

Abstract
RAGE, a receptor for advanced glycation endproducts, is an immunoglobulin-like cell surface receptor that is often described as a pattern recognition receptor due to the structural heterogeneity of its ligand. RAGE is an important cellular cofactor for amyloid β-peptide (Aβ)-mediated cellular perturbation relevant to the pathogenesis of Alzheimer's disease (AD). The interaction of RAGE with Aβ in neurons, microglia, and vascular cells accelerates and amplifies deleterious effects on neuronal and synaptic function. RAGE-dependent signaling contributes to Aβ-mediated amyloid pathology and cognitive dysfunction observed in the AD mouse model. Blockade of RAGE significantly attenuates neuronal and synaptic injury. In this review, we summarize the role of RAGE in the pathogenesis of AD, specifically in Aβ-induced cellular perturbation.
Keywords: Amyloid beta, RAGE, Alzheimer Disease, Inflammation, Review
2. Introduction
Alzheimer's disease (AD) is the most common form of dementia affecting the elderly. Clinically, AD is characterized by a progressive decline in cognitive function characterized by memory loss as well as personality changes. AD is pathologically characterized by the presence of senile plaques containing amyloid-β (Aβ) protein and neurofibrillary tangles that consist mainly of intracellular and abnormally phosphorylated tau protein (1-6), as well as severe gliosis in the cerebral cortex and the hippocampus (7). Recent studies suggest an increase in inflammatory responses such as an increase in production of proinflammatory mediators, microglial infiltration and activation, and levels of several SI00 calcium-binding proteins (S100B, S100A6, S100A9, and S100A12) in the AD brain.
Aβ is a neurotoxic peptide, the accumulation of which leads to cellular perturbations including impaired energy metabolism and mitochondrial respiratory function, increased oxidative stress, neuroinflammation. synaptic failure, and neurodegeneration (6, 8-15). Proteolytic cleavage of the transmembrane β-amyloid precursor protein (APP) leads to generation of 1-40 or 1-42 amino acid Aβ, which can form soluble oligomers, beta sheets containing fibrils, and insoluble aggregates (7). Accumulation of Aβ in the brain plays a key role in the development of AD and the mutation in APP increases Aβ accumulation in the brains of AD patients and transgenic AD mice (9, 16). In fact, multiple lines of evidence frame Aβ oligomers and Aβ fibrils as the main culprits behind the synaptic dysfunction and neuronal death observed in AD patients (7). Oligomer Aβ interferes with long term potentiation (LTP) and cognitive processes, suggesting the causal role of Aβ peptides in the neuronal dysfunction that characterizes the early stages of AD (17-21). However, the mechanisms underlying Aβ-mediated neuronal and synaptic dysfunction have not yet been fully elucidated.
As expected for a pleiotropic peptide such as Aβ, many cell surface interaction sites and intracellular proteins have been reported, ranging from cell surface receptors, such as receptor for advanced glycation endproducts (RAGE) (10, 17-22), α-7-acetylcholine receptor (α7-nAChRs) (23), type A macrophage scavenger receptor (MSR) (24), integrins (25). insulin receptor (26), and P75 neurotrophin receptor (P75NTR) (27-28), LRP-1 (29) to cell-associated proteoglycans (30), mitochondrial proteins, such as amyloid-β (Aβ) peptide-binding alcohol dehydrogenase (ABAD) (31-35) and cyclophilin D (5, 9). The pathogenic significance of these interactions remains to be further defined. It is likely that several cell cofactors contribute to the various cellular effects of AB. Thus, to gain further insight into this complex system, these interactions must be dissected at the level of the individual cellular cofactor.
2.1. Structure of RAGE
RAGE has several isoforms, each with a distinct tissue-specific expression pattern (7). Among the various alternatively spliced isoforms of RAGE, two prevalent isoforms are full-length RAGE and the secreted isoform of soluble RAGE (7). Full-length RAGE is composed of an extracellular element consisting of one Ig-like V-domain followed by two Ig-like C-type domains, a transmembrane domain consisting of a single helix and a short cytosolic domain, which allows for signal transduction (7-8). Soluble RAGE (sRAGE) consists solely of the extracellular part including V, CI, and C2 domains with no transmembrane domain, and is thus released into the extracellular space (7). X-ray crystal structure analysis demonstrated that V and CI domains comprise the S100B binding domain through a highly electropositive surface (36, 37). The same domain is presumed to also bind Aβ, which is known to be negatively charged. Additionally, the structural data suggest that RAGE has features typical of I-set topology, which is characteristic for cell adhesion molecules and ligand-induced oligomerization within cytoplasmic membrane might be the mechanism for its activation (37).
3. The Role of Rage in the Aβ-Induced Cellular and Molecular Events Relevant to the Pathogenesis of Alzheimer's Disease
3.1. Increased expression of RAGE in Alzheimer's disease
Studies on human AD brains reveal increased RAGE expression in neuronal, microglial and endothelial cells when compared to age-matched control subjects without AD (7, 18, 38-39). Cells around senile plaques express higher levels of RAGE during disease progression (7). Increased RAGE expression was found in microglia from AD brains when compared to those from age-matched, nondemented control brains (18, 38). Lue and colleagues reported that microglia in AD-affected regions, including the hippocampus, had higher levels of RAGE in the AD brains than that in the age-matched non-AD controls. Furthermore, expression levels of RAGE are correlated to the severity of the disease (clinical score of the amyloid plaque and tangle, respectively) (38). Similarly, vasculature with Aβ deposits from an AD patient also displayed increased RAGE antigen as compared to age-matched non-AD controls (18, 38-41). In the recent study, Lue and colleagues reported a 60% increase in RAGE protein levels in the blood vessels from cerebral amyloid angiopathy (CAA) patients, compared with vessels from age-matched non-AD controls (42).
