

Abstract
There is significant interest in understanding the contribution of intracellular signaling and synaptic substrates to memory flexibility, which involves new learning and suppression of obsolete memory. Here, we report that enhancement of Ca2+-stimulated cAMP signaling by overexpressing type 1 adenylyl cyclase (AC1) facilitated long-term potentiation (LTP) but impaired long-term depression (LTD) at the hippocampal Shaffer collateral-CA1 synapses. However, following the induction of LTP, low-frequency stimulation caused comparable synaptic depotentiation in both wild type and AC1 transgenic (AC1 TG) mice. Although previous studies have suggested the function of LTD in spatial memory flexibility, AC1 TG mice showed not only better initial learning in the Morris water maze, but also faster acquisition and increased ratio of new memory formation to old memory retention during the reversal platform training. In the memory extinction test, which requires suppression of old memory without involving the acquisition of the new platform information, AC1 TG and wild type mice showed comparable performance. Our results demonstrate new functions of Ca2+-stimulated AC1, and also suggest that certain aspects of hippocampus-dependent behavioral flexibility may not require intact LTD.
Previous studies with animal models have demonstrated important roles of cAMP signaling in regulating synaptic plasticity and memory formation (Nguyen and Woo 2003; Abel and Nguyen 2008). The activation of cAMP-PKA signaling may be mediated by the Ca2+-stimulated adenylyl cyclases (AC), which respond to the activity-dependent rise in intracellular calcium and generate cAMP. Among all ACs, type 1 and type 8 AC (AC1 and AC8) are the major Ca2+-stimulated ACs in the brain (Wong et al. 1999). Compared to AC8, AC1 is neurospecific and has major contribution to cAMP production (Wang and Storm 2003). Genetic deletion of AC1 causes impairments in hippocampus-dependent spatial memory retention and mild defects in long-term potentiation (LTP) (Wu et al. 1995).
In addition to memory retention, suppression of obsolete memory and acquisition of new information are critical for behavioral flexibility and adaptation. Suppression of obsolete memory or memory extinction is not a passive process, and usually requires activity-dependent neuronal modification (Myers and Davis 2007). Although the function of cAMP in memory retention has been extensively investigated, only a handful of studies implicate the role of cAMP signaling in memory flexibility. It has been suggested that, while PKA activity facilitates fear memory formation, it acts as a molecular constraint for fear extinction (Isiegas et al. 2006). Lowering PKA activity enhances extinction. Supportively, overexpression of AC1, which causes elevation in PKA activity, suppresses fear memory extinction (Wang et al. 2004).
Intriguingly, different mechanisms may be involved in regulating spatial memory flexibility. Our recent study demonstrates that genetic deletion of Ca2+-stimulated ACs hampers reversal platform learning and the extinction of old spatial memory (Zhang et al. 2011). Thus, we hypothesize that enhancing Ca2+-stimulated AC activity may facilitate certain aspects of behavioral flexibility such as reversal spatial learning. Here, we used AC1 transgenic (AC1 TG) mice that overexpress AC1 in the forebrain regions. AC1 TG mice showed superior performance in both the initial hidden platform and the reversal platform training. Intriguingly, AC1 TG mice displayed enhanced in vivo long-term potentiation (LTP) but impaired long-term depression (LTD). The synaptic depotentiation was effectively induced by low-frequency stimulation (LFS) in AC1 TG mice. Our data demonstrate new functions of AC1, and also suggest no absolute requirement of intact synaptic LTD for memory flexibility.
Results
Overexpression of AC1 facilitates LTP in vivo
Previous studies have demonstrated that reduction of cAMP-dependent protein kinase activity leads to LTP deficits (Abel et al. 1997). Furthermore, mice lacking Ca2+-stimulated AC show impaired LTP both in vitro and in vivo (Wong et al. 1999; Zhang et al. 2011). These results suggest that enhancing Ca2+-stimulated cAMP signaling may facilitate LTP. Consistent with this possibility, our previous work with acute hippocampal slices demonstrated enhanced in vitro LTP in AC1 TG mice (Wang et al. 2004). Here, we further examined high-frequency stimulation (HFS)-induced LTP in vivo with anesthetized mice (2–4 mo of age). At the Schaffer collateral/CA1 synapses, AC1 TG and wild type (WT) mice showed comparable basal neural transmission, as indicated by the comparable output field excitatory postsynaptic potential (fEPSP) responses to different stimulation intensities (genotype, F(1,44) = 0.4; genotype by intensity, F(4,176) = 0.2; both P > 0.5, two-way repeated ANOVA) (Fig. 1A). AC1 TG mice also showed normal paired-pulse facilitation (genotype, F(1,19) = 0.1; genotype by paired-pulse interval, F(9,171) = 0.6; both P > 0.7) (Fig. 1B). Next, we stimulated the Schaffer collateral pathway with a single train of HFS (at 100 Hz, 1-sec duration). To our surprise, AC1 TG and WT mice showed similar degree of synaptic potentiation (TG, 123.2 ± 4.4%; WT, 124.8 ± 3.2%; genotype, F(1,10) = 0.1; P =0.8, two-way repeated measures ANOVA) (Fig. 1C).
