

Abstract
Ellis-van Creveld (EVC) syndrome is a genetic disorder with autosomal recessive transmission, which may clinically present as small stature, short limbs, fine sparse hair, hypoplastic fingernails, multiple musculofibrous frenula, conical teeth, hypoplasia of the enamel, hypodontia, and malocclusion. Heart defects, especially abnormalities of atrial septation, have been found in about 60% of cases. The mutation in EVC and EVC2 gene is responsible for this syndrome. The presence of multiple orodental findings makes this syndrome important for dentists. The aim of this article is to present a rare case of EVC syndrome in a 10-year-old girl along with the review of literature.
Keywords: Chondroectodermal dysplasia, Ellis-van Creveld syndrome, polydactyly
INTRODUCTION
Ellis-van Creveld (EVC) syndrome is an autosomal recessive skeletal dysplasia, which is characterized by short stature, retarded growth, polydactyly, and ectodermal and heart defects. The first case of EVC syndrome was reported by McIntosh in 1933, but Richard W.B. Ellis of Edinburgh and Simon van Creveld of Amsterdam in 1940, first described this condition and defined it as EVC syndrome. It is also known as chondroectodermal dysplasia and mesoectodermal dysplasia.
Birth prevalence of EVC has been estimated to be 7 per 10,00,000 population and more than 300 cases has been reported in the literature.[1] Baujat G et al. described that about 150 cases were reported between 1940 and 2008,[2] whereas Zanwill KM et al. documented that among 300 reported cases of EVC syndrome, 93% had ectodermal dysplasia.[3] EVC syndrome is found predominantly in Amish community, Pennsylvania, US, where largest pedigree has been described, that is, 52 cases in 30 sib ships. In this community, the incidence is estimated to be 5 cases per 1000 live births and the frequency of carriers may be as high as 13%.[4] The exact prevalence of this rare syndrome is still unknown. This syndrome doesn’t have any gender predilection[5] but parental consanguinity was reported in 30% of cases.[1]
EVC syndrome is one of a group of diseases called ciliopathies, which is caused by abnormalities in the primary cilia (these are vibrating, hair-like projections on the surface of cells). Cilia dysfunction in EVC syndrome has been linked to a mutation in two adjacent genes on chromosome 4. The genes are EVC and EVC2, which play a role in the development of cilia. The EVC gene, which was identified in 2000, controls the development of (codes for) EVC protein, while EVC2, which was identified in 2002, codes for a protein called limbin. It has been found that the affected EVC individuals with mutations in EVC or EVC2 are phenotypically indistinguishable.[6]
CASE REPORT
A 10-year-old girl of Indian origin reported to our institution with the chief complaint of malformed teeth in front region of upper and lower jaws. The patient was the first child of non-consanguineous and normally developed parents. Her birth was at full term normal delivery with birth weight around 2.5 kg. There was no history of presence of natal or neonatal teeth. She had a younger brother with no congenital abnormality. Medical history revealed that she had some atrial septal defect and was undergoing treatment for that problem. Her psychosomatic and mental developmental were within normal limits.
On general physical examination, the patient had short stature with 110 cm height. Examination of the hands and feet revealed bilateral postaxial polydactyly [Figures 1 and 2]. In right foot, polydactyly with syndactyly was present. Lower limbs were deformed with outward bending of knee (genu valgum) [Figure 3]. Nails were hypoplastic and hairs were thin and sparse.
Figure 1.
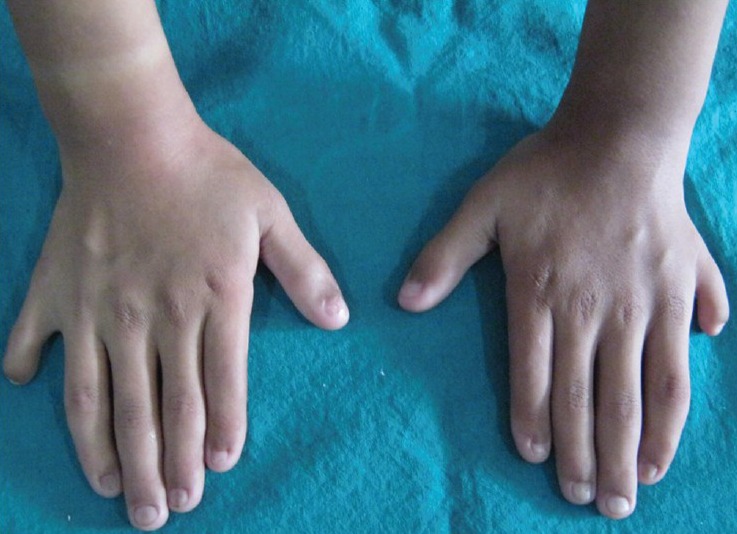
Hands revealing postaxial polydactyly
Figure 2.