Meanwhile, expression of RAGE_v1, the prevalent isoform of RAGE in endothelial cells and in the human brain, is reduced in hippocampal neurons of AD patients (43), since RAGE_vl is the dominant negative (ND) form of RAGE. Reduction of RAGE_vl may lead to sustained RAGE activation. On the other hand, the soluble isoform of RAGE (sRAGE) can prevent the adverse effects of RAGE signaling by acting as a decoy that binds to RAGE ligands thereby preventing the interaction of RAGE with its ligands. Recent studies comparing the concentration of sRAGE in the serum of patients versus controls in various pathophysiological condition, demonstrate both positive and negative correlation between the concentration of sRAGE and the severity of the disease (7). sRAGE levels were significantly reduced in the plasma of patients with AD or mild cognitive impairment (MCI) (44-45). Levels of sRAGE in the plasma are related to the level of cognitive impairment in AD and MCI patients (46). These results suggest that change in RAGE expression levels might impact the cellular perturbation relevant to the development and progression of AD. Recent studies suggest that the RAGE G82S polymorphism is associated with the AD. The plasma sRAGE levels were also lower in AD than in normal elderly controls and the presence of the risk allele was associated with further plasma sRAGE reduction and quicker/faster cognitive deterioration (47). Thus, the RAGE G82S variant might be involved in genetic susceptibility to AD.
In a transgenic AD mouse model, RAGE expression was elevated in mice expressing a mutant form of human APP (mAPP). Increased levels of RAGE were observed in the neurons and microglia of these animals as they age and accumulate Aβ (8, 19, 38).
3.2. RAGE-Aβ interaction mediates neuroinflammation
Deficient as well as excessive responses in neuroinflammation and microglial activation can result in pathological conditions (8, 48-50). The innate responses of glia to injury, foreign pathogen, or activating stimuli generally lead to beneficial outcomes, such as phagocytosis or production of reparative or protective factors; however, sustained activation of microglia and overproduction of proinflammatory mediators disturbs homeostasis, resulting in disease progression and exacerbation of induction factors that influence the severity of neuronal dysfunction and the progression of neuropathology (8, 48, 51-55). The precise mechanisms by which Aβ mediates activation of microglia and astrocytes remain to be elucidated. It appears that there is an important role for RAGE-mediated signaling in the microglial activation and neuronal dysfunction relevant to AD pathology.
Microglia play a critical role in Aβ-mediated neuronal perturbation and death present in the pathogenesis of AD. Many inflammatory mediators detected in AD brains are of microglial origin (8, 50, 55-59). Increased microglial activation, microglial association with senile plaques, and elevated levels of proinflammatory mediators (i.e. cytokines, chemokines and free radicals) have all been observed in the AD brain and AD mouse models and evidence indicates that the aforementioned contribute to neuronal damage (8, 49, 61). Pathological analyses of brains from AD patients and AD-type-mouse models (including transgenic mouse systems and direct infusion of Aβ into brain parenchyma) show that activated microglia and astrocytes accumulate to the greatest extent in the proximity of amyloid plaques (8). Studies carried out with an in vitro cell culture model show that direct administration of Aβ to multiple cell types can induce cellular stress, and this effect is probably increased in the presence of activated microglia (8, 38).
RAGE triggers the generation of proinflammatory cytokines at the blood brain barrier (7). Lue and colleagues cultured microglia from AD and control brains to show that increases in macrophage-colony stimulating factor (M-CSF) production in the presence of Aβl-42 in AD-derived microglia was a RAGE-dependent process (40). Further, in gene array studies of microglia retrieved from AD brains, there was an up-regulation of multiple proinflammatory cytokines and matrix metalloproteinases (40).
In the transgenic mouse model of AD, RAGE dependent signaling in microglia stimulates inflammatory responses and processes that exacerbate neuronal damage. ultimately impairing neuronal function (8, 38). Fang and colleagues demonstrated that overexpression of microglia RAGE in Tg APP mice exacerbates neuroinflammation, as evidenced by increased proinflammatory mediator production, Aβ accumulation, impaired learning and memory, and neurotoxicity (8). Transgenic mice expressing human mutant APP in neurons and RAGE in microglia (mAPP/RAGE mice) display: 1) age-dependent enhancement of interleukin -1 β (IL-1β) and tumor necrosis factor- alpha (TNF-α) production in the cerebral cortex, two months prior to that observed in mAPP mice; 2) increased plaque-associated microglial clusters and astrocyte infiltration when compared to mAPP mice at 9-10 months of age; 3) Aβ accumulation; 4) reduced acetylcholine esterase activity; 5) and accelerated deterioration of spatial learning/memory.
Neuropathological changes are accelerated in mAPP/RAGE mice and occur as early as 4-5 months of age, whereas transgenic mutant APP mice do not display such abnormalities at that age. Introduction of the DN-RAGE transgene, a signal transduction-defective mutant form of RAGE, into the microglia of mAPP mice delayed and attenuated increases in brain cytokine levels as well as the deterioration induced by Aβ from 2 to 10 months of age. Notably, in the absence of RAGE ligands, it does not appear that DN-RAGE or RAGE has an effect on cytokine production, as evidenced by the comparable levels of IL-1β and TNF-alpha among single Tg RAGE, DN-RAGE and nonTg littermate controls (8). mAPP/DN-RAGE mice do not show significant microglial and astrocyte infiltration in the cerebral cortex and hippocampus. Introduction of DN-RAGE into mAPP mice may exert a protective effect against accelerated learning and memory deterioration.
Extensive evidence supports the theory that the p38 MAPK (mitogen-activated protein kinase) signaling cascade contributes to cytokine overproduction and the neurodegenerative effects seen in AD (8, 20, 54-55, 62-63). For instance, activation of p38 MAPK was found in early stage AD brain and in an AD mouse model, while inhibition of p38 MAPK activation blocks Aβ-mediated cytokine production and neuronal death (8, 19-20, 51-52, 64-65). In brains of mAPP mice with increased expression of microglial RAGE, levels of p38 and extracellular-signal-regulated kinases 1/2 (ERK1/2) phosphorylation were significantly higher when compared to the levels found in the brains of transgenic mAPP mice without over-expression of RAGE; cytokine production and exaggerated neuronal stress are also seen with increased microglial RAGE expression (8).
