

Abstract
Introduction:
Verapamil hydrochloride (VH) is a calcium channel blocking agent used in the treatment of hypertension, cardiac arrhythmia and angina pectoris. The short half-life and high frequency of administration of VH makes it a suitable candidate for designing sustained drug delivery system. The aim of the present investigation was to develop a sustained release matrix tablet of verapamil hydrochloride (VH) using ethyl cellulose, methyl cellulose, Eudragit RS 100, hydroxypropyl methylcellulose and carboxymethyl cellulose and to evaluate the drug release kinetics.
Materials and Methods:
In order to achieve the required sustained release profile, the tablets were prepared by a wet granulation method using avicel PH 101 and magnesium stearate as binder and lubricant, respectively.
Results:
The formulated tablets were characterized for pre-compression and post-compression parameters and they were in the acceptable limits. The drug release data obtained after an in vitro dissolution study was fitted to various release kinetic models in order to evaluate the release mechanism and kinetics. The criterion for selecting the best fit model was linearity (coefficient of correlation). Drug release mechanism was found to follow a complex mixture of diffusion, swelling and erosion. Furthermore, to minimize the initial burst drug release, batches were coated by using Eudragit RS100 polymer. After coating the tablets, a better release profile of the formulated tablets was expected and the release rate of the drug was compared with the marketed SR tablet of VH.
Conclusion:
The dosage form holds the potential to control the release rate of drug and extend the duration of action of a drug.
Keywords: Ethyl cellulose, eudragit RS 100, hydroxypropyl methylcellulose, matrix tablet, methyl cellulose, verapamil HCL
INTRODUCTION
Controlled release dosage forms are mainly designed to maintain therapeutic blood or tissue levels of the drugs that have a short elimination half-life.[1,2] The controlled release dosage forms offer a number of advantages over immediate release products, such as better patient compliance due to decrease in dosing frequency, portability, convenience and fewer side-effects. Such dosage forms exhibit better pharmacological effect and prolonged therapeutic activity. Matrix tablets are one of the most commonly used controlled release dosage forms as they release the drug in a controlled manner. Polymers commonly used in sustained release matrices are hydrophilic polymers (cellulosic and non-cellulosic) or hydrophobic polymers (Ethyl cellulose [EC], hypromellose acetate succinate, cellulose acetate propionate, methacylic acid copolymers, polyvinyl acetate, etc.).[3]
Verapamil hydrochloride (VH) is a calcium channel blocking agent used in the treatment of hypertension, cardiac arrhythmia and angina pectoris. The biological half-life is 4-6 h, and it is completely absorbed from the gastrointestinal tract. The usual dose of the drug is 40-240 mg 3 times a day.[4] Hence, due to the short half-life and high frequency of administration, VH was considered as a suitable candidate for designing sustained release tablets. Therefore, the present study was aimed toward formulation and in vitro evaluation of the sustained release matrix tablet of VH by the wet granulation method using various polymers EC, methyl cellulose (MC), Eudragit RS100, hydroxypropyl methylcellulose (HPMC) K100 and carboxymethyl cellulose (CMC) to reduce the dosage regimen, better therapeutic efficacy and improved patient compliance with less toxicity.[5–7]
MATERIALS AND METHODS
Materials
VH was procured as a gift sample from Ranbaxy Laboratories Ltd., India. HPMC, EC, MC, Eudragit RS100, CMC, Polyvinylpyrrolidone (PVP) K 30, Avicel PH 101 and magnesium stearate and talc were of suitable analytical grade and were used as received.
Methods
Preparation of granules
Matrix tablets of VH were prepared by the wet granulation method. For preparing granules, the specified amount of each component was passed through sieve no #60 [Table 1]. Then, the drug was mixed properly with the polymer and the granulating agent. The wet mass was passed through sieve no. #10 to obtain granules. The granules were oven-dried at 40°C for 30 min. After drying, the granules were passed through sieve no. #36 and sieve no. #22. The granules were then lubricated with magnesium stearate and finally, talc was added to the blend.