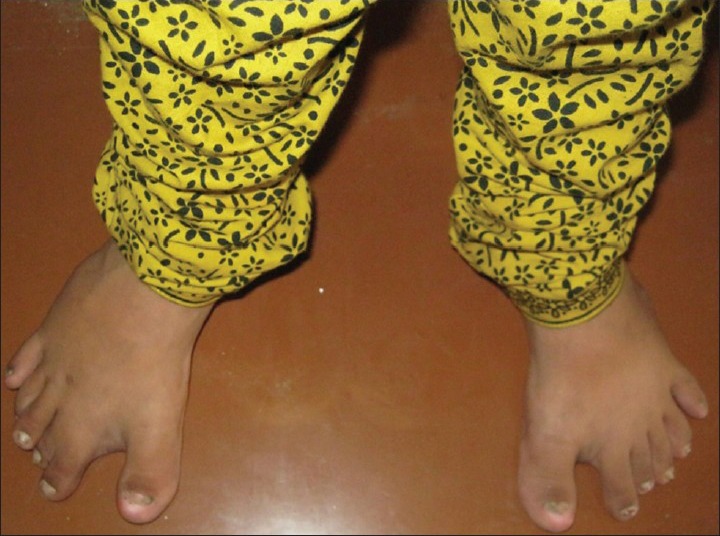
Feet revealing postaxial polydactyly
Figure 3.
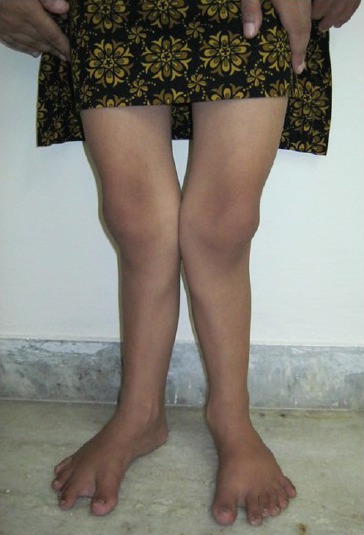
Outward bending of knees (genu valgum)
Intra-oral examination along with panoramic radiography revealed multiple missing teeth (12, 22, 31, 32, 41, 42) in mandibular and maxillary anterior region and a retained mandibular deciduous incisor (81) [Figures 4 and 5]. Maxillary central incisors (12, 21) were malformed and 21 had a deep carious labial fissure. Multiple frenulum and labiogingival frenum hypertrophy were present in maxillary and mandibular anterior region.
Figure 4.
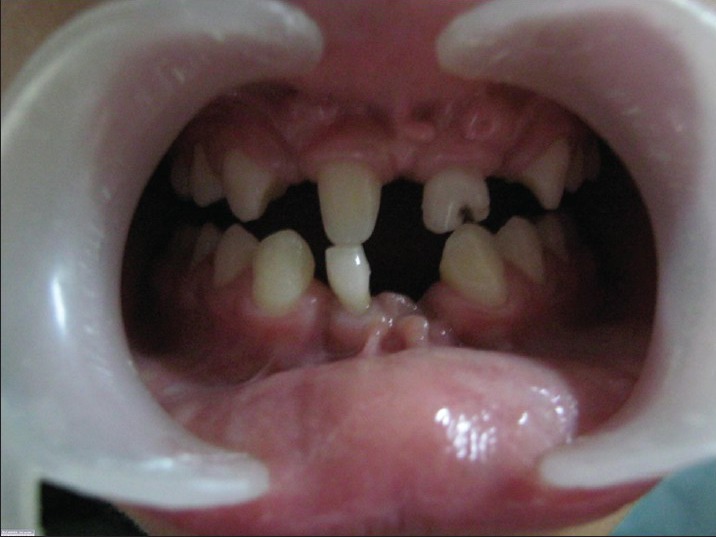
Intraoral photograph revealing multiple missing and malformed teeth in both maxillary and mandibular arches
Figure 5.
