

Abstract
Malignant peripheral nerve sheath tumor (MPNST) is very uncommon tumor of parotid gland and it is an uncommon spindle cell sarcoma accounting for approximately 5% of all soft-tissue sarcoma. There is strong association between MPNSTs and neurofibromatosis (NF-1) and previous irradiation. Structural abnormality of chromosome 17 is associated with NF-1 and so MPNST. We present a case of a 78-year-old male presenting with slowly growing parotid mass who underwent tumor resection.
Keywords: Facial nerve, malignant peripheral nerve sheath tumor, parotid gland
INTRODUCTION
Malignant peripheral nerve sheath tumor (MPNST) of neck region is an extremely rare entity, especially, when presenting as a parotid mass. However, it is of diagnostic importance because it is one of the most aggressive malignant soft-tissue sarcomas and it is also called as neurofibrosarcoma as it arises from the neuroectodermal lining of the peripheral nerves.
MPNSTs are often associated with Von-Recklinghausen's disease of skin. The incidence of MPNSTs with Von-Recklinghausen's disease of skin is 30-50% and with previous irradiation is 4-11% while rest of the cases arise de novo.[1,2]
CASE REPORT
A 78-year-old male presented with a gradually increasing painful swelling over left side of the neck since 8 years. Physical examination revealed a mass in the lateral region of the neck behind the sternocleidomastoid muscle. The mass measured 7 cm in diameter and was tender and non-mobile. The overlying skin was unremarkable. Facial nerve function and the remainder of head and neck examination were normal.
Fine needle aspiration cytology (FNAC) was performed, which was suggestive of a cystic lesion of the salivary gland. Computed tomography (CT) scan showed centrally necrotic rounded mass lesion arising from parotid gland [Figure 1]. The lesion was almost completely replacing the outer lobe of left parotid gland and the inner lobe was normal. Anteriorly the lesion was seen abutting the masseter muscle and fat planes. CT findings suggested neoplastic lesion of the left parotid gland with involvement of bilateral submandibular nodes and left level II lymph nodes. Patient was taken up for surgery. Intra-operatively a 7 × 6 cm mass was seen involving the left parotid gland and all the branches of the facial nerve except the cervical branch. The facial nerve along with its branches was traced anteriorly. The entire tumor along with the parotid gland and the facial nerve was excised en block. The cervical branch of facial nerve was preserved.
Figure 1.
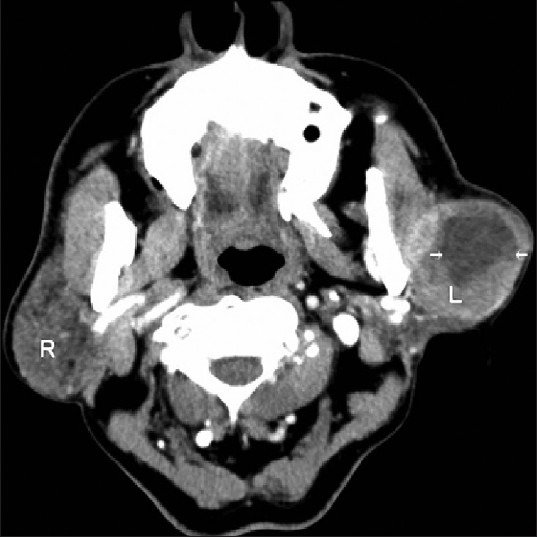
Computed tomography scan-neoplastic lesion of left parotid gland (arrows)
Histopathological examination revealed a tumor composed of spindle shaped cells with elongated hyperchromatic nuclei, arranged in long and short fascicles [Figure 2]. At places cells were arranged around hyaline bands in the form of cords and at places they were arranged in whorls. Some of the tumor cells had epitheloid appearance, while some cells had marked nuclear pleomorphism with tumor giant cells. In some areas, chondroid differentiation was seen. The stroma showed extensive areas of necrosis and hemorrhage. The blood vessels showed hyalinized walls. Mitotic figures were more than five per high-power field and areas of compressed parotid gland were present at the periphery of tumor.
Figure 2.