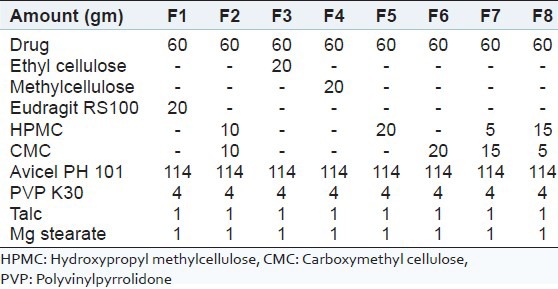
Table 1.
Specification of different batches prepared by wet granulation method
Flow properties of granules
The prepared granules were evaluated for bulk density (BD) (1), tapped density (TD) (2), compressibility index (CI/Carr's index) (3), Hausner's ratio (HR) (4) and angle of repose (AR). The BD and TD were determined using a 25 mL measuring cylinder. The TD was determined after tapping the volumetric flask 100 times.

Where, W = Weight of the powder, Vo = Initial volume, Vf = Final volume after tapping.
Tablet compression
Compression was performed on a single punch tablet punching machine. For tablet compression, an 8 mm punch was used. Eight batches were formulated using different polymers and combinations of polymers [Table 1].
Coating of tablets
Coating of tablets of different batches was done using Eudragit RS100 polymer. The coating solution was prepared by dissolving Eudragit RS100 in acetone using a magnetic stirrer. On complete solubilization of the polymer, castor oil (10% w/w of dry polymer) was added followed by addition of talc (0.1% w/v) as anti-adherent and titanium dioxide (0.5% w/v) as opacifier. The solution was stirred for 15 min at room temperature. The tablets were coated with this solution over the tablets using the dip method and dried in hot air.
Evaluation of tablets
Physical characterization
The formulated tablets were characterized for weight variation, hardness, thickness and friability.[7]
Weight variation test
For the weight variation test, 20 tablets from each batch were selected at random and their average weight was determined using an electronic balance. Then, the average weight was calculated and compared with the individual weight of each tablet.[7]
Hardness test
A Monsanto hardness tester (Cad Mach) was used to determine the hardness of the tablets. Ten tablets were selected at random from each batch for the study. Each tablet was placed between the plungers and the handle was pressed, and the force of the fracture was recorded. Their crown to crown thickness was also determined using a vernier caliper.[7]
Friability test
The friability was determined by placing 10 tablets in a Roche friability tester for 4 min at 25 rpm. The tablets were dropped at a height of 6 inches in each revolution. Tablets were de-dusted using a soft muslin cloth and reweighed. The friability was given by the formula:
![]()
Where, Wo is the weight of the tablets before the test and W is the weight of the tablet after the test.[7]
In vitro drug release study
In vitro drug release study for the prepared coated matrix tablets was performed for the 12 h sample using a eight-station United State Pharmacopoeia (USP)-22 Type I dissolution apparatus at 37 ± 0.5°C and at 50 rpm speed in 0.1 N HCl (900 mL) as dissolution media. From the dissolution medium, 5 mL of the sample was withdrawn at the specific time intervals and replaced with an equal volume of fresh medium (5 mL) to maintain constant media volume. After filtration, each sample was analyzed for VH using a UV-Visible spectrophotometer (λmax = 230 nm). This study was performed in triplicate for each batch.
Release kinetics
The in vitro drug release kinetics were characterized by fitting the data obtained from in vitro release studies of the coated matrix tablet from various batches to standard drug release kinetics equations (zero order, first-order, Higuchi (Mt/M∞ <0.6), Korsmeyer-Peppas model (Mt/M∞ <0.6) and Hixson-Crowell model.[8]
Release profile comparison
Similarity factor (f2)
This factor was introduced by Moore and Flanner, and has been adopted by the Center for Drug Evaluation and Research (US Food and Drug Administration FDA) and by European Medicines Evaluation Agency (EMEA) as a criterion for the assessment of the similarity between two dissolution profiles. The similarity factor, f2, as defined by the FDA and EMEA, is a logarithmic reciprocal square root transformation of one plus the mean squared (the average sum of squares) differences of drug percent dissolved between the test and the references products.
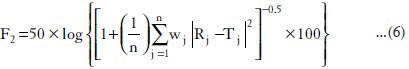
where,
Rj = Cumulative percentage release of drug from reference product at time t.
Tj = Cumulative percentage release of drug from test product at time t.
