392. [DOI] [PubMed] [Google Scholar]
- 9.Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63. doi: 10.1007/s10555-008-9168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manser E, Leung T, Salihuddin H, Zhao Z-s, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–6. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- 11.Guo F, Debidda M, Yang L, Williams DA, Zheng Y. Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. J Biol Chem. 2006;281:18652–9. doi: 10.1074/jbc.M603508200. [DOI] [PubMed] [Google Scholar]
- 12.Tang Y, Chen Z, Ambrose D, Liu J, Gibbs JB, Chernoff J, et al. Kinase deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol Cell Biol. 1997;17:4454–64. doi: 10.1128/mcb.17.8.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q, Mullins SR, Sloane BF, Mattingly RR. p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia. 2008;10:314–29. doi: 10.1593/neo.07970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dadke D, Fryer BH, Golemis EA, Field J. Activation of p21-activated kinase 1-nuclear factor kappaB signaling by Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor during cellular transformation. Cancer Res. 2003;63:8837–47. [PubMed] [Google Scholar]
- 15.Ong CC, Jubb AM, Haverty PM, Zhou W, Tran V, Truong T, et al. Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7177–82. doi: 10.1073/pnas.1103350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arias-Romero LE, Villamar-Cruz O, Pacheco A, Kosoff R, Huang M, Muthuswamy SK, et al. A Rac-Pak signaling pathway is essential for ErbB2-mediated transformation of human breast epithelial cancer cells. Oncogene. 2010;29:5839–49. doi: 10.1038/onc.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shrestha Y, Schafer EJ, Boehm JS, Thomas SR, He F, Du J, et al. PAK1 is a breast cancer oncogene that coordinately activates MAPK and MET signaling. Oncogene. 2012;31:3397–408. doi: 10.1038/onc.2011.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balasenthil S, Sahin AA, Barnes CJ, Wang RA, Pestell RG, Vadlamudi RK, et al. p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J Biol Chem. 2004;279:1422–8. doi: 10.1074/jbc.M309937200. [DOI] [PubMed] [Google Scholar]
- 19.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–92. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh RR, Song C, Yang Z, Kumar R. Nuclear localization and chromatin targets of p21-activated kinase 1. J Biol Chem. 2005;280:18130–7. doi: 10.1074/jbc.M412607200. [DOI] [PubMed] [Google Scholar]
- 21.Lightcap CM, Kari G, Arias-Romero LE, Chernoff J, Rodeck U, Williams JC. Interaction with LC8 is required for Pak1 nuclear import and is indispensable for zebrafish development. PLoS One. 2009;4:e6025. doi: 10.1371/journal.pone.0006025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slack-Davis JK, Eblen ST, Zecevic M, Boerner SA, Tarcsafalvi A, Diaz HB, et al. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J Cell Biol. 2003;162:281–91. doi: 10.1083/jcb.200212141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King AJ, Sun H, Diaz B, Barnard D, Miao W, Bagrodia S, et al. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature. 1998;396:180–3. doi: 10.1038/24184. [DOI] [PubMed] [Google Scholar]
- 24.Zhu G, Wang Y, Huang B, Liang J, Ding Y, Xu A, et al. A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene. 2012;31:1001–12. doi: 10.1038/onc.2011.294. [DOI] [PubMed] [Google Scholar]
- 25.He H, Huynh N, Liu KH, Malcontenti-Wilson C, Zhu J, Christophi C, et al. P-21 activated kinase 1 knockdown inhibits beta-catenin signalling and blocks colorectal cancer growth. Cancer Lett. 2012;317:65–71. doi: 10.1016/j.canlet.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 26.Murray BW, Guo C, Piraino J, Westwick JK, Zhang C, Lamerdin J, et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci U S A. 2010;107:9446–51. doi: 10.1073/pnas.0911863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khalil S, Tan GA, Giri DD, Zhou XK, Howe LR. Activation status of Wnt/β-catenin signaling in normal and neoplastic breast tissues: relationship to HER2/neu expression in human and mouse. PLoS One. 2012;7:e33421. doi: 10.1371/journal.pone.0033421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroeder JA, Adriance MC, McConnell EJ, Thompson MC, Pockaj B, Gendler SJ. ErbB-beta-catenin complexes are associated with human infiltrating ductal breast and murine mammary tumor virus (MMTV)-Wnt-1 and MMTV-c-Neu transgenic carcinomas. J Biol Chem. 2002;277:22692–8. doi: 10.1074/jbc.M201975200. [DOI] [PubMed] [Google Scholar]
