

Abstract
Chronic pain is a debilitating condition with major socioeconomic impact, whose neurobiological basis is still not clear. An involvement of the neurovascular unit (NVU) has been recently proposed. In particular, the blood-brain barrier (BBB) and blood-spinal cord barrier (BSCB), two NVU key players, may be affected during the development of chronic pain; in particular, transient permeabilization of the barrier is suggested by several inflammatory- and nerve-injury-based pain models, and we argue that the clarification of molecular BBB/BSCB permeabilization events will shed new light in understanding chronic pain mechanisms. Possible biases in experiments supporting this theory and its translational potentials are discussed. Moving beyond an exclusive focus on the role of the endothelium, we propose that our understanding of the mechanisms subserving chronic pain will benefit from the extension of research efforts to the NVU as a whole. In this view, the available evidence on the interaction between analgesic drugs and the NVU is here reviewed. Chronic pain comorbidities, such as neuroinflammatory and neurodegenerative diseases, are also discussed in view of NVU changes, together with innovative pharmacological solutions targeting NVU components in chronic pain treatment.
1. Introduction
According to the International Association for the Study of Pain (IASP), pain is an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage [1].
Chronic pain onset can be sudden or slow and progressive, varies in intensity from mild to severe, and its end cannot be predicted. The diagnosis of chronic pain requires that the condition lasts longer than 3–6 months. Chronic pain can be a debilitating condition with potentially devastating impact on the quality of life [2]. It occurs in a wide variety of conditions, including peripheral neuropathy, stump pain, phantom pain, complex regional pain syndrome, central pain, polymyalgia rheumatica, fibromyalgia, pain of psychological origin, and epilepsy. The recently revised taxonomy includes several new conditions, such as chronic paroxysmal hemicrania: remitting form, hemicrania continua, postlumbar puncture headache, and so forth [1].
According to a report released in June 2011 by the Institute of Medicine of the National Academies, chronic pain affects about 100 million American adults—more than the total affected by heart disease, cancer, and diabetes combined [3]. The 2010 Patient Protection and Affordable Care Act required the Department of Health and Human Services of the United States of America to consider pain as a public health problem.
A 2006 study in 15 European countries and Israel indicates that chronic pain of moderate to severe intensity occurs in 19% of adult Europeans, seriously affecting the quality of their social and working lives [4]. A more recent evaluation of chronic pain in the European Union reports an even higher impact on the general adult population, with an average prevalence of 27%, similar to the that of other common chronic conditions [5].
Understanding the biological, cognitive, and psychological underpinnings of chronic pain represents a major research challenge. From a neurobiological standpoint, the cellular and molecular communication between the central nervous system (CNS) parenchyma and the circulating mediators of the immune and inflammatory response is at the core of such challenge. Indeed, an increasingly compelling body of evidence highlights a major role for the role of nonneuronal cells and diffusible mediators in the functional state of the brain, including neuronal excitability. The concept is captured in the term “neurovascular unit” (NVU), an ensemble of cellular and noncellular players (neurons, endothelial cells, glial cells, pericytes, the extracellular matrix, immune cells, inflammatory mediators) which form an integrated functional unit [6, 7].
In the context of the NVU, an obviously crucial role is played by the blood-brain barrier (BBB) and of the blood-spinal cord barrier (BSCB), both in general and with respect to the pathophysiology of chronic pain.
The purpose of this review is to explore the role played in the establishment and maintenance of chronic pain by the NVU, emphasizing (but not limited to) BBB and/or BSCB permeabilization phenomena. Chronic pain has a significant prevalence in neurodegenerative and neuroinflammatory pathologies, and BBB/BSCB permeabilization is discussed in this extended context. Finally, novel strategies targeting the NVU are considered for chronic pain relief.
2. BBB and BSCB in the Neurovascular Unit
The importance of a full understanding of BBB/BSCB function is emphasized by its well-known role in regulating paracellular and transcellular drug transport, thus preventing or allowing CNS-acting drugs for chronic pain relief to reach their intended target [8]. In addition, there is a possibility that BBB/BSCB permeability may be altered in association with the development of chronic pain [9–12].