Difference factor (f1)
Difference factor measures the percent error between two drug release curves over all time points.

where,
Rj = Cumulative percentage release of drug from reference product at time t.
Tj = Cumulative percentage release of drug from test product at time t.
Fourier transforms infrared studies
To examine the chemical interaction between the drug and other components of the tablet, infrared (IR) spectra for VH and the physical mixture of all the components were recorded using a spectrophotometer (FTIR 1615, Perkin Elmer, and USA).
Differential scanning calorimetry studies
DSC was performed for pure VH, pure polymers and crushed tablets of each batch using DSC 60, Shimadzu, Japan. Sealed and perforated aluminum pans were used in the experiments. Empty pan was used as the reference. The DSC scans of the samples were performed at a scanning rate of 10°C/min from 50°C to 300°C.[9]
RESULTS
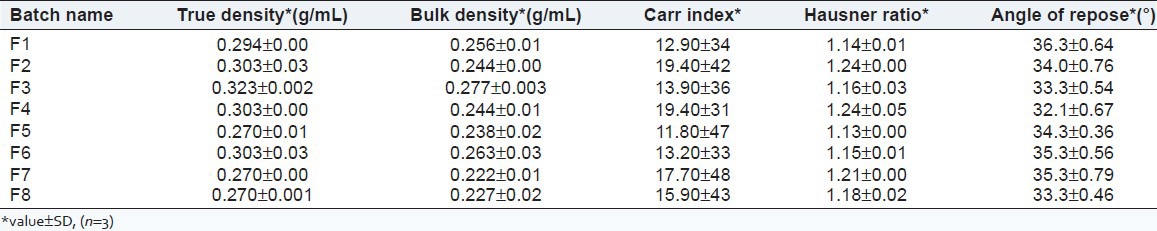
Flow properties of granules
Table 2 shows the flow properties of the granules of different batches. Various tests have been performed on these batches such as TD, BD, CI, HR and AR. The AR of granules varied from 32.6 to 36.3; CI from 11.8 to 19.4; and HR from 1.13 to 1.24.
Table 2.
Flow properties of granules for the different batches
Evaluation of tablets
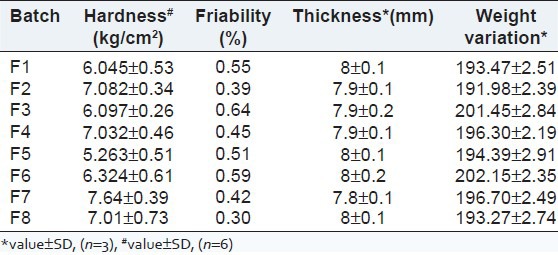
The formulated tablets were evaluated for hardness, friability, weight variation and thickness, and were found to be within the acceptable official limits [Table 3].
Table 3.
Evaluation of tablets for various parameters like hardness, friability, thickness
In vitro drug release study
Table 4 indicates the in vitro drug release of VH from the various batches. In vitro drug release study revealed that batches F1, F2 and F3 release 0.94%, 0.94% and 1.78% of drug, respectively, at the end of 2 h and F1 and F3 release 31.48% and 27.23% of the drug after 12 h. Batch F2 releases 92.58% of drug after 9 h. At the end of 12 h, 100% release of drug was not observed for batches F1 and F3. Marketed tablet of VH shows 61.65% of drug release after 12 h.
Table 4.