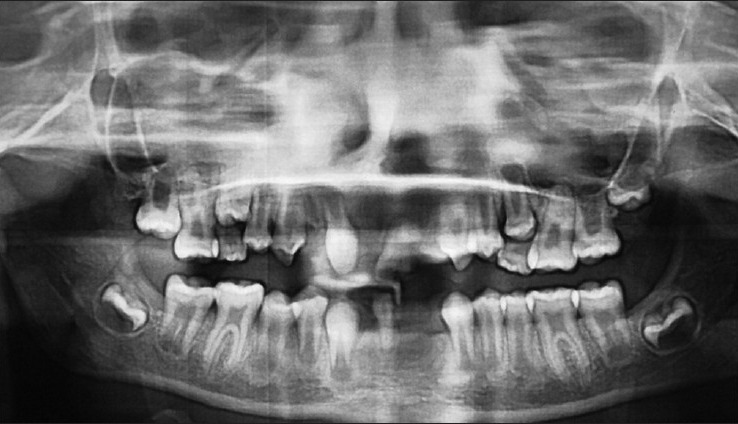
Orthopantomogram showing missing teeth i.r.t 12,22, 31,32,41,42 and retained deciduous tooth i.r.t 81
Routine blood investigations like hemoglobin (11.5 g%), total leukocyte count (7000/cu mm), Blood urea (13.0 mg%), serum creatinine (0.6 mg%), serum calcium (9.9 mg%), serum protein (6.4 g%), serum albumin (3.8 mg%), and serum globulin (2.6 g%) were within normal limits with exception of slightly elevated serum alkaline phosphatase (135.7 I.U/L).
DISCUSSION
The diagnosis of EVC syndrome can be made prenatally or immediately after birth, but our case was diagnosed at the age of 10 years because our patient belonged to a rural hilly area where medical services were lacking.
Some important clinical features of this syndrome are: 1) disproportionate small stature with increasing severity from the proximal to distal portions of the limbs, and shortening of the middle and distal phalanges 2) polydactyly affecting hands, which can be unilateral or bilateral and occasionally, the feet 3) hidrotic ectodermal dysplasia mainly affecting the nails, hair, and teeth 4) congenital heart malformations occurring in about 50-60% of cases and comprising of single atrium, defects of the mitral and tricuspid valves, patent ductus, ventricular septal defect, atrial septal defect, and hypoplastic left heart syndrome.[2,7] 5) There may be a valgus deformity of the knees or lumbar lardosis. Other uncommon anomalies may include urinary tract anomalies, strabismus, congenital cataracts, cryptorchidism, and epi- and hypospadias.
Oral manifestations of EVC syndrome may include enamel hypoplasia, hypodontia, conical teeth, diastema, multiple labial gingival frenulae, labiogingival adherence, submucosal clefts, short upper lip, presence of neonatal teeth, premature eruption, and/or exfoliation, etc., The diagnosis in our case became easy because of presence of most of the classical signs of EVC. The involvement of all the three embryonic layers can be seen by the presence of malformed and missing teeth, hypoplastic nails, thin and sparse hair, short stature, genu valgum, bilateral polydactyly of hand and feet, and cardiac abnormality. Only in 10% of cases of EVC, polydactyly is present in feet bilaterally[6,8] and ours is one among them.
The presence of typical oral manifestations, which helped us to distinguish it from several other syndromes, were malformed and missing teeth, multiple frenum, labio-gingival adherence, maxillary and mandibular labial vestibule obliteration, etc., The clinical variability of the oral abnormalities in EVC syndrome could be due to the fact that its genetic effect on the teeth and other oral structure development occurs during a relatively long period, and could be the result of other genetic and environmental phenotype-modifying factors.
The differential diagnosis of EVC syndrome includes Saldino-Noonan syndrome, Majewski syndrome, Verma-Naumoff syndrome, Beemer-Langer syndrome, Jeune Dystrophy, McKusick-Kaufman syndrome, and Weyers syndrome.[2] Final diagnosis is usually made after careful clinical and skeletal radiographic examination. DNA mapping is the most reliable method in diagnosing the syndrome.
Prenatal diagnosis of EVC syndrome is usually made by ultrasonography after the 18th gestation week, which shows narrow thorax, marked shortening of the long bones, hexadactyly of hands and feet, and cardiac defects.[9,10] Increased first-trimester fetal nuchal translucency thickness in association with EVC has been described at 13th week of gestation.[10]
Diagnosis at birth can be made by observing the typical symptoms of the disease and an X-ray of the skeleton. Chest radiography, ECG, and echocardiography may also help in diagnosis of EVC syndrome. The definitive diagnosis can be made by molecular diagnostic methods, which is based on homozygosity for a mutation in the EVC and EVC2 genes by direct sequencing.