Figure 1.

Overexpression of AC1 enhances LTP in vivo without affecting basal transmission and paired-pulse facilitation. (A) WT and AC1 TG mice show comparable basal neural transmission. The slopes of fEPSP stimulated by different stimulation intensities are plotted. (B) WT and AC1 TG mice show similar degree of paired-pulse facilitation, which was examined by comparing the slopes of two fEPSPs triggered by pair stimulation with intervals ranging from 20 to 500 msec. (C) WT and AC1 TG mice show comparable LTP induced by HFS at 100 Hz. (D) HFS at 50 Hz induces significant LTP in AC1 TG but not in WT mice. Representative fEPSPs before (indicated as 1) and 60 min after (indicated as 2) HFS are shown.
When we decreased the stimulation frequency to 50 Hz, both WT and AC1 TG animals showed robust and comparable post-tetanic potentiation (PTP). However, the potentiation in WT decayed to the baseline level (106.6 ± 2.4%, P > 0.1 when compared to the baseline fEPSP value) (Fig. 1D) at 60 min after the HFS. In contrast, this weaker stimulation protocol induced persistent LTP in AC1 TG mice (123.3 ± 4.2%) (genotype, F(1,9) = 13.0, P < 0.01) (Fig. 1D). These data suggest that the threshold regulating the transition from PTP to the maintenance phase of LTP may be reduced in AC1 TG mice.
AC1 TG mice show defective LTD but effective synaptic depotentiation
In addition to synaptic potentiation, neurons also show activity-dependent synaptic depression. Interestingly, reduction of PKA activity or Ca2+-stimulated AC impairs LTD in vitro and in vivo (Qi et al. 1996; Zhang et al. 2011). Here, we found that 900 pulses at 1 Hz successfully induced robust in vivo LTD in WT adult mice (2–4 mo of age) (83.3 ± 4.7%) (Fig. 2). The same low-frequency stimulation (LFS) protocol failed to induce measurable LTD in AC1 TG mice (100.3 ± 5.4%, F(1,10) = 5.0, P < 0.05 between WT and AC1 TG mice) (Fig. 2).
Figure 2.
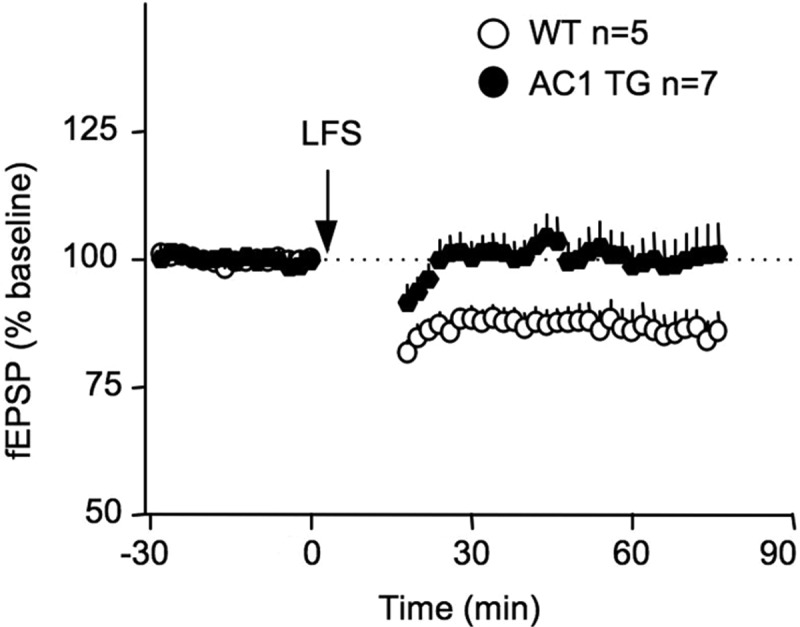
Mice overexpressing AC1 show impaired LTD in vivo. The extracellular fEPSP at the Shaffer collateral/CA1 synapses was recorded from anesthetized animals. LFS (900 pulses at 1 Hz) induces significant synaptic depression in WT but not AC1 TG mice.