Since expression of microglial DN-RAGE in mAPP mice attenuates Aβ-mediated detrimental effects, it has been suggested that RAGE-dependent activation of p38 and ERK1/2 MAPK pathways is at least partially responsible for Aβ-mediated microglial activation and the induction of proinflammatory mediators (8). Fang and colleagues showed that phosphorylated p38 and ERK1/2 levels were significantly increased in brain extracts of mAPP and mAPP/RAGE mice, as compared to nonTg mice; further, mAPP/RAGE brains exhibited even higher levels of phosphorylated p38 and ERK1/2 than the mAPP brain. mAPP/DN-RAGE mice showed significantly less p38 and ERK1/2 phosphorylation, as compared to mAPP/RAGE mice. Taken together, these findings indicate the existence of RAGE-dependent activation of MAPK p38 and ERK1/2 in microglia (8). Importantly, findings by Fang and colleagues also indicate that microglial RAGE contributes significantly to increases in neuroinflammation, Aβ accumulation, and neuronal perturbation in an Aβ-rich environment (8), suggesting that RAGE-dependent signal transduction in MAP kinase (i.e., p38 and ERK1/2) activation is an important mechanism underlying Aβ-involved neuronal inflammation and neuronal injury.
Elevated levels of AGEs or Aβ at sites of inflammation can trigger RAGE-dependent oxidative stress and NF-κB activation, which in turn leads to further increased RAGE expression because of the presence of NF-κB response elements within the promoter region of RAGE; thus, activation of RAGE sustains NF-κB activation (7, 19, 41). This process represents a potential positive feedback loop between RAGE, oxidative stress and inflammation. In various cell types, including neurons, endothelial cells and microglia, RAGE-Aβ interaction can lead to the formation of reactive oxygen species (ROS), the activation of NF-κB, or the expression of cell adhesion molecules mediating the recruitment of inflammatory cells (7, 40-41). Interestingly, administration of sRAGE to cells in vitro or animals in vivo attenuates RAGE-mediated cellular perturbation. For example, transgenic APP mice that received sRAGE show significantly reduced amyloid accumulation in the brain and improved vascular and synaptic function (22, 66).
3.3. RAGE mediates neuronal and synaptic stress
The interaction of RAGE with Aβ affects neuronal function as evidenced by in vitro cell culture (18, 41, 67-68) and in vivo animal model studies (17, 19-22). Studies on the AD-type mouse model provide substantial evidence that RAGE serves as a cellular cofactor in Aβ-induced neuronal stress. Transgenic APP mice overexpressing neuronal RAGE and Aβ (Tg RAGE/mAPP) exhibited earlier onset of spatial learning/memory function abnormalities and neuropathological changes as compared to animals expressing only mutant APP (19).
Analysis of synaptic function by measurement of synaptic transmission including both basal synaptic transmission (BST) and LTP states in the hippocampus including CA1 stratum radiatum, revealed that BST was abnormal in both transgenic mAPP mice and transgenic RAGE/mAPP mice, whereas LTP abnormality was observed in transgenic RAGE/mAPP but not in transgenic mAPP mice. These results imply that overexpression of neuronal RAGE in transgenic mAPP mouse brain accelerates synaptic dysfunction further impairing synaptic plasticity. In experiments to control for the effect of APP over-expression independent of Aβ production, we tested learning memory performance with the radial-arm water-maze in transgenic RAGE/wild-type (wt) APP mice. These mice are known to produce much lower amounts of Aβ 1 -40 and Aβ 1-42. We found that overexpression of wtAPP did not further impair learning and memory in transgenic RAGE/wtAPP mice. Thus, the effect of RAGE on spatial working memory is likely due to its interaction with the overproduction of Aβ. Consistent with the results from behavioral and electrophysiological experiments, transgenic RAGE/mAPP mice displayed early changes in neuropathology and showed a decrease in acetylcholinesterase-positive neurites in AD-affected regions including subiculum, entorhinal cortex and CA1 region as early as 3-4 months of age, that then proceeded to develop cerebral Aβ deposition. Notably, introduction of DN-RAGE into transgenic mAPP mice significantly improved learning/memory, preserved LTP, and alleviated neuropathology, suggesting the involvement of RAGE-dependent signal transduction in neuronal damage due to Aβ (19).
Synaptic dysfunction is an early pathological feature of AD (69). Impaired memory and synaptic loss occur before extensive deposition of Aβ in the brains of AD-type murine models and in AD patients (70-78). These observations suggest that early in AD, when levels of Aβ are low, mechanisms amplifying and focusing the effects of Aβ on cellular targets contribute to neuronal dysfunction. Our studies have demonstrated that blockade of RAGE reversed Aβ-induced synaptic dysfunction in AD-affected regions including hippocampus, entorhinal cortex, and visual cortex (17, 19-20). Thus, it appears that RAGE-triggered signal transduction contributes to synaptic dysfunction.
3.4. RAGE and amyloid accumulation in Alzheimer's disease brain
RAGE has been shown to mediate the transport of Aβ through the neuronal cell membrane and the blood brain barrier (7, 22). Administration of sRAGE to animals overexpressing cerebral Aβ significantly reduced Aβ accumulation in the brain. In parallel, Aβ levels were increased in the plasma of sRAGE-treated APP mice. Furthermore, Aβ-sRAGE complex was found in the plasma of those mice (22). These data suggest that sRAGE functions as a decoy peptide: binding to circulating Aβ in the plasma, regulating equilibrium of Aβ between brain parenchyma and the peripheral circulation, thereby enhancing clearance of Aβ.