There is no definite cure of EVC syndrome. Treatment is usually symptomatic, which can be accomplished with multidisciplinary approach. Patients generally need the consultation of Pulmonologist, Cardiologist, Orthopedist, Physiotherapist, Plastic surgeon, Dentist, Psychologist, etc.
Patients with cardiac abnormality should be first referred to pediatric cardiologist for surgical or non-surgical management of the defect. An early referral to pediatric orthopedic may play an important role in improving the quality of life. Treatment of polydactyly, i.e. surgical removal of extra finger or toe is usually carried out in 1st year of life. Orthopedic surgeon along with physiotherapist improves the strength and movement of fingers/toes. Surgical intervention of knee valgus alleviates pain and improves walking.
Patients with EVC syndrome should seek dental treatment in early years of their life. A dentist not only improves the aesthetics by replacing missing teeth, treating cleft lip, and aligning the teeth in dental arch, but also improves the masticatory efficiency of dentition. Dental health education for patients and their parents should include diet counseling, methods of plaque control, oral hygiene instructions, and importance of timely dental check-up.
The treatment of present case includes the extraction of retained deciduous mandibular incisor, restoration of carious fissure on 21, and replacement of edentulous spans with removable artificial prosthesis. The patient was already under treatment for her cardiac defect. Therefore, we have planned to refer the patient to orthopedic surgeon and physiotherapist for treatment of polydactyly and knock-knee.
CONCLUSION
EVC syndrome is a rare autosomal recessive disorder, which can be diagnosed by its clinical features like postaxial polydactyly of the hands, short limb dwarfism, and dysplastic fingernails and teeth. In year 2000, EVC, and in year 2002, EVC2 gene was identified to be associated with this syndrome. Moreover, along with some constant clinical features, this syndrome also has some variable components in different patients. Therefore, further studies are needed to explore other genes involved in EVC manifestations.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Atasu M, Biren S. Ellis-van Creveld syndrome: Dental, clinical, genetics, and dermatoglyphic finding of a case. J ClinPaediatr Dent. 2000;24:141–5. [PubMed] [Google Scholar]
- 2.Baujat G, Le Merrer M. Ellis-Van Creveld syndrome. Orphanet J Rare Dis. 2007;2:27. doi: 10.1186/1750-1172-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zanwill KM, Boal DK, Ladda RL. Dandy-Walker malformation in Ellis-vanCreveld syndrome. Am J Med Genet. 1998;31:123–9. doi: 10.1002/ajmg.1320310114. [DOI] [PubMed] [Google Scholar]
- 4.McKusick VA. Ellis-van Crefeld syndrome and the Amish. Nat Genet. 2000;24:203–4. doi: 10.1038/73389. [DOI] [PubMed] [Google Scholar]
- 5.Shilpy S, Nikhil M, Samir D. Ellis-van Creveld syndrome. J Indian Soc Pedo Prev Dent. 2007;25:S5–7. [PubMed] [Google Scholar]
- 6.Ruiz-Perez VL, Tompson SW, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet. 2003;72:728–32. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahuana A, Palma C, Gonzales W, Gean E. Oral manifestations in Ellis-vanCreveld syndrome: Report of five cases. Paediatr Dent. 2004;26:277–82. [PubMed] [Google Scholar]
- 8.Howard TD, Guttmacher AE, McKinnon W, Sharma M, McKusick VA, Jabs EW. Autosomal dominant postaxial polydactyly, nail dystrophy, and dental abnormalities map to chromosome 4p16, in theregion containing the Ellis-van Creveld syndrome locus. Am J Hum Genet. 1997;61:1405–12. doi: 10.1086/301643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horigome H, Hamada H, Sohda S, Oyake Y, Kurosaki Y. Prenatal ultrasonic diagnosis of a case of Ellis-van Creveld syndrome with a single atrium. Pediatr Radiol. 1997;27:942–4. doi: 10.1007/s002470050277. [DOI] [PubMed] [Google Scholar]
- 10.Venkat-Raman N, Sebire NJ, Murphy KW, Carvalho JS, Hall CM. Increased first-trimester fetal nuchal translucenty thickness in association with chondroectodermal dysplasia (Ellis-van Creveld) Ultrasound Obstet Gynecol. 2005;25:412–4. doi: 10.1002/uog.1849. [DOI] [PubMed] [Google Scholar]