2.1. Anatomical Structure of Blood-Brain Barrier and Blood-Spinal Cord Barrier
The BBB is the regulating interface between circulating blood and brain parenchyma. Endothelial cells of brain capillaries, unlike those of the peripheral circulation, are characterized by the absence of cell membrane fenestrations, the presence of tight junctions, having a high number of cytosolic mitochondria, and minimal pinocytotic activity [7]. As an exception, the so-called circumventricular organs (CVOs) do possess fenestrated vasculature. In particular, secretory CVOs (median eminence and neurohypophysis) present a higher vascular permeability for low-molecular-mass tracers compared to sensory CVOs (organum vasculosum of lamina terminalis, subfornical organ, and area postrema) [13].
The surface area of the BBB, depending on the anatomical region, is between 150 and 200 cm2/g of tissue, resulting in a total area for blood-brain exchange between 12 and 18 m2 for the average human adult [14].
A functional equivalent of the BBB is the blood-spinal cord barrier (BSCB), constituted by nonfenestrated endothelial cells, basement membrane, pericytes, and astrocytic feet processes [15]. Several aspects distinguish BSCB from BBB, such as the glycogen deposits in the superficial vessels of the spinal cord [16], increased permeability to tracers and cytokines [17–19], decreased expression of tight-junction proteins and adherens junction proteins [20]. Such differences should be taken into account when these barriers are targeted for chronic pain treatment.
2.2. Mechanisms of Transport through Blood-Brain Barrier
The BBB has low passive permeability to many essential water soluble nutrients and metabolites required by the nervous tissue. However, in healthy conditions, the BBB shows temporary increases in permeability, allowing access to nutrients and oxygen. Since no brain cell is farther than about 15 μm from a capillary [21], drugs and other solutes can rapidly reach all neurons and glial cell bodies, once the BBB has been crossed. Exchange of small organic compounds between blood and brain is regulated by plasma membrane transporters working either in the blood-to-brain direction, the brain-to-blood direction, or both. The directionality of transport is set by the subcellular location of the transport system (blood-facing or brain-facing membrane of the endothelial cells) and by the transport mechanism [8]. Several transport pathways have been identified in the BBB, such as (i) passive diffusion into brain of lipid soluble molecules (e.g., oxygen and carbon dioxide); (ii) ATP-binding cassette transporters (ABC-transporter, see below) efflux (P-glycoprotein (P-gp), multidrug resistance protein (MRP) 1–6, and breast cancer resistance protein transporters (BCRP)); (iii) solute carriers—SLC (transporters of glucose, amino acids, nucleosides, monocarboxylic acids, thyroid hormone, organic anions, organic cations, amine, and choline); (iv) transcytosis of macromolecules by receptor-mediated or adsorptive-mediated mechanism (transport of transferrins, lipoproteins, glycosylated proteins, IgG, insulin, leptin, tumor necrosis factor-alpha (TNF-α), EGF, LIF, cationised albumin, cell penetrating peptides); and (v) mononuclear leukocyte migration [22, 23]. In this review, particular attention will be devoted to ABC transporters with regard to chronic pain and BBB, as the majority of the analgesics are substrates for these transporters, especially for P-gp transporter [24–28].
The endothelial cells of capillary vessels play a major role in BBB physiology. The flattened cells present a luminal and an abluminal surface, separated by a 300–500 nm thick cytoplasm in human brain microvessels [29]. The tight junctions (TJs) connecting adjacent cells represent the most significant BBB structure and serve a dual purpose. On one hand, by sealing the intercellular space, they control the paracellular transport pathway (“gate function”). On the other hand, they effectively subdivide the membrane into two distinct functional domains (“fence function”) [30]. The endothelial cell polarization arises in particular from the differential expression of specific transporter proteins on either surface. The TJ-associated membrane proteins comprise occludin, tricellulin (also called marvelD2), cingulin, claudins (CL-1, CL-3, CL-5), junction-associated molecules of the immunoglobulin superfamily (JAMs), zona occludens proteins (ZO-1, ZO-2, ZO-3), 7H6, and AF-6 [7, 31, 32]. Signaling pathways involved in TJs regulation include G-proteins, serine-, threonine- and tyrosine-kinases, extra- and intracellular calcium levels, cAMP levels, proteases, and cytokines, and all these pathways share the modulation of cytoskeletal elements and the connection of TJs' transmembrane molecules to the cytoskeleton [31].