Different from LTD at naive synapses, depotentiation involves activity-dependent reversal of the previously potentiated synapses. Previous studies have demonstrated that synaptic potentiation can be reversed by LFS (1–5 Hz) in a time-dependent manner (Huang and Hsu 2001). Effective depotentiation only occurs when LFS is delivered shortly (i.e., within a 3- to 15-min window) after LTP induction both in vitro (Huang et al. 1999) and in vivo (Staubli and Lynch 1990; Staubli and Scafidi 1999). Here, we first induced potentiation by HFS at 100 Hz, waited for 5 min, and then delivered 900 stimulation pulses at 1 Hz. WT and AC1 TG mice showed effective and comparable depotentiation (F(1,9) = 0.3, P = 0.6 between WT and AC1 TG) (Fig. 3).
Figure 3.
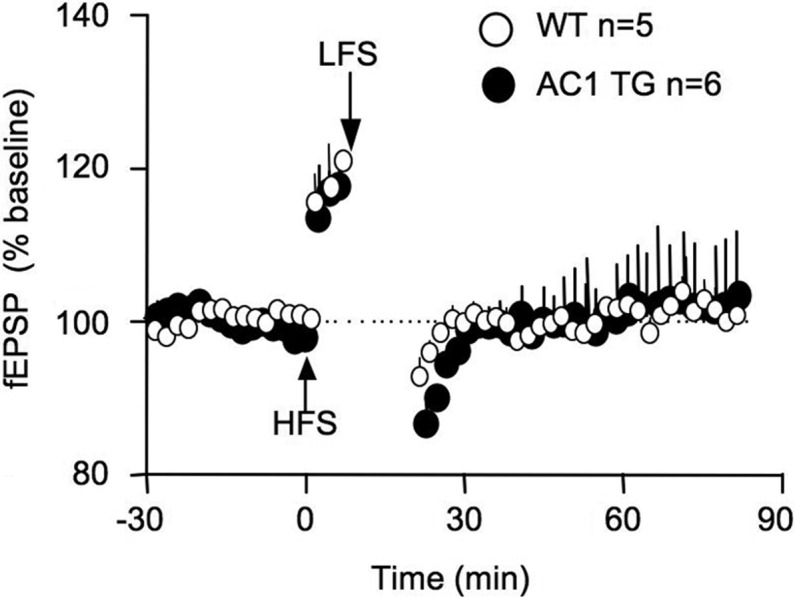
Mice overexpressing AC1 show effective synaptic depotentiation in vivo. The extracellular fEPSP at the Shaffer collateral/CA1 synapses was recorded from anesthetized animals. LFS (900 pulses at 1 Hz) was delivered 5 min after the HFS (100 Hz for 1 sec).
Overexpression of AC1 facilitates the flexibility of spatial learning
The function of LTD, as well as intact bidirectional synaptic plasticity, has been implicated in regulating both initial and reversal spatial learning (Migaud et al. 1998; Nicholls et al. 2008). Having identified impaired hippocampal LTD in AC1 TG mice, we decided to investigate its impact on hippocampus-dependent spatial learning. We trained AC1 TG and WT mice with the Morris water maze (as illustrated in Fig. 4A). We first subjected mice to 6 d of hidden platform training (one trial/day) and performed a probe test on day 7. While WT mice improved gradually in escape latency, AC1 TG showed significant faster acquisition to find the hidden platform (F(1,21) = 17.0, P < 0.001 between the two genotypes, two-way repeated measures ANOVA) (Fig. 4B1). During the probe test, both WT (F(3,30) = 19.5, P < 0.0001) and AC1 TG mice (F(3,33) = 15.30, P < 0.0001) showed preference to the target quadrant (WT, 52.0 ± 5.6%; TG, 50.0 ± 4.7%) (Fig. 4B2). There was no significant difference between WT and AC1 TG mice in the time spent in the target quadrant (P = 0.63) (Fig. 4B2) and in the numbers of platform crossing (P = 0.33) (Fig. 4B3). These data implicate that enhancing AC1 activity facilitates the rate of acquisition.
Figure 4.

Mice overexpressing AC1 show superior initial as well as reversal learning in the Morris water maze. (A) Illustration of the Morris water maze training protocol. WT and AC1 TG mice were first trained to find the hidden platform (placed in the center of the arbitrarily defined quadrant 1) by one trial daily for 6 d, and tested by the first probe test on day 7. Starting from day 8, the same animals were subjected to the reversal platform training, during which the platform was moved from quadrant 1 to 3 (as illustrated). Animals were trained by one trial daily for 6 d (from day 8 to day 13), tested by the second probe test on day 14, further trained by one trial daily for another 4 d (i.e., from day 15 to day 18), and tested by the third probe test on day 19. Animals’ performance in escape latency (B1,C1), percentage of time spent in each quadrant (B2,C2), and number of crossings over the target platform location (B3,C3) during the hidden platform (B1–B3) and the reversal platform (C1–C3) training are expressed as average ± SEM. (*) P < 0.05; (ns) not significant.