Percent drug release of different batches at different time intervals
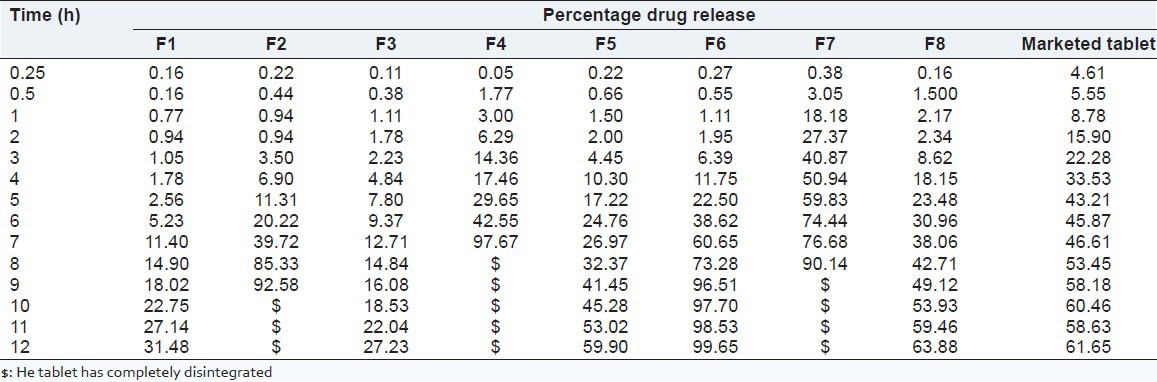
The batches F4, F5, F6, F7 and F8 released 6.29%, 2.00%, 1.95%, 27.37% and 2.35% of VH, respectively, at the end of 2 h and batch F4 showed 97.67% of drug release after 7 h. After 12 h, F5 showed 59.90%; F6 showed 99.65%; and F8 showed 63.88% drug release.
Release kinetics
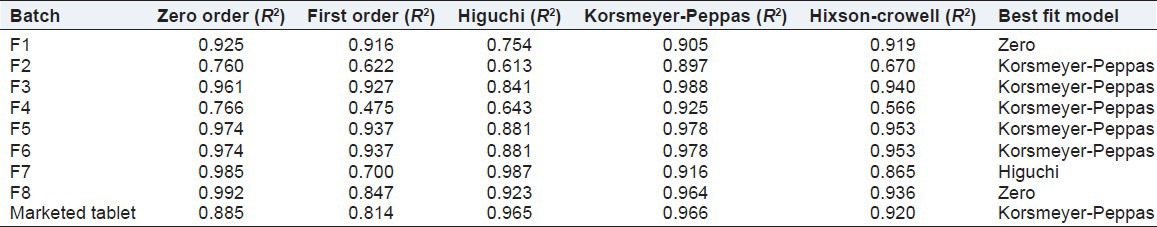
Zero order was the best fit model of drug release from batches F1 and F8. Batches F2, F3, F4, F5 and F6 and the marketed tablets followed the Korsmeyer Peppas model of drug release and batch F7 showed higuchi release kinetics. The value of coefficient of correlation (R2) for various models is depicted in Table 5.
Table 5.
Release kinetics of different batches
Release profile comparison
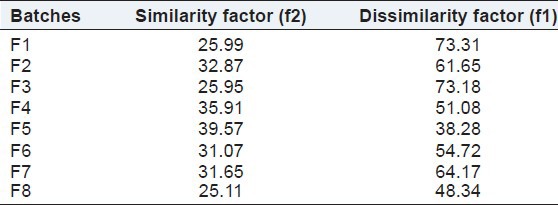
The similarity and the dissimilarity factors depict that the drug release from the prepared batches were significantly different from the release of drug from the marketed tablet. The significance of using similarity factor was to compare the solubility and release profile of the prepared tablets with that of the marketed tablets. The f2 value was found to vary from 25.11 for batch F8 to 39.57 for batch F%. The f1 value ranged from 38.28 for F5 to 73.31 for F1. Table 6 represents the similarity and the dissimilarity factors for the various batches.
Table 6.
Similarity and dissimilarity factor of different batches
FTIR analysis
The FTIR spectra of all the combinations containing drug and one or more polymer showed similar or slight shift in peak values when compared with the characteristics peak values of the pure drug [Figure 1]. The spectrum of VH showed characteristic peaks of NH group at 3041 cm-1, 2953.02 cm-1 and 2839.22 cm-1 (C-H stretching vibration in methyl); 2578.83 cm-1 and 2540.25 cm-1 (aldehydic C-H stretching vibration); 2237.43 cm-1 (C-N stretching vibration); 1597.06 cm-1, 1517.98 cm-1 and 1458.18 cm-1 (C = C in aromatic ring); 1261.45 cm-1 and 1151.5 cm-1 (C-O stretching in aromatic and aliphatic); and 1024.2 cm-1 (C-N aliphatic stretching vibration), 813.96 cm-1 and 767.67 cm-1 (meta substituted benzene).
Figure 1.