The role of RAGE-Aβ interaction in exacerbating amyloid pathology was further investigated in transgenic mice overexpressing neuronal RAGE and Aβ. Transgenic RAGE/mAPP mice demonstrated significantly higher Aβ accumulation, as shown by ELISA and quantitative immunohistochemistry for amyloid plaque load in the cortex and hippocampus (39). Importantly, young RAGE/mAPP mice (3-4 months old) displayed functional and pathological evidence of neuronal perturbation, preceding accumulation of cerebral Aβ and plaque formation (19). Recently. Vodopivec reported that there were lower levels of Aβ extractable by SDS and formic acid (insoluble Aβ) in the brains of RAGE-/-/arcAβ mice at the age of 6 months, but that this effect disappeared by the age of 12 months (79), suggesting the significance of RAGE in an early stage of amyloid accumulation. However, lack of RAGE did not significantly improve cognitive function in arcAβ animals using Y maze performance (79). This could be explained by experimental methodologies and animal models. Interestingly, serum levels of Aβ40 in RAGE-/-/arcAβ mice were significantly lower than those in the arcAβ animal at the age of 12 months (79), suggesting a possible relevance of RAGE to A(3 transport across the blood-brain barrier for cerebral Aβ accumulation, though deficiency in RAGE does not affect extracellular amyloid deposits in arcAβ animal model (79)
Microglia are believed to play an active role in regulating Aβ levels and the amyloid burden in the brain. Overexpression of RAGE in microglia provokes significant increases in Aβ levels in the hippocampus and cortex (8). Fang and colleagues assessed the levels of neocortical and hippocampal Aβ by ELISA in brain extracts prepared from transgenic mice at 5 and 9-10 months of age; at 5 months, levels of Aβ were relatively low in all groups, but these same levels were observed to be significantly higher in mAPP/RAGE mice as compared to single mAPP mice; at 9-10 months a significant increase in Aβ levels was observed in the brains of mAPP mice, but mAPP/RAGE mice displayed significantly more Aβ in the hippocampus and cortex when compared to mAPP mice (8). Introduction of the DN-RAGE transgene into mAPP mice resulted in lower Aβ levels (8). Additionally, in the hippocampus and cerebral cortex of mAPP/RAGE mice, immunoreactive Aβ deposits occupied a larger area and were more extensively distributed as compared to mAPP mice at 9-10 months of age (8). However, the amyloid burden in the hippocampus and cerebral cortex of mAPP/DN-RAGE mice was notably reduced as compared to that observed in both mAPP and mAPP/RAGE mice (8). Studies indicate that the overexpression of microglial RAGE increases plaque load in animals and that RAGE signal transduction in microglia plays a critical role in amyloid accumulation in the hippocampus and cerebral cortex (8).
It has been suggested that in an Aβ-rich environment, RAGE action favors Aβ accumulation; this could result from increased production of Aβ or from mechanisms preventing its clearance (8). It has been suggested that RAGE-mediated neuroinflammation impairs protective clearance mechanisms, perhaps by sequestering pathogenic Aβ species in an inflammatory environment or down-regulating specific clearance mechanisms (8, 80-81). Since there were no significant differences in hAPP and endogenous mouse APP protein expression among mice carrying mAPP, mAPP/RAGE or mAPP/DN-RAGE transgenes, Aβ accumulation in Tg mAPP/RAGE mice is not simply due to increased hAPP transgene or endogenous APP expression (8). Data from Fang and colleagues suggest that microglial RAGE expression does not significantly affect hAPP transgene expression or Aβ clearance by insulin degrading enzyme, a process that is involved in cerebral Aβ accumulation in mice (82); instead, other RAGE-dependent processes might be involved. An increase in Aβ generation as an indirect effect of microglial mediators affecting neurons or other cell types can not be ruled out (8). RAGE-mediated induction of proinflammatory mediators enhances Aβ accumulation through a positive feedback loop stimulating the RAGE receptor, which further exaggerates neuroinflammation and amyloid pathology (8). These results are consistent with an increasing body of evidence that correlates inflammation with Aβ levels in transgenic mice expressing mutant APP and in AD patients (59, 61, 83). In this context, anti-inflammatory drugs, such as ibuprofen, have been shown to reduce plaque pathology and brain Aβ (61) levels in animal models of AD. Increased induction of proinflammatory mediators such as TNF-α, IL-lβ, or interferon-γ is also associated with neuronal damage in an Aβ rich environment, as well as Aβ accumulation (8, 19, 50, 81, 84-86). In fact, polymorphisms in the regulatory regions of these cytokines are associated with a higher risk for developing AD (83).
3.5. Other RAGE ligands in Alzheimer's disease
Other RAGE ligands might also be associated with pathogenesis of AD. RAGE was initially identified as a receptor for the advanced glycation endproduct (AGE) (87-88). AGE is increased in diabetes, and it also occurs in normal aging and neurodegenerative diseases including AD. Both Aβ and phosphorylated tau, which are key components of AD pathology (amyloid and tau pathology), have been found to be present in non-enzymatic glycation with AGE formation (89-97). AGE modified proteins produce high levels of oxidative stress, increase release of Aβ, and exaggerate neuronal injury (90-92, 97-98).
Endogenous RAGE ligand, SI00 could have potential effects on the proinflammatory response in Aβ rich environments. The RAGE ligand S100B can be secreted into the extracellular space where it takes on the role of a cytokine (7). Two other cytokine-like SI00 proteins, S100A9 and S100A12, which interact with RAGE and trigger RAGE dependent cellular signaling leading to sustained inflammation, have been identified at elevated levels in the microglia of patients suffering from sporadic AD (7). S100A12 mediates chemotaxis of human peripheral blood-derived mononuclear phagocytes (40). Additionally, in RAGE expressing Bv2 microglia-type cells, S100A12 stimulates production of IL-iβ and TNF-alpha and activates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); such effects are suppressed in the presence of a dominant negative (DN) form of RAGE (RAGE with the cytoplasmic domain deleted) and thus unable to engage in signal transduction (39). Overexpression of S100B in APP mice (Tg2576), one of the AD mouse models, accelerates brain inflammation and neuronal dysfunction as shown by astrogliosis and microgliosis, induction of proinflammatory mediators and AD-like pathology (99-100). Thus, we anticipate that RAGE/S100 interaction might be an important mechanism underlying AD.