We next subjected the same animals to reversal platform training. As illustrated in Figure 4A, we moved the platform to the opposite quadrant. During the reversal training, animals needed to learn the new platform location and in the meantime suppress the memory for the old platform location. AC1 TG mice displayed better improvement in escape latency (F(1,21) = 6.7, P = 0.02 between AC1 TG and WT mice) (Fig. 4C1). In the second (on day 14) and third (on day 19) probe tests, WT and AC1 TG mice did not show significant difference in the time spent in each quadrant (Fig. 4C2) and number of crossing over the new or the old platform location (Fig. 4C3). However, AC1 TG but not WT animals showed preference to the new target quadrant over the old target quadrant (Fig. 4C2). WT mice only spent significantly more time in the new target quadrant than in the non-target quadrants (i.e., quadrant 2 and 4) during the third probe test (P = 0.02) (Fig. 4C2). Also, AC1 TG (P < 0.05) but no WT (P > 0.05) animals showed more preference to the new quadrant in the third probe test than in the second probe test. When comparing the numbers of platform crossing, only AC1 TG mice showed more crossing over the new than the old platform location in both probe tests (AC1 TG mice, P = 0.02 for the second and P = 0.015 for the third test; WT mice: P = 0.59 for the second and P = 0.16 for the third test) (Fig. 4C3). These data demonstrate that overexpression of AC1 enhanced spatial memory flexibility by facilitating both initial and reversal learning.
Because WT mice did not show robust reversal learning in the one trial/day training paradigm, we next trained mice with a more intensive protocol (Fig. 5A). Following 2 d of visible platform training (Fig. 5B), WT and AC1 TG mice were trained by four trials daily for 6 d (from day 3 to day 8) to find the hidden platform. The two groups of animals showed similar improvement in escape latency (genotype, F(1,19) = 0.4, P = 0.52; genotype by training day, F(5,95) = 1.0, P = 0.43) (Fig. 5B). During the probe test performed on day 9, WT and AC1 TG mice showed comparable preference for the target quadrant (TG, 56.2 ± 5.1%; WT, 47.6 ± 5.0%; genotype, F(1,19) = 3.3, P = 0.08; genotype by quadrant, F(3,57) = 1.8, P = 0.3) (Fig. 5C) and similar crossing number over the hidden platform location (P = 0.2) (Fig. 5D).
Figure 5.

AC1 TG mice show stronger new memory formation than WT mice. (A) Morris water maze training protocol. (B) Improvement of escape latency during the visible platform, hidden platform, and reversal platform training. (C) Percentage of time spent in each quadrant during the first probe test (after hidden platform training) and the second probe test (after the reversal platform training). (D) Number of crossing over the target platform location during the first and the second probe tests. As indicated by the filled circle, the platform location was in quadrant 1 during the hidden platform training, and in quadrant 3 during the reversal training. (*) P < 0.05; (ns) not significant.
The same groups of animals were then subjected to a four trials/day reversal learning for 6 d (from day 10 to day 15) and examined by a probe test on day 16 (Fig. 5A). The reversal learning as indicated by the improvement of escape latency was significant for both groups, and not distinguishable between WT and AC1 TG mice (genotype, F(1,19) < 0.1, P = 1.0; genotype by training day, F(5,95) = 0.9, P = 0.5) (Fig. 5B). Although both groups showed preference to the new target quadrant, AC1 TG mice displayed higher preference (TG, 54.4 ± 5.8%; WT, 40.7 ± 4.0%; genotype by quadrant, F(3,57) = 3.0, P = 0.03; TG vs. WT in quadrant 3, P < 0.05, Bonferroni post hoc) (Fig. 5C) and more crossings over the new platform location than WT mice (TG, 4.8 ± 0.7; WT, 2.6 ± 0.4; genotype by quadrant, F(1,19) = 8.1, P = 0.01; TG vs. WT in quadrant 3, P < 0.01, Bonferroni post hoc) (Fig. 5D).