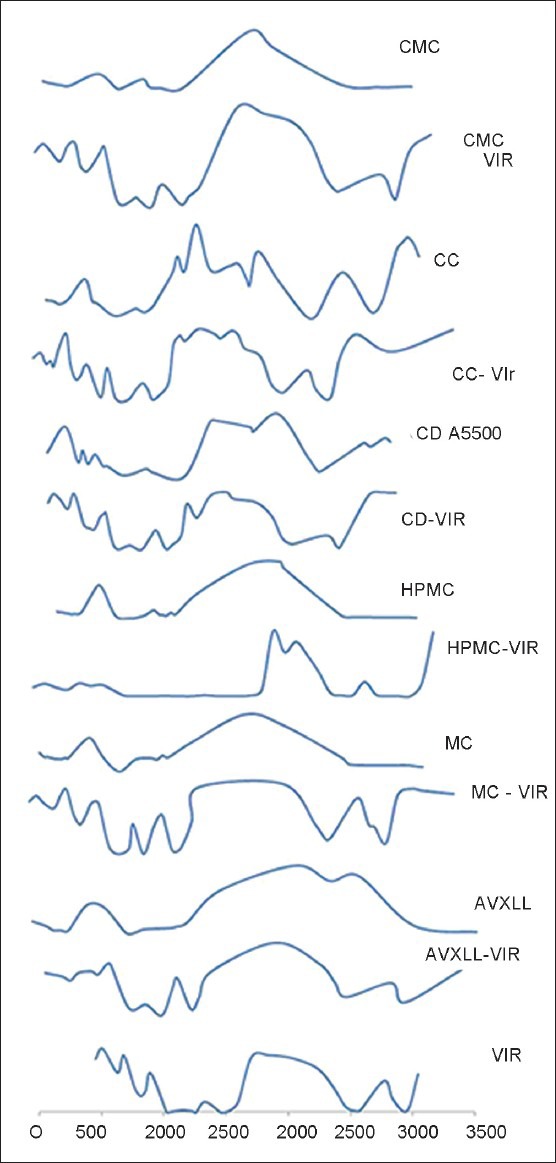
Fourier transforms infrared spectra of pure verapamil hydrochloride and its physical mixture with the polymer
EC showed the characteristic peak absorption bands for C-O-C-stretching vibration at 1093 cm-1, C-H stretching bands at 2873.94 cm-1 and 2974.23 cm-1 and C-H bending at 1371.39 cm-1. It also showed that absorption at 3483.44 cm-1 corresponded to O-H stretching.[10]
HPMC showed characteristic O-H vibrational stretching peaks at 3500 cm-1 -3400 cm-1, symmetric stretching mode of methyl and hydroxypropyl at 2900 cm-1, stretching vibration of C-O for six membered cyclic rings at 1600 cm-1 and 1650 cm-1, the symmetric vibration of methoxy group at 1400 cm-1 -1350 cm-1 and asymmetric 1500 cm-1 -1450 cm-1.[11]
CMC showed the peaks at 3288 cm-1 of O-H stretching, methyl (1419.61 cm-1) and hydroxyl (1321.1 cm-1), absorption band at 1598.99 cm-1 showed presence of carboxyl group, C-H stretching vibration at 2902.87 cm-1 and band at 1419.61 cm-1 and 1325.1 cm-1 due to CH scissoring and hydroxyl group bending vibration.
MC shows characteristic peak of CH stretching at 2899.01 cm-1. The IR interpretation of the tablet showed that the drug and the polymers show similar IR peaks, respectively, even after mixing with the polymers, which indicate that there is no interaction between the drug and the polymers.[6] Hence, the drug and the polymers remained in the pure state even after mixing.
DSC
DSC was performed to determine the physiochemical compatibilities and to study the interaction of the drug and the used excipient. DSC thermograms of VH and various batches revealed that the melting point of VH in the pure form was 146.50°C, and varied from 139.70°C to 141.29°C in the tablet form, which indicated that there is no significant difference in the melting point of the drug.[6] Figure 2 represents the DSC of the drug and its batches.
Figure 2.