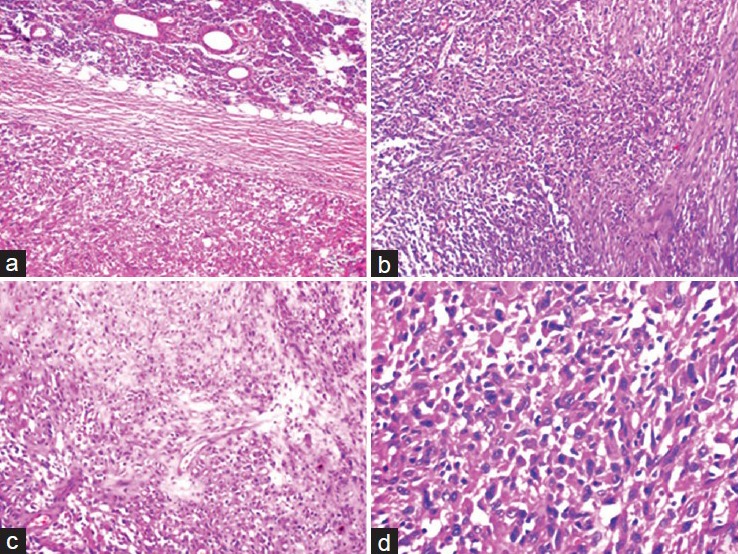
(a) Tumor with normal parotid gland (H and E, ×100), (b) Tumor showing spindle shaped cells arranged in long and short fascicles (H and E, ×100), (c) Tumor showing chondroid differentiation in some areas (H and E, ×100). (d) High power view of the tumor cells showing pleomorphism. tumor giant cells and abnormal mitosis (H and E, ×400)
Immunohistochemically, the tumor cells showed diffuse and strong positivity for S-100, (Dako), however, were negative for Pan-cytokeratin (Dako, AE1/AE3), Desmin (Dako, D33), and Human Melanoma Black (HMB) 45 (Dako) [Figure 3] and Vimentin (Dako). With the above features, a diagnosis of MPNST was made.
Figure 3.
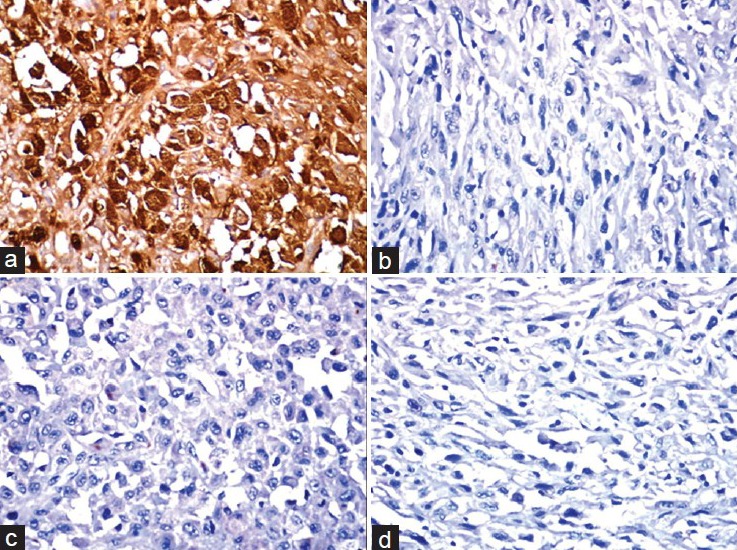
Immunohistochemical staining showing (a) Positivity of tumor cells with S-100 and negativity for (b) Pan cytokeratin, (c) HMB 45, and (d) Vimentin (×400)
Post-operatively, the patient had facial palsy [Figure 4] and distant metastases were not found on whole body scan after 6 months of follow-up.
Figure 4.
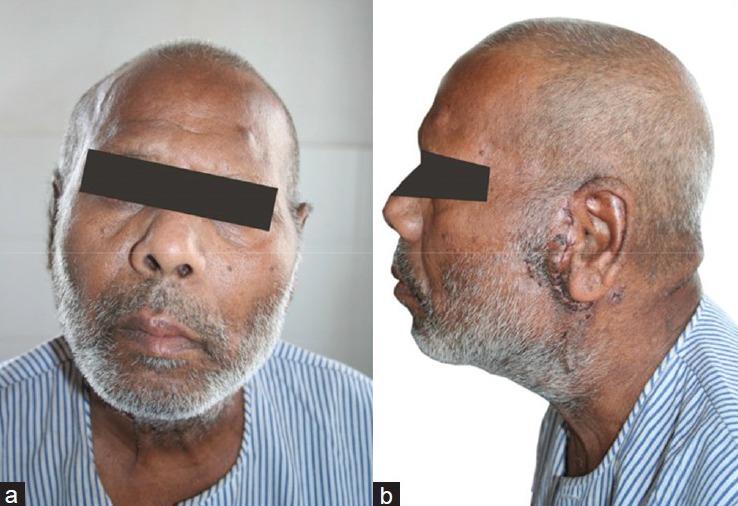
Post-operative photograph showing (a) Location of excised tumor, (b) Patient with post-operative facial palsy
DISCUSSION
MPNSTs also called as neurofibrosarcomas are malignant tumors developing from cells present in peripheral nerve tissue.[3] The MPNST accounts for approximately 5-10% of all soft-tissue sarcomas and about one-fourth to one-half occur in the setting of neurofibromatosis (NF-1).[3]
The MPNST is typically a disease of adult life, as most tumors occur in patients of 20-50 years of age.[3] The age of onset tends to be lower in cases associated with NF-1.[4]
The most common anatomical sites for MPNST include extremities, head and neck region and trunk. Parotid gland involvement is very rare in this disease and it is very difficult to differentiate other parotid tumors from MPNSTs on clinical or gross examination. FNAC is also not very contributory in this respect.