AC1 TG and WT mice show comparable spatial memory extinction
To better examine activity-dependent old memory suppression, we performed memory extinction tests. New cohorts of animals were first trained by hidden platform protocol (four trials/day for 6 d) following the visible platform training (Fig. 6A,B). In the first extinction trial, animals were introduced to the maze without any platform (this is equivalent to a probe test). Both groups showed similar preference to the target quadrant (Fig. 6C) and crossings over the hidden platform location (Fig. 6D). During the subsequent extinction trials, WT and AC1 TG mice showed similar suppression of the old memory. The time spent in the target quadrant (genotype, F(1,20) = 0.7; genotype by extinction trial, F(7,140) = 1.1, both P > 0.4) (Fig. 6C) and platform crossing (genotype, F(1,20) = 0.1; genotype by extinction trial, F(7,140) = 0.3, both P > 0.8) (Fig. 6D) effectively decreased in both groups.
Figure 6.

AC 1 TG mice show effective and comparable spatial memory extinction to that of WT animals. (A) Water maze training and extinction protocol. After 6 d of hidden platform training (B), animals were subject to the pool without the platform four times a day for 2 d. The time that animals spent in the target quadrant (C) and the number of crossing over the target platform location (D) were recorded.
Discussion
Regulation of LTP by the cAMP-PKA cascade has been demonstrated by numerous studies (Nguyen and Woo 2003). Previous reports also show that genetic deletion of Ca2+-stimulated ACs impairs certain forms of LTP both in vitro and in vivo (Wong et al. 1999; Zhang et al. 2011). The data from this study is, in general, consistent with the conclusion that enhancing AC1 facilitates LTP (Wang et al. 2004). Our previous study showed that HFS at 100 Hz resulted in higher LTP in hippocampal slices from AC1 TG mice. However, 100-Hz HFS induced comparable in vivo LTP in AC1 TG and WT mice. This may be mainly attributed to the intrinsic feature of in vivo recording, which does not involve significant disruption of the intact network and circuitry. The magnitude of in vivo LTP is generally smaller and a certain degree of saturation in synaptic potentiation may be reached after the 100-Hz HFS. The data from this study suggest a novel function of AC1 in facilitating the transition from PTP to LTP. As shown in Figure 1D, lowering the stimulation frequency from 100 to 50 Hz induced robust PTP in both WT and AC1 TG mice. However, while the potentiation decayed in WT mice, long-lasting potentiation was maintained in AC1 TG animals.
Garelick et al. (2009) have reported that young adult AC1 TG and WT mice show comparable spatial learning and memory retention. In their study, spatial learning was examined by the performance in a Barnes maze, during which animals were trained by four trials daily for 6 d. Similarly, we also found no difference between AC1 TG and WT mice when they were trained by the four trials/day water maze protocol. However, when trained by the one trial/day protocol, AC1 TG mice showed enhanced spatial learning. Such training/stimulation-dependent difference is also reflected in LTP. AC TG mice showed higher LTP than WT mice following 50-Hz but not 100 Hz-HFS.
In addition to LTP, cAMP and PKA also contribute to bidirectional regulation of synaptic plasticity. Decreasing Ca2+-stimulated AC or PKA activity impairs both LTD and depotentiation (Brandon et al. 1995; Qi et al. 1996; Zhang et al. 2011; but also see Malleret et al. 2010). To our knowledge, this study is the first to demonstrate that increasing Ca2+-stimulated cAMP causes selective deficits in LTD but not depotentiation. The differential requirement of cAMP in LTD and depotentiation implicates different mechanisms involved in activity-dependent weakening of naive synapses vs. previously potentiated synapses. Such possibility is supported by previous findings showing that induction of LTD and depotentiation leads to different changes of GluR1 phosphorylation at distinct serine residuals (Lee et al. 1998, 2000). The enhancement of LTP and defective LTD in AC1 TG mice is consistent with previous studies showing that cAMP-enhancing neuromodulators promote LTP but suppress LTD (Hu et al. 2007; Huang et al. 2012).
Although the contribution of cAMP and PKA to memory retention has been extensively studied, their role in relearning and memory extinction is largely unclear. Several investigations have demonstrated that increasing cAMP impairs fear memory extinction (Wang et al. 2004; Monti et al. 2006). Intriguingly, the extinction mechanisms appear to be different between fear memory and spatial memory. While decreasing PKA activity facilitates fear memory extinction (Isiegas et al. 2006), deletion of Ca2+-stimulated AC impairs reversal learning and memory extinction in the Morris water maze (Zhang et al. 2011). This study, for the first time, demonstrates that enhancing Ca2+-stimulated AC activity facilitates both initial and reversal spatial learning.