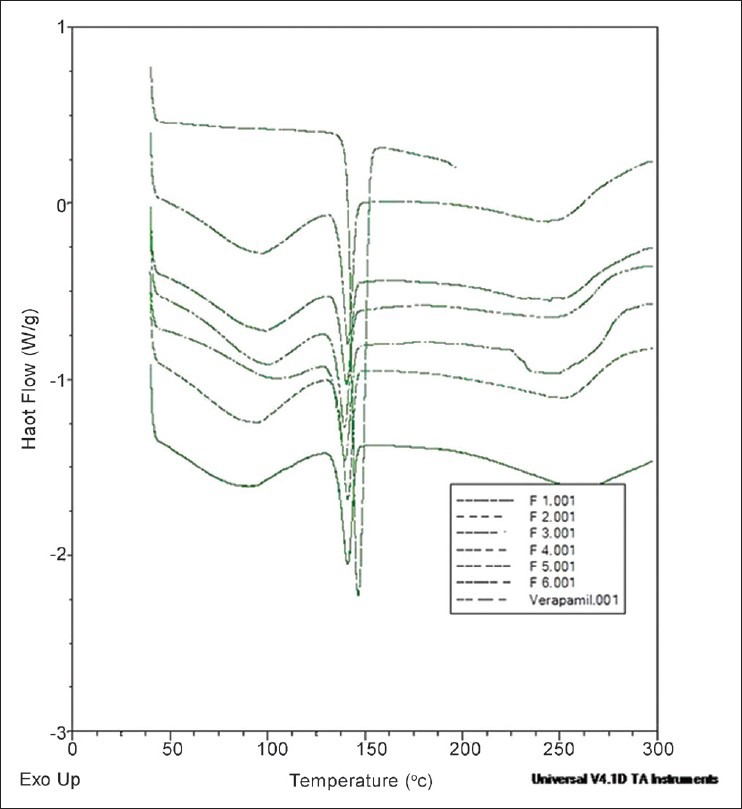
Overlay of differential scanning calorimetry thermogram of pure verapamil hydrochloride and different batches
DISCUSSION
The flow properties of the granules indicated good compliance with the official standards. The AR of granules varied from 32.1°C to 36.3°C, and the flow property was fair. According to USP 2007, granules of batches F1, F3, F5 and F6 showed good and batches F2, F4, F7 and F8 granules showed fair CI (USP 2007). HR of granules of batches F1, F3, F5, F6 and F8 falls within the good range and that of batches F2, F4 and F7 falls within the fair range. Hardness, friability, weight variation and thickness of the formulated tablets were acceptable.[5,6,7]
The IR spectra study revealed no chemical interaction between the drug and the other formulation components. DSC thermogram indicated that the drug remains intact even in the formulation, and that there was no interaction of drug with the polymers.
To minimize the friability losses as well as to improve the sustaining effect of the matrix tablet, all tablet batches were film coated using Eudragit RS100. Because all the batches were uniformly coated by Eudragit RS100, the release retardant effect of this polymer will be uniform for all the batches. Batch F1 was prepared using Eudragit RS100, F2 using a combination of HPMC and CMC in the ratio of 1:1, F3 using EC, F4 using MC, F5 using HPMC, F6 using CMC, F7 using HPMC: CMC: 1:3 ratio and F8 using HPMA: CMC: 3:1 ratio.
The batch F1 (having Eudragit RS100) exhibited a delayed release of the drug. Batches F1 and F3 (having EC) prolonged the release of drug and did not show complete dissolution after a study period of 12 h. This may be attributed to sustained release of the drug from the polymer matrix (due to slow erosion of these polymers). The comparison of the drug release from the various batches with the marketed product indicated better sustained release of VH from batches F1, F3 and F5. Batch F8 showed an equivalent drug release as shown by the marketed tablet. The batches F4, F5 and F6 were composed of MC, HPMC and CMC polymers, respectively. Batches F7 and F8 were composed of a combination of polymers HPMC and CMC in the ratio of 1:3 and 3:1, respectively. Batches F7 and F4 showed earlier drug release, which may be attributed to improper coating of the batch or may be due to improper mixing of the polymers. The result of the release profile comparison indicates the success of the formulation along with the achievement of better results.