- 29.Wang K, Ma Q, Ren Y, He J, Zhang Y, Zhang Y, et al. Geldanamycin destabilizes HER2 tyrosine kinase and suppresses Wnt/beta-catenin signaling in HER2 overexpressing human breast cancer cells. Oncol Rep. 2007;17:89–96. [PubMed] [Google Scholar]
- 30.Allen JD, Jaffer ZM, Burgin S, Hofmann C, Sells MA, Derr-Yellin E, et al. p21-activated kinase 1 is required in mast cells for FcεRI-mediated inflammatory responses. Blood. 2009;113:2695–705. doi: 10.1182/blood-2008-06-160861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–15. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Shao Y, Tong Y, Shen T, Zhang J, Li Y, et al. Nucleo-cytoplasmic shuttling of PAK4 modulates beta-catenin intracellular translocation and signaling. Biochim Biophys Acta. 2012;1823:465–75. doi: 10.1016/j.bbamcr.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 33.Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S, et al. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A. 2011;108:5954–63. doi: 10.1073/pnas.1017496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waaler J, Machon O, Tumova L, Dinh H, Korinek V, Wilson SR, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Research. 2012;72:2822–32. doi: 10.1158/0008-5472.CAN-11-3336. [DOI] [PubMed] [Google Scholar]
- 35.Scaltriti M, Serra V, Normant E, Guzman M, Rodriguez O, Lim AR, et al. Antitumor activity of the Hsp90 inhibitor IPI-504 in HER2-positive trastuzumab-resistant breast cancer. Mol Cancer Ther. 2011;10:817–24. doi: 10.1158/1535-7163.MCT-10-0966. [DOI] [PubMed] [Google Scholar]
- 36.Rayala SK, Molli PR, Kumar R. Nuclear p21-activated kinase 1 in breast cancer packs off tamoxifen sensitivity. Cancer Res. 2006;66:5985–8. doi: 10.1158/0008-5472.CAN-06-0978. [DOI] [PubMed] [Google Scholar]
- 37.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rennefahrt UE, Deacon SW, Parker SA, Devarajan K, Beeser A, Chernoff J, et al. Specificity profiling of Pak kinases allows identification of novel phosphorylation sites. J Biol Chem. 2007;282:15667–78. doi: 10.1074/jbc.M700253200. [DOI] [PubMed] [Google Scholar]
- 39.Huang Z, Ling J, Traugh JA. Localization of p21-activated protein kinase gamma-PAK/Pak2 in the endoplasmic reticulum is required for induction of cytostasis. J Biol Chem. 2003;278:13101–9. doi: 10.1074/jbc.M212557200. [DOI] [PubMed] [Google Scholar]
- 40.Dharmawardhane S, Sanders LC, Martin SS, Daniels RH, Bokoch GM. Localization of p21-activated kinase 1 (Pak1) to pinocytotic vesicles and cortical actin structures in stimulated cells. J Cell Biol. 1997;138:1265–78. doi: 10.1083/jcb.138.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Incassati A, Chandramouli A, Eelkema R, Cowin P. Key signaling nodes in mammary gland development and cancer: beta-catenin. Breast Cancer Res. 2010;12:213. doi: 10.1186/bcr2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsu MC, Chang HC, Hung WC. HER-2/neu transcriptionally activates Jab1 expression via the AKT/beta-catenin pathway in breast cancer cells. Endocr Relat Cancer. 2007;14:655–67. doi: 10.1677/ERC-07-0077. [DOI] [PubMed] [Google Scholar]
- 43.Hallett RM, Kondratyev MK, Giacomelli AO, Nixon AM, Girgis-Gabardo A, Ilieva D, et al. Small molecule antagonists of the wnt/beta-catenin signaling pathway target breast tumor-initiating cells in a her2/neu mouse model of breast cancer. PloS one. 2012;7:e33976. doi: 10.1371/journal.pone.0033976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun J, Khalid S, Rozakis-Adcock M, Fantus IG, Jin T. P-21-activated protein kinase-1 functions as a linker between insulin and Wnt signaling pathways in the intestine. Oncogene. 2009;28:3132–44. doi: 10.1038/onc.2009.167. [DOI] [PubMed] [Google Scholar]
- 45.Park MH, Kim DJ, You ST, Lee CS, Kim HK, Park SM, et al. Phosphorylation of beta-catenin at serine 663 regulates its transcriptional activity. Biochem Biophys Res Commun. 2012;419:543–9. doi: 10.1016/j.bbrc.2012.02.056. [DOI] [PubMed] [Google Scholar]
- 46.Chan PM, Manser E. PAKs in human disease. Prog Mol Biol Transl Sci. 2012;106:171–87. doi: 10.1016/B978-0-12-396456-4.00011-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.