In pathological states, such as neurodegenerative diseases (including stroke, multiple sclerosis, rheumatoid arthritis, and AIDS dementia) or neuroinflammation, BBB has an uncontrolled and prolonged increase in permeability that results in vasogenic edema and leakage of neurotoxic plasma constituents [33].
2.3. ABC Transporters in BBB and BSCB
ABC transporters represent the largest family of transmembrane proteins. Upon binding ATP, these proteins translocate a wide variety of substrates across extra- and intracellular membranes, including metabolic products, lipids and sterols, and drugs [34]. Tight junctions and ABC transporters expressed in the brain and spinal cord endothelial capillaries represent the major “guardians” of the transport through BBB/BSCB endothelium. ABC transporters, such as P-gp, MRP 1–6, and BCRP, are expressed in the barriers endothelium both in humans and rodents [27].
It is presumed that only efflux transporters located on the luminal (apical) side of the endothelium can restrict drug uptake into the brain [25]. However, transport balance (influx/efflux) is dramatically affected by pathological stressors, such as status epilepticus and neurodegenerative diseases [47–49], and has been suggested to be also partly modified in inflammatory pain [26, 36, 37]. P-gp and BCRP expression in the BBB is regulated during early inflammatory stages by TNF-α and IL-1β [50].
Table 1 summarizes some of the links established so far between ABC transporters in BBB/BSCB and chronic pain.
Table 1.
ABC transporters presence in BBB and BSCB and their potential role in pain.
| ABC transporter (gene) | BBB | BSCB | Localization in brain capillary endothelium | Direction of efflux/influx | Implications in pain/analgesics or anti-inflammatory drugs versus ABC transporters |
|---|---|---|---|---|---|
| P-gp (ABCB1) | Yes [24] | Yes [35] | Luminal [25] | Blood [25] | There is an increased P-gp expression and dynamic redistribution between membrane domains of P-glycoprotein and caveolin-1 in peripheral inflammatory pain [26, 36, 37]. P-gp is involved in pain control with opioid analgesics [38]. Diclofenac is not transported by P-gp [39]. |
|
| |||||
| MRP1 (ABCC1) | Yes [24, 25, 40] | Yes [40] | Luminal [25] Abluminal [24] |
Blood [25] Brain [24] |
The nonsteroidal anti-inflammatory drug indomethacin, an efficient analgesic in some forms of trigeminal autonomic cephalalgias (e.g., paroxysmal hemicrania) [41], was proved to inhibit MRP1 function and expression in cancer cell lines [42]. Most probably indomethachin inhibits MRP1 in BBB. Diclofenac, rofecoxib, and celecoxib are poor inhibitors of MRP1 in HEK293 cells [43]. |
|
| |||||
| MRP2 (ABCC2) | Yes [24, 27] | Yes [35] | Luminal [25] | Blood [25] | Diclofenac is not transported by MRP2 [39]. |
|
| |||||
| MRP3 (ABCC3) | Yes [24] | ? | Abluminal [24] | Brain [24] | Mice lacking MRP3 show altered morphine pharmacokinetics and morphine-6-glucuronide antinociception [44]. |
|
| |||||
| MRP4 (ABCC4) | Yes [24, 27] | ? | Luminal [25] Abluminal [24, 25] |
Blood [25] Brain [25] |
Mice lacking MRP4 show increases in inflammatory pain threshold compared to wild-type mice [45]. MRP4 acts as a prostaglandin efflux transporter and is inhibited by nonsteroidal anti-inflammatory drugs (e.g., indomethacin, indoprofen, ketoprofen, and flurbiprofen) [43]. Diclofenac, rofecoxib, and celecoxib are poor inhibitors of MRP4 [43]. |
|
| |||||
| MRP5 (ABCC5) | Yes [24, 27] | ? | Luminal [25] Abluminal [24] |
Blood [25] Brain [24] |
? |
|
| |||||