It is surprising that AC1 TG mice showed better memory flexibility but impaired LTD, which has been suggested as a possible synaptic substrate for reversal spatial learning and old memory suppression. Normal new memory formation and less suppression of the old memory after reversal training have been observed in a transgenic model that shows normal LTP and depotentiation but defective LTD (Nicholls et al. 2008). A study with D-serine, which facilitates LTD without affecting LTP and depotentiation, has revealed a slightly different outcome. During the probe test following reversal trainings, animals receiving D-serine showed stronger new memory formation but normal suppression of old memory (Duffy et al. 2008). Because another study found that D-serine also modulates LTP (Panatier et al. 2006), it is not clear whether the in vivo dose of D-serine selectively affects LTD but not LTP or depotentiation in free-moving animals. In our previous study, we found that mice lacking Ca2+-stimulated AC had better old spatial memory retention after both reversal and extinction training. However, we could not determine the functional relevance of LTD and depotentiation, because both of them were impaired in the AC mutant mice (Zhang et al. 2011). In this study, our data demonstrate that effective spatial memory extinction and reversal learning may not require intact LTD.
How could AC1 TG mice achieve superior reversal learning and normal extinction in the absence of intact LTD? Previous studies have implicated the involvement of synaptic depotentiation. Similar to extinction, which involves activity-dependent weakening of the previously formed memory, depotentiation requires activity-dependent weakening of the previously established potentiation. There are some implications for the function of depotentiation in amygdala and fear memory extinction. Kim et al. (2007) showed that synaptic depression could only be induced in amygdala slices obtained from trained but not naive animals. Such synaptic depression was referred to as depotentiation, because it was postulated that the synapses from the trained animal had already been potentiated. Strikingly, slices obtained after memory extinction did not show synaptic depression (Kim et al. 2007). Another study demonstrated that delivering LFS to the amygdala of trained animals decreases fear memory (Lin et al. 2003). Moreover, post-training infusion of the PKMζ inhibitor ZIP, which selectively causes depotentiation but not LTD (Serrano et al. 2005), to dorsal hippocampus disrupts the consolidated spatial memory (Serrano et al. 2008). Interestingly, defective reversal learning is very often associated with schizophrenia, and selective depotentiation defects have been reported in both the neurodevelopmental and genetic models of schizophrenia (Sanderson et al. 2012; Shamir et al. 2012).
One possible explanation for the enhanced behavioral flexibility in AC1 TG mice is that the balance of LTP and synaptic depotentiation contributes to effective reversal learning. While LTP at naive synapses may be related to the learning of new platform location, depotentiation at the experienced synapses may be required for old memory suppression. Thus, enhanced LTP and normal depotentiation in AC1 TG mice correlate with the higher ratio of new memory to old memory. Such possibility is also implicated by the poor reversal learning in mice with enhanced LTP and defective depotentiation (Errico et al. 2008). One complication is that effective depotentiation only occurs when electric stimulations (i.e., LFS) are delivered shortly after the onset of LTP (Huang et al. 1999; Staubli and Scafidi 1999). Consolidated synaptic potentiation is resistant to LFS (Huang and Hsu 2001). The reversal learning and extinction measurement in this study were examined a long time (at least more than 24 h) after the consolidation of the old memory. Future studies should investigate whether behavioral stimuli such as reversal learning and extinction can effectively reverse the consolidated synaptic potentiation.
Materials and Methods
Animals
The expression of AC1 transgene is driven by α-CaMK II promoter (Wang et al. 2004). The AC1 TG mice have been bred into C57BL/6 background for more than 12 generations. Animals had free access to food and water, and were kept in the 12-h:12-h light/dark condition. Animal behavior and electrophysiology experiments were performed between ZT 3 to 8. For both behavioral and electrophysiological experiments in this study, we used 2- to 4-mo-old male mice. All manipulations were in compliance with the guidelines and approved by the Institutional Animal Care and Use Committee at Michigan State University.
Morris water maze task
Animal activities were measured by a video-based tracking system (WaterMaze; Coulbourn Instruments). The training apparatus was a 125-cm-diameter pool. The water surface was covered with white polypropylene beads (generous gifts from local Quality Dairy bottling facility). A platform (10 cm in diameter) was placed 1 cm below the water surface. Three water maze protocols were used to examine spatial memory. For the first protocol (as illustrated in Fig. 4A), mice were trained by one trial daily for 6 d to find the hidden platform, and examined by a probe test on day 7. On day 8, we moved the hidden platform to the opposite quadrant and started reversal training. Mice were trained by one trial daily for 6 d to find the new platform position. On day 14, mice were examined by a probe test (the second probe test). Then, the mice were further trained by one trial daily for four additional days (from day 15 to day 18). The third probe test was performed on day 19.