4. Conclusion
As shown herein, multiple lines of evidence indicate that RAGE is an important cellular target for Aβ-mediated perturbation. Aβ interacts with RAGE on the surface of microglia, neurons, and cells in the vasculature. These interactions exacerbate AD-type pathology including impaired blood brain barrier and vascular function, neuronal stress, activation of microglia and proinflammatory pathways, and deficits in learning/memory in mouse models of Alzheimer's disease. RAGE-dependent signaling in microglia contributes to neuroinflammation and Aβ accumulation, which in turn enhances neuropathological and behavioral changes in the animal models. Blockade of RAGE significantly attenuates Aβ-mediated, sustained neuronal and microglial stress, and improves cognitive and vascular function in AD mouse models (Figure 1). Taken together, the studies described here provide substantial support for targeting RAGE as a therapeutic approach in AD; indeed a RAGE antagonist has already been developed and has demonstrated a protective effect in an animal model (100). The RAGE inhibitor has an excellent safety profile, and has been well-tolerated for over 10 weeks in patients with AD in oral treatments according to results of a Phase II clinical study (102). RAGE inhibitors thus hold a potential for therapeutic advance in halting AD.
Figure 1.
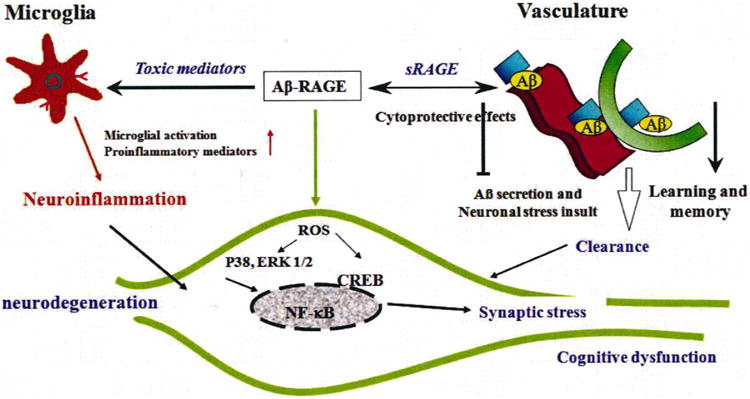
Evidence of RAGE-mediated cellular perturbation in Alzheimer's brain. Aβ -RAGE interaction exerts its toxic effects on vascular cells, microglia, and neurons. RAGE is involved in Aβ transport across the blood-brain barrier and accumulation of Aβ in brain, leading to a decrease in Aβ clearance and neuronal insults. Inhibition of RAGE-ligand interaction via sRAGE suppresses accumulation of Aβ in brain parenchyma in an AD mouse model. Microglial RAGE interaction with Aβ leads to increased production of proinflammatory mediators and microglia migration/infiltration, which increases neuroinflammation and neuronal damage. Aβ can directly interact with neuronal RAGE and provoke oxidative stress through the generation of reactive oxygen species (ROS) and activation of MAP kinase (P38 and Erk1/2) signalling pathways, subsequently triggering activation of nuclear transcription NF-kB and CREB. Together, these events eventually initiate synaptic and neuronal injury and cognitive dysfunction.
Acknowledgments
This work was supported by grants from the USPHS (POlAG 17490 and AG037319) and Alzheimer Association.
Abbreviations
- AD
Alzheimer Disease
- Aβ
Amyloid beta
- LTP
long term potentiation
- RAGE
receptor for advanced glycation endproducts
- MSR
macrophage scavenger receptor
- CAA
cerebral amyloid angiopathy
- sRAGE
soluble isoform of RAGE
- mAPP
mutant form of human APP and Aβ
- IL-1β
interleukin -1 β
- TNF-alpha
tumor necrosis factor- alpha
- DN-RAGE
signal transduction-defective mutant form of RAGE
- MAPK
mitogen-activated protein kinase
- ERK
extracellular-signal-regulated kinases
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- TGF-beta
transforming growth factor-beta
- ROS
reactive oxygen species
- BST
basal synaptic transmission
References
- 1.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 3.Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckert A, Hauptmann S, Scherping I, Meinhardt J, Rhein V, Drose S, Brandt U, Fandrich M, Muller WE, Gotz J. Oligomeric and fibrillar species of beta-amyloid (A beta 42) both impair mitochondrial function in P301L tau transgenic mice. Journal of molecular medicine (Berlin, Germany) 2008;86:1255–1267. doi: 10.1007/s00109-008-0391-6. [DOI] [PubMed] [Google Scholar]
- 5.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. Faseb J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 7.Leclerc E, Sturchler E, Vetter S. The S100B/RAGE Axis in Alzheimer's Disease. Cardiovasc Psychiatry Neurol. 2010;2010:1–11. doi: 10.1155/2010/539581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Schmidt AM, Chen JX, Yan SS. RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease. FASEB J. 2010;24:1043–1055. doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, Sosunov A, McKhann G, Funatsu Y, Nakamichi N, Nagai T, Mizoguchi H, Ibi D, Hori O, Ogawa S, Stern DM, Yamada K, Yan SS. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci USA. 2009;106:20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer's disease. J Alzheimers Dis. 2010;20(Suppl 2):S569–578. doi: 10.3233/JAD-2010-100357. [DOI] [PubMed] [Google Scholar]
- 12.Du H, Yan SS. Mitochondrial permeability transition pore in Alzheimer's disease: cyclophilin D and amyloid beta. Biochim Biophys Acta. 2010;1802:198–204. doi: 10.1016/j.bbadis.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen JX, Yan SD. Pathogenic role of mitochondrial amyloid-beta peptide. Expert Rev Neurother. 2007;7:1517–1525. doi: 10.1586/14737175.7.11.1517. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, Yan SD. Mitochondrial Abeta: a potential cause of metabolic dysfunction in Alzheimer's disease. IUBMB Life. 2006;58:686–694. doi: 10.1080/15216540601047767. [DOI] [PubMed] [Google Scholar]