The factors f1 and f2 play a very important role in comparing the formulations’ release profile. When the two dissolution profiles are identical, the value of f2 is 100 and when the dissolution of one product (test or reference) is completed before the other begins, f2 can be rounded to zero. Thus, the value of f2 ranges from 0 to 100. If a difference between the test and the reference products is 10%, and this average absolute difference is substituted in the equation, f2 becomes 50. Two dissolution profiles are considered “similar” when the f2 value is between 50 and 100. A higher f2 value indicates closeness between the two dissolution profiles. However, the equation is only applicable in comparing curves in which the average differences between the reference and the test formulation profiles is less than 100 and the amount of drug released in percent. The percent error is zero when the test and the drug reference profiles are identical, and increases proportionally with the dissimilarity between the two dissolution profiles. It is generally accepted that values of F1 between 0 and 15 do not indicate dissimilarity. Thus, the dissolution profiles of batches of matrix tablet prepared in the present investigation were significantly different from that of the marketed tablet, indicating the success of the formulation.
CONCLUSION
The sustained release matrix tablet of VH using various hydrophilic polymers like EC and Eudragit RS100 and polymers like HPMC, MC and CMC, and the combination of HPMC and CMC were successfully prepared by the wet granulation method. The prepared granules were found to be free flowing. The DSC and FTIR study indicated that there is no physical or chemical interaction/incompatibility between the drug and the polymer used for the study. The batches prepared using polymers Eudragit RS100 and EC were found to extend the time of release of drug release. On the other hand, the batches prepared using polymers like MC, HPMC and CMC and a combination of HPMC and CMC significantly retarded the release of drug. The similarity and the dissimilarity factors indicate that the sustained release formulations are quite different from the marketed tablet, and more sustained than the marketed tablet. It may thus be concluded that the sustained release formulation can be achieved using both hydrophilic and hydrophobic polymers, which can also maintain the sustained release profile over an extended period of time.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Mishra B, Bansal A, Sankar C. Development and In Vitro evaluation of hydrophilic matrix tablets of diltiazem hydrochloride. Acta Pharm Turc. 2005;47:115–26. [Google Scholar]
- 2.George M, Grass IV, Robinson JR. Newyork: Marcel Dekker; 1978. Sustained and controlled release drug delivery systems; pp. 124–7. [Google Scholar]
- 3.Maderuelo C, Zarzuelo A, Lanao JM. Critical factors in the release of drugs from sustained release hydrophilic matrices. J Control Release. 2011;154:2–19. doi: 10.1016/j.jconrel.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Bhagwat DA, Kawtikwar PS, Sakarkar DM. Sustained release matrices of verapamil HCl using glyceryl monosterate and stearic acid. Res J Pharm Tech. 2008;1:405–09. [Google Scholar]
- 5.Singh SK, Pandit JK, Mishra DN. Formulation and in vitro evaluation of carbopol 934P matrix tablets: Influence of drug solubility and co-excipients on release rate of the drug. J Pharm Res. 2007;6:20–4. [Google Scholar]
- 6.Singh SK, Pandit JK, Mishra DN. Influence of drug solubility, drug polymer ratio, nature of co-excipients and thermal treatment on drug release from carbopol 974P matrix tablet. Acta Pharma Sciencia. 2006;48:167–78. [Google Scholar]
- 7.Singh SK, Vijay Kumar SG, Pandit JK, Mishra DN. HPMC based matrix tablet of nimodipine: Effect of viscosity of polymer, type of diluents and sintering on in vitro release. J Sci Pharm. 2006;7:101–9. [Google Scholar]
- 8.Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–33. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 9.Deshmukh G, Ruikar D, Seth AK, Ghelani T, Patel H, Patel J, et al. Formulation and statistical optimization of Verapamil Hydrochloride floating drug delivery system by response surface methodology. Pharm sci monitor Int J Pharm Sci. 2011;2:26–42. [Google Scholar]
- 10.Suthar V, Pratap A, Raval H. Studies on poly hydroxy alkanoates/ethylcellulose blends. Bull Mater Sci. 2000;23:215–9. [Google Scholar]
- 11.Subhashree S, Kanti CC, Chandra MS, Sharmistha N. Qualitative analysis of controlled release ofloxacin/HPMC mucoadhesive suspension. Int J Drug Dev Res. 2011;3:217–32. [Google Scholar]