For the second protocol (as illustrated in Fig. 5A), animals were trained by four trials every day for 2 d to find the visible platform, during which the platform was marked with a flag. The visible platform was randomly placed at different locations in each trial. Then, the animals were trained with four trials every day for 6 d (from day 3 to day 8) to find the hidden platform followed by a probe test on day 9. From day 10 to day 15, animals were trained by four trials every day for 6 d to find the reversal hidden platform location. A second probe test was performed on day 16.
For the third protocol (as illustrated in Fig. 6A), animals were trained to find the visible (four trials/day for 2 d) and hidden (four trials/day for 6 d) platform, after which animals were subjected to extinction training. The animals were subjected to eight probe tests (four tests/day with 30-min intervals for 2 d). In each hidden platform trial, mice were allowed to stay on the platform for 30 sec after they found the platform. If mice were not able to find the platform within 90 sec, they were manually guided to the platform. For protocols 2 and 3, the interval between trials was 30 min. During the probe tests, the platform was removed from the pool. Mice were allowed to search the maze for 60 sec. The time that animals spent in each of the four quadrants, as well as the number of crossings over the hidden platform location, was recorded.
In vivo field potential recordings at the CA1 synapses
Mice were deeply anesthetized by pentobarbital (at 100 mg/kg, i.p. administration), and mounted to a stereotaxic frame (David Kopf Instruments). The body temperature was monitored by an anal probe, and maintained at 37°C by a heating pad connected to the Physitemp Controller. Medical oxygen was continuously supplied to the mouse snout during the whole recording procedure. Recording and stimulating electrodes were made of a pair of Teflon-coated wires with 100-μm outer diameter (WPI Inc.). The recording electrode was placed in the striatum radiatum of hippocampal CA1 subarea (AP 1.7–1.9 mm, ML 1.2 mm, and DV 1.5–1.9 mm from skull surface), and the stimulating electrode was placed at the ipsilateral Schaffer/collateral fibers (AP 1.7–1.9 mm, ML 1.7 mm, and DV 1.6–2.0 mm from skull surface). The placements of the electrodes were slowly adjusted to obtain optimal amplitude of the field excitatory postsynaptic potential (fEPSP) according to electrophysiological criteria (Sakata et al. 2000; Wang et al. 2009) and stereotaxic coordinates. Stimulating pulses (0.10-msec duration) with current intensities of 25–300 μA were applied to obtain the input–output curve. An intensity that evoked half of the maximum response was delivered at 0.033 Hz and used to obtain the baseline field potentials. Signals were amplified and recorded by a Powerlab System (AD Instruments).
Induction of LTP and LTD, and depotentiation at the CA1 synapses
After the baseline fEPSP was stabilized for at least 30 min, one train of high-frequency stimulation (HFS, 100 pulses at 50 or 100 Hz) was delivered to induce LTP. As described in our previous study (Zhang et al. 2011), LTD was induced by 900 pulses of stimulation at 1 Hz. To examine synaptic depotentiation, potentiation was first induced by HFS (100 Hz, 1 sec). Five minutes later, LFS (900 pulses at 1 Hz) was delivered.
Data analysis
All data are reported as average ± SEM. Two-way repeated ANOVA and post-ANOVA Tukey multiple comparison tests were used.
Acknowledgments
This work was supported by NIH grant (MH093445 to H.W.).
References
- Abel T, Nguyen PV 2008. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res 169: 97–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R 1997. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88: 615–626 [DOI] [PubMed] [Google Scholar]
- Brandon EP, Zhuo M, Huang YY, Qi M, Gerhold KA, Burton KA, Kandel ER, McKnight GS, Idzerda RL 1995. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI β subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci 92: 8851–8855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S, Labrie V, Roder JC 2008. D-serine augments NMDA-NR2B receptor-dependent hippocampal long-term depression and spatial reversal learning. Neuropsychopharmacology 33: 1004–1018 [DOI] [PubMed] [Google Scholar]
- Errico F, Nistico R, Palma G, Federici M, Affuso A, Brilli E, Topo E, Centonze D, Bernardi G, Bozzi Y, et al. 2008. Increased levels of d-aspartate in the hippocampus enhance LTP but do not facilitate cognitive flexibility. Mol Cell Neurosci 37: 236–246 [DOI] [PubMed] [Google Scholar]
- Garelick MG, Chan GC, DiRocco DP, Storm DR 2009. Overexpression of type I adenylyl cyclase in the forebrain impairs spatial memory in aged but not young mice. J Neurosci 29: 10835–10842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R 2007. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 131: 160–173 [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS 2001. Progress in understanding the factors regulating reversibility of long-term potentiation. Rev Neurosci 12: 51–68 [DOI] [PubMed] [Google Scholar]