- 15.Du H, Yan SS. Mitochondrial permeability transition pore in Alzheimer's disease: Cyclophilin D and amyloid beta. Biochimica et biophysica acta. 2009 doi: 10.1016/j.bbadis.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Origlia N, Capsoni S, Cattaneo A, Fang F, Arancio O, Yan SD, Domenici L. Abeta-dependent Inhibition of LTP in different intracortical circuits of the visual cortex: the role of RAGE. J Alzheimers Dis. 2009;17:59–68. doi: 10.3233/JAD-2009-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stem D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 19.Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stem A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, Stern DM, Du Yan SS. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. Embo J. 2004;23:4096–4105. doi: 10.1038/sj.emboj.7600415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci. 2008;25:3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Origlia N, Bonadonna C, Rosellini A, Leznik E, Arancio O, Yan SS, Domenici L. Microglial Receptor for Advanced Glycation End Product-Dependent Signal Pathway Drives {beta}-Amyloid-Induced Synaptic Depression and Long-Term Depression Impairment in Entorhinal Cortex. J Neurosci. 2010;30:11414–11425. doi: 10.1523/JNEUROSCI.2127-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nature medicine. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 23.Liu Q, Kawai H, Berg DK. Beta -Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4734–4739. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 25.Sabo S, Lambert MP, Kessey K, Wade W, Krafft G, Klein WL. Interaction of beta-amyloid peptides with integrins in a human nerve cell line. Neuroscience letters. 1995;184:25–28. doi: 10.1016/0304-3940(94)11159-g. [DOI] [PubMed] [Google Scholar]
- 26.Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer's beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22:RC221. doi: 10.1523/JNEUROSCI.22-10-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. The Journal of clinical investigation. 1997;100:2333–2340. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fombonne J, Rabizadeh S, Banwait S, Mehlen P, Bredesen DE. Selective vulnerability in Alzheimer's disease: amyloid precursor protein and p75(NTR) interaction. Annals of neurology. 2009;65:294–303. doi: 10.1002/ana.21578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deane R, Wu Z, Sagare A, Davis J, Yan SD, Hamm K, Xu F, Parisi M, LaRue B, Xu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron. 2004;43(3):333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 30.Snow AD, Kinsella MG, Parks E, Sekiguchi RT, Miller JD, Kimata K, Wight TN. Differential binding of vascular cell-derived proteoglycans (perlecan, biglycan, decorin, and versican) to the beta-amyloid protein of Alzheimer's disease. Archives of biochemistry and biophysics. 1995;520:84–95. doi: 10.1006/abbi.1995.1345. [DOI] [PubMed] [Google Scholar]
- 31.Yan SD, Shi Y, Zhu A, Fu J, Zhu H, Zhu Y, Gibson L, Stem E, Collison K, Al-Mohanna F, Ogawa S, Roher A, Clarke SG, Stem DM. Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Abeta-induced cytotoxicity. J Biol Chem. 1999;274:2145–2156. doi: 10.1074/jbc.274.4.2145. [DOI] [PubMed] [Google Scholar]
- 32.Yan Y, Liu Y, Sorci M, Belfort G, Lustbader JW, Yan SS, Wang C. Surface plasmon resonance and nuclear magnetic resonance studies of ABAD-Abeta interaction. Biochemistry. 2007;46:1724–1731. doi: 10.1021/bi061314n. [DOI] [PubMed] [Google Scholar]
- 33.Lustbader JW, Cirilli M, Lm C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stem D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 34.Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stem E, Saido T, Tohyama M, Ogawa S, Roher A, Stem D. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 1997;389:689–695. doi: 10.1038/39522. [DOI] [PubMed] [Google Scholar]
- 35.Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, Trillat AC, Stem DM, Arancio O, Yan SS. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. Faseb J. 2005;19:597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 36.Park H, Boyington JC. The 1.5 Å crystal structure of human receptor for advanced glycation endproducts (RAGE) ectodomains reveals unique features determining ligand binding. J Biol Chem. 2010 Dec 24;285(52):40762–70. doi: 10.1074/jbc.M110.169276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koch M, Chitayat S, Dattilo BM, Schiefiner A, Diez J, Chazin WJ, Fritz G. Structural Basis for Ligand Recognition, Activation of RAGE. Structure. 2010 Oct 13;18(10):1342–52. doi: 10.1016/j.str.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stem DM, Yan SD. Involvement of microglial receptor for advanced glycation end products (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol. 2001;171:29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 39.Chen X, S D, Yan SD. Cellular targets of amyloid a peptide: potential roles in neuronal cell stress and toxicity. Third. Oxford University Press; 2007. [Google Scholar]
- 40.Yan SF, Du Yan S, Ramasamy R, Schmidt AM. Tempering the wrath of RAGE: An emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Annals of medicine. 2009:1–15. doi: 10.1080/07853890902806576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stem D, Schmidt AM. Amyloid-beta peptide-receptor for advanced glycation end product interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:5296–5301. doi: 10.1073/pnas.94.10.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lue LF, Yan SD, Stem DM, Walker DG. Preventing activation of receptor for advanced glycation endproducts in Alzheimer's disease. Curr Drug Targets CNS Neurol Disord. 2005;4:249–266. doi: 10.2174/1568007054038210. [DOI] [PubMed] [Google Scholar]
- 43.Leclerc E, Sturchler E, Vetter SW. The S100B/RAGE Axis in Alzheimer's Disease. Cardiovasc Psychiatry Neurol 2010. 2010:539581. doi: 10.1155/2010/539581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emanuele E, D'Angelo A, Tomaino C, Binetti G, Ghidoni R, Politi P, Bernardi L, Maletta R, Bruni AC, Geroldi D. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol. 2005;62:1734–1736. doi: 10.1001/archneur.62.11.1734. [DOI] [PubMed] [Google Scholar]
- 45.Ghidoni R, Benussi L, Glionna M, Franzoni M, Geroldi D, Emanuele E, Binetti G. Decreased plasma levels of soluble receptor for advanced glycation end products in mild cognitive impairment. J Neural Transm. 2008;115:1047–1050. doi: 10.1007/s00702-008-0069-9. [DOI] [PubMed] [Google Scholar]