Das Gupta et al.,[5] in their study on a series of 303 benign nerve sheath tumors found that 44.9% of the tumors occurred in head and neck region. In the neck, they may arise medially or laterally. Medially, they arise from the last four cranial nerves (glossopharyngeal, vagus, accessory, and hypoglossal) or the sympathetic chain. Laterally, they arise from cutaneous or muscular branches of the cervical plexus or the brachial plexus.
MPNSTs have to be differentiated from neurofibromas sand schwannomas. Schwannomas are encapsulated tumors of nerve sheath and their growth usually does not involve the nerve per se. There is palisading of nuclei in schwannomas. MPNSTs are hyper-cellular and show pleomorphism.[3]
It is difficult to differentiate MPNST from neurofibromas. However, neurofibromas are cellular; have low or no mitotic activity and show minimal nuclear atypia. Low-grade MPNSTs arising from neurofibroma is diagnosed when there is generalized nuclear atypia, increased cellularity and unusually low-levels of mitotic activity.[3]
Wide surgical excision of tumor and radical neck dissection is the treatment of choice for MPNSTs arising in head and neck region.
According to Wise et al.[6] the prognosis of MPNSTs of head and neck region is poor and have 5 year survival rate of 20-50%.
Large tumor size (>5 cm), presence of neurofibromatosis, and total resection are most important prognostic indicators for MPNST.[7]
Histologic findings such as cellularity, pleomorphism, mitotic activity, and size also contributes in assessing the prognosis of the tumor.[8] For surgeons the invasion of facial nerve and parotid gland is suggestive of malignant tumor of parotid gland.
Absence of neurofibromatosis and complete removal of tumor with radical neck dissection is probably the reason for better prognosis in our case.
CONCLUSION
We present this case to show that an unusual tumor can be encountered when dealing with a parotid mass. MPNSTs arising as parotid mass are very rare. The pathologists and surgeons should keep this in mind when dealing with a patient of parotid mass. Wide surgical resection and adjuvant radiotherapy is the choice of treatment.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Neville H, Corpron C, Blakely ML, Andrassy R. Pediatric neurofibrosarcoma. J Pediatr Surg. 2003;38:343–6. doi: 10.1053/jpsu.2003.50105. [DOI] [PubMed] [Google Scholar]
- 2.Kenali MS, Bridger PG, Baldwin M, Smee R. Malignant peripheral nerve sheath tumour of the tongue. Aust N Z J Surg. 1999;69:243–6. doi: 10.1046/j.1440-1622.1999.01541.x. [DOI] [PubMed] [Google Scholar]
- 3.Weiss SW, Goldblum JR. Malignant tumors of the peripheral nerves. In: Weiss SW, Goldblum JR, editors. Enzinger and Weiss's Soft Tissue Tumors. 2nd ed. Philadelphia: Mosby; 1998. pp. 903–44. [Google Scholar]
- 4.Hruban RH, Shiu MH, Senie RT, Woodruff JM. Malignant peripheral nerve sheath tumors of the buttock and lower extremity. A study of 43 cases. Cancer. 1990;66:1253–65. doi: 10.1002/1097-0142(19900915)66:6<1253::aid-cncr2820660627>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 5.Das Gupta TK, Brasfield RD, Strong EW, Hajdu SI. Benign solitary schwannomas (neurilemomas) Cancer. 1969;24:355–66. doi: 10.1002/1097-0142(196908)24:2<355::aid-cncr2820240218>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 6.Wise JB, Patel SG, Shah JP. Management issues in massive pediatric facial plexiform neurofibroma with neurofibromatosis type 1. Head Neck. 2002;24:207–11. doi: 10.1002/hed.10001. [DOI] [PubMed] [Google Scholar]
- 7.Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006–21. doi: 10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Colreavy MP, Lacy PD, Hughes J, Bouchier-Hayes D, Brennan P, O’Dwyer AJ, et al. Head and neck schwannomas: A 10 year review. J Laryngol Otol. 2000;114:119–24. doi: 10.1258/0022215001905058. [DOI] [PubMed] [Google Scholar]