- Huang CC, Liang YC, Hsu KS 1999. A role for extracellular adenosine in time-dependent reversal of long-term potentiation by low-frequency stimulation at hippocampal CA1 synapses. J Neurosci 19: 9728–9738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Trevino M, He K, Ardiles A, Pasquale R, Guo Y, Palacios A, Huganir R, Kirkwood A 2012. Pull-push neuromodulation of LTP and LTD enables bidirectional experience-induced synaptic scaling in visual cortex. Neuron 73: 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isiegas C, Park A, Kandel ER, Abel T, Lattal KM 2006. Transgenic inhibition of neuronal protein kinase A activity facilitates fear extinction. J Neurosci 26: 12700–12707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, et al. 2007. Amygdala depotentiation and fear extinction. Proc Natl Acad Sci 104: 20955–20960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF 1998. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21: 1151–1162 [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL 2000. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405: 955–959 [DOI] [PubMed] [Google Scholar]
- Lin CH, Lee CC, Gean PW 2003. Involvement of a calcineurin cascade in amygdala depotentiation and quenching of fear memory. Mol Pharmacol 63: 44–52 [DOI] [PubMed] [Google Scholar]
- Malleret G, Alarcon JM, Martel G, Takizawa S, Vronskaya S, Yin D, Chen IZ, Kandel ER, Shumyatsky GP 2010. Bidirectional regulation of hippocampal long-term synaptic plasticity and its influence on opposing forms of memory. J Neurosci 30: 3813–3825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, et al. 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396: 433–439 [DOI] [PubMed] [Google Scholar]
- Monti B, Berteotti C, Contestabile A 2006. Subchronic rolipram delivery activates hippocampal CREB and arc, enhances retention and slows down extinction of conditioned fear. Neuropsychopharmacology 31: 278–286 [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M 2007. Mechanisms of fear extinction. Mol Psychiatry 12: 120–150 [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH 2003. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol 71: 401–437 [DOI] [PubMed] [Google Scholar]
- Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S, Kandel ER 2008. Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron 58: 104–117 [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH 2006. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125: 775–784 [DOI] [PubMed] [Google Scholar]
- Qi M, Zhuo M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL 1996. Impaired hippocampal plasticity in mice lacking the Cβ1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci 93: 1571–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata K, Tokue A, Kawai N 2000. Altered synaptic transmission in the hippocampus of the castrated male mouse is reversed by testosterone replacement. J Urol 163: 1333–1338 [PubMed] [Google Scholar]
- Sanderson TM, Cotel MC, O’Neill MJ, Tricklebank MD, Collingridge GL, Sher E 2012. Alterations in hippocampal excitability, synaptic transmission and synaptic plasticity in a neurodevelopmental model of schizophrenia. Neuropharmacology 62: 1349–1358 [DOI] [PubMed] [Google Scholar]
- Serrano P, Yao Y, Sacktor TC 2005. Persistent phosphorylation by protein kinase Mζ maintains late-phase long-term potentiation. J Neurosci 25: 1979–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P, Friedman EL, Kenney J, Taubenfeld SM, Zimmerman JM, Hanna J, Alberini C, Kelley AE, Maren S, Rudy JW, et al. 2008. PKMζ maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biol 6: 2698–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamir A, Kwon OB, Karavanova I, Vullhorst D, Leiva-Salcedo E, Janssen MJ, Buonanno A 2012. The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J Neurosci 32: 2988–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Lynch G 1990. Stable depression of potentiated synaptic responses in the hippocampus with 1–5 Hz stimulation. Brain Res 513: 113–118 [DOI] [PubMed] [Google Scholar]
- Staubli U, Scafidi J 1999. Time-dependent reversal of long-term potentiation in area CA1 of the freely moving rat induced by θ pulse stimulation. J Neurosci 19: 8712–8719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Storm DR 2003. Calmodulin-regulated adenylyl cyclases: Cross-talk and plasticity in the central nervous system. Mol Pharmacol 63: 463–468 [DOI] [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR 2004. Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci 7: 635–642 [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang M, Moon C, Hu Q, Wang B, Martin G, Sun Z, Wang H 2009. The APP-interacting protein FE65 is required for hippocampus-dependent learning and long-term potentiation. Learn Mem 16: 537–544 [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR 1999. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 23: 787–798 [DOI] [PubMed] [Google Scholar]
- Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, Palmiter RD, Storm DR 1995. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci 92: 220–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Storm DR, Wang H 2011. Bidirectional synaptic plasticity and spatial memory flexibility require Ca2+-stimulated adenylyl cyclases. J Neurosci 31: 10174–10183 [DOI] [PMC free article] [PubMed] [Google Scholar]