- 46.Hemanz A, De la Fuente M, Navarro M, Frank A. Plasma aminothiol compounds, but not serum tumor necrosis factor receptor II and soluble receptor for advanced glycation end products, are related to the cognitive impairment in Alzheimer's disease and mild cognitive impairment patients. Neuroimmunomodulation. 2007;14:163–167. doi: 10.1159/000110641. [DOI] [PubMed] [Google Scholar]
- 47.Li K, Dai D, Zhao B, Yao L, Yao S, Wang B, Yang Z. Association between the RAGE G82S polymorphism and Alzheimer's disease. J Neural Transm. 2010;117:97–104. doi: 10.1007/s00702-009-0334-6. [DOI] [PubMed] [Google Scholar]
- 48.Harper SJ, Wilkie N. MAPKs: new targets for neurodegeneration. Expert Opin Ther Targets. 2003;7:187–200. doi: 10.1517/14728222.7.2.187. [DOI] [PubMed] [Google Scholar]
- 49.Griffin WS, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dudal S, Krzywkowski P, Paquette J, Morissette C, Lacombe D, Tremblay P, Gervais F. Inflammation occurs early during the Abeta deposition process in TgCRND8 mice. Neurobiology of aging. 2004;25:861–871. doi: 10.1016/j.neurobiolaging.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 51.Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Martin Watterson D. Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol. 2007;82:277–296. doi: 10.1016/S0074-7742(07)82015-0. [DOI] [PubMed] [Google Scholar]
- 52.Munoz L, Ranaivo HR, Roy SM, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watterson DM. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer's disease with human postmortem microglial cultures. Neurobiology of aging. 2001;22:945–956. doi: 10.1016/s0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- 54.Johnson GV, Bailey CD. The p38 MAP kinase signaling pathway in Alzheimer's disease. Exp Neurol. 2003;183:263–268. doi: 10.1016/s0014-4886(03)00268-1. [DOI] [PubMed] [Google Scholar]
- 55.Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesbery WR, Patel E, Johnson GV, Bing G. p38 kinase is activated in the Alzheimer's disease brain. J Neurochem. 1999;72:2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- 56.Britschgi M, Wyss-Coray T. Immune cells may fend off Alzheimer disease. Nat Med. 2007;13:408–409. doi: 10.1038/nm0407-408. [DOI] [PubMed] [Google Scholar]
- 57.Fan R, Xu F, Previti ML, Davis J, Grande AM, Robinson JK, Van Nostrand WE. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J Neurosci. 2007;27:3057–3063. doi: 10.1523/JNEUROSCI.4371-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walker DG, Lue LF. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer's disease and other neurodegenerative diseases. Journal of neuroscience research. 2005;81:412–425. doi: 10.1002/jnr.20484. [DOI] [PubMed] [Google Scholar]
- 59.Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–753. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- 60.Li M, Pisalyaput K, Galvan M, Tenner AJ. Macrophage colony stimulatory factor and interferon-gamma trigger distinct mechanisms for augmentation of beta-amyloid-induced microglia-mediated neurotoxicity. J Neurochem. 2004;91:623–633. doi: 10.1111/j.1471-4159.2004.02765.x. [DOI] [PubMed] [Google Scholar]
- 61.McGeer PL, Rogers J, McGeer EG. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271–276. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- 62.Schieven GL. The biology of p38 kinase: a central role in inflammation. Current topics in medicinal chemistry. 2005;5:921–928. doi: 10.2174/1568026054985902. [DOI] [PubMed] [Google Scholar]
- 63.Kim SH, Smith CJ, Van Eldik LJ. Importance of MAPK pathways for microglial proinflammatory cytokine IL-1 beta production. Neurobiol Aging. 2004;25:431–439. doi: 10.1016/S0197-4580(03)00126-X. [DOI] [PubMed] [Google Scholar]
- 64.Zhu X, Mei M, Lee HG, Wang Y, Han J, Perry G, Smith MA. P38 activation mediates amyloid-beta cytotoxicity. Neurochemical research. 2005;30:791–796. doi: 10.1007/s11064-005-6872-x. [DOI] [PubMed] [Google Scholar]
- 65.Pyo H, Jou I, Jung S, Hong S, Joe EH. Mitogen-activated protein kinases activated by lipopolysaccharide and beta-amyloid in cultured rat microglia. Neuroreport. 1998;9:871–874. doi: 10.1097/00001756-199803300-00020. [DOI] [PubMed] [Google Scholar]
- 66.Chen X, Walker DG, Schmidt AM, Arancio O, Lue LF, Yan SD. RAGE: a potential target for Abeta-mediated cellular perturbation in Alzheimer's disease. Current molecular medicine. 2007;7:735–742. doi: 10.2174/156652407783220741. [DOI] [PubMed] [Google Scholar]
- 67.Hadding A, Kaltschmidt B, Kaltschmidt C. Overexpression of receptor of advanced glycation end products hypersensitizes cells for amyloid beta peptide-induced cell death. Biochim Biophys Acta. 2004;1691:67–72. doi: 10.1016/j.bbamcr.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 68.Onyango IG, Tuttle JB, Bennett JP., Jr Altered intracellular signaling and reduced viability of Alzheimer's disease neuronal cybrids is reproduced by beta-amyloid peptide acting through receptor for advanced glycation end products (RAGE) Mol Cell Neurosci. 2005;29:333–343. doi: 10.1016/j.mcn.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 69.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 70.D'Hooge R, Nagels G, Westland CE, Mucke L, De Deyn PP. Spatial learning deficit in mice expressing human 751-amino acid beta-amyloid precursor protein. Neuroreport. 1996;7:2807–2811. doi: 10.1097/00001756-199611040-00080. [DOI] [PubMed] [Google Scholar]
- 71.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li QX, Maynard C, Cappai R, McLean CA, Chemy RA, Lynch T, Culvenor JG, Trevaskis J, Tanner JE, Bailey KA, Czech C, Bush AI, Beyreuther K, Masters CL. Intracellular accumulation of detergent-soluble amyloidogenic A beta fragment of Alzheimer's disease precursor protein in the hippocampus of aged transgenic mice. J Neurochem. 1999;72:2479–2487. doi: 10.1046/j.1471-4159.1999.0722479.x. [DOI] [PubMed] [Google Scholar]
- 73.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 74.Larson J, Lynch G. Induction of synaptic potentiation in hippocampus by patterned stimulation involves two events. Science. 1986;232:985–988. doi: 10.1126/science.3704635. [DOI] [PubMed] [Google Scholar]
- 75.Giacchino J, Criado JR, Games D, Henriksen S. In vivo synaptic transmission in young and aged amyloid precursor protein transgenic mice. Brain Res. 2000;876:185–190. doi: 10.1016/s0006-8993(00)02615-9. [DOI] [PubMed] [Google Scholar]
- 76.Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001;306:116–120. doi: 10.1016/s0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- 77.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 78.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 79.Vodopivec I, Galichet A, Knobloch M, Bierhaus A, Heizmann CW, Nitsch RM. RAGE does not affect amyloid pathology in transgenic Arc Abeta mice. Neurodegener Dis. 2009;6:270–280. doi: 10.1159/000261723. [DOI] [PubMed] [Google Scholar]
- 80.Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ, Huh K, Mook-Jung I. RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer's disease animal model. FASEB J. 2009;23:2639–2649. doi: 10.1096/fj.08-126383. [DOI] [PubMed] [Google Scholar]
- 81.Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fanns W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blalock EM, Chen KC, Stromberg AJ, Norris CM, Kadish I, Kraner SD, Porter NM, Landfield PW. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer's disease: statistical reliability and functional correlation. Ageing Res Rev. 2005;4:481–512. doi: 10.1016/j.arr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 84.Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-lbeta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- 85.Akiyama H, Barger S, Bamum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiology of aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:1200–1205. doi: 10.1136/jnnp.74.9.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. 1992;267:14987–14997. [PubMed] [Google Scholar]
- 88.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 89.Luth HJ, Ogunlade V, Kuhla B, Kientsch-Engel R, Stahl P, Webster J, Arendt T, Munch G. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer's disease brains. Cereb Cortex. 2005;15:211–220. doi: 10.1093/cercor/bhh123. [DOI] [PubMed] [Google Scholar]
- 90.Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, et al. Non-enzymatically glycated tau in Alzheimer's disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med. 1995;1:693–699. doi: 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- 91.Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, Scott CW, Caputo C, Frappier T, Smith MA, et al. Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A. 1994;91:7787–7791. doi: 10.1073/pnas.91.16.7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smith MA, Tabaton M, Perry G. Early contribution of oxidative glycation in Alzheimer disease. Neurosci Lett. 1996;217:210–211. [PubMed] [Google Scholar]
- 104.Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:4766–4770. doi: 10.1073/pnas.91.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:4766–4770. doi: 10.1073/pnas.91.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smith MA, Monnier VM, Sayre LM, Perry G. Amyloidosis, advanced glycation end products and Alzheimer disease. Neuroreport. 1995;6:1595–1596. doi: 10.1097/00001756-199508000-00001. [DOI] [PubMed] [Google Scholar]
- 95.Castellani RJ, Harris PL, Sayre LM, Fujii J, Taniguchi N, Vitek MP, Founds H, Atwood CS, Perry G, Smith MA. Active glycation in neurofibrillary pathology of Alzheimer disease: N(epsilon)-(carboxymethyl) lysine and hexitol-lysine. Free Radic Biol Med. 2001;31:175–180. doi: 10.1016/s0891-5849(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 96.Tabaton M, Perry G, Smith M, Vitek M, Angelini G, Dapino D, Garibaldi S, Zaccheo D, Odetti P. (1997) Is amyloid beta-protein glycated in Alzheimer's disease? Neuroreport. 1997;8:907–909. doi: 10.1097/00001756-199703030-00018. [DOI] [PubMed] [Google Scholar]
- 97.Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C, Catalano MG, Smith MA, Perry G, Danni O, Boccuzzi G, Tabaton M. (2010) AGEs/RAGE complex upregulates BACE1 via NF-kappaB pathway activation. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 98.Guglielmotto M, Giliberto L, Tamagno E, Tabaton M. Oxidative stress mediates the pathogenic effect of different Alzheimer's disease risk factors. Front Aging Neurosci. 2010;2:3. doi: 10.3389/neuro.24.003.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Craft JM, Watterson DM, Marks A, Van ldik LJ. Enhanced susceptibility of S-100B transgenic mice to neuroinflammation and neuronal dysfunction induced by intra cerebro ventricular infusion of human beta-amyloid. Glia. 2005;51:209–216. doi: 10.1002/glia.20194. [DOI] [PubMed] [Google Scholar]
- 100.Mori T, Koyama N, Arendash GW, Horikoshi-Sakuraba Y, Tan J, Town T. Overexpression ofhuman S100B exacerbates cerebral amyloidosis and gliosisin the Tg2576 mouse model of Alzheimer's disease. Glia. 2010;58:300–314. doi: 10.1002/glia.20924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Webster J, Andrews R, Shen J, Majalli AMM, Sahagan BG, Nelson RB, Bronk BS, Rothlein R. (July 2008) Small-molecular inhibitors of the receptor for advanced glycation end-products (RAGE) are an effective therapy in animal models of Alzheimer's disease. Program and abstracts of the ICAD 2008: Alzheimer's Association International Conference on Alzheimer's disease; Chicago, IL, USA. 2008. pp. 26–31. [Google Scholar]
- 102.Sabbagh MN, B J, Agro A, Asien P, Galasko D. (July 2008) An oral anatagonist of the receptor for advanced glycation end-products (RAGE) is safe and well-tolerated in the treatment of Alzheimer's disease: Results from a Phase II study. Program and abstracts of the ICAD Alzheimer's Association International Conference on Alzheimer's disease; Chicago, IL, USA. 2008. pp. 26–31. [Google Scholar]