| MRP6 (ABCC6) | Yes [24, 27] | Possible [46] | Abluminal [24] | Brain [24] | ? |
|
| |||||
| BCRP (ABCG2) | Yes [24] | Yes [35] | Luminal [25] |
Blood [25] | Diclofenac, an analgesic mainly used against cancer- associated chronic pain, is efficiently transported by murine BCRP1 and moderately by human BCRP [39]. |
In conclusion, ABC transporters appear to play an important role in inflammatory pain and in analgesia (opioids or nonsteroidal anti-inflammatory drugs). Knocking out genes encoding ABC transporters has consequences in inflammatory pain or analgesic profile. For example, knocking out the gene encoding for MRP4 increases inflammatory pain threshold [45] and knocking out the gene encoding for MRP3 alters morphine pharmacokinetics [44]. Therefore, these transporters, in particular P-gp, represent key molecules that might contribute to BBB/BSCB permeabilization induced by inflammation-like stimuli in various pain syndromes [26, 36, 37].
3. Cross Talk between NVU Partners in Chronic Pain
In vitro and in vivo animal studies have confirmed NVU cellular crosstalk in inflammation-induced hyperalgesia or nerve injury models and results can be extrapolated to chronic pain.
3.1. Glia-Neuron Interactions
Glia are significantly activated in response to trauma, ischemia, and invading pathogens by means of cytokine release (IL-1β, TNF-α) and may contribute to the maintenance of chronic pain [51, 52]. In addition to proinflammatory cytokine release at the peripheral site of injury, release also takes place in the CNS (spinal cord, brainstem, and forebrain) [53–55]. Released cytokines together with activated glia have been proved to influence and modulate neurons in the trigeminal nucleus region in a trigeminal model of inflammatory hyperalgesia [56]. On the other hand, different signaling pathways mediate IL-1β actions in hippocampal neurons compared to astrocytes [57].
Glia activation within the CNS has been suggested to maintain the pain sensation, even after the original injury or inflammation has healed, and convert it into chronic pain by altering neuronal excitability [58]. In a peripheral nerve injury pain model, the inhibition of microglia after four weeks from nerve injury normalized the pain threshold, while removing the inhibitor immediately restored pain-related phenomena [52].
3.2. Microglia-Astrocytes Interactions
Both in vitro and in vivo data provide clues on how the crosstalk between microglia and astrocytes may play a role in chronic pain maintenance [59–63]. The activation of microglia has been shown to cause astrocytic activation, with a delay of about 4 days [54, 64]. Preventing microglial activation (and subsequent astrocyte activation) inhibits hyperalgesia or allodynia [59, 61]. Once the astrocytes are activated, inhibiting microglia has no effect on pain [59, 60]. On the other hand, brain astrocytes can be activated in response to peripheral nerve injury without prior microglia differentiation [65]. A dialogue between microglia expressing IL-18 and astrocytes expressing its receptor (IL-18R) was suggested to be important in tolerance to morphine analgesia, by means of a P2X7R/IL-18/D-serine/N-methyl-D-aspartate receptor (NMDAR)/PKCγ-mediated signaling pathway [62], but also for tactile allodynia after nerve injury [66].
Increased monocyte chemotactic protein 3 (MCP-3, known as CCL7) expression associated with IL-6-dependent epigenetic modification at the MCP-3 promoter after nerve injury, mostly in spinal astrocytes, may serve to facilitate astrocyte-microglia interaction in the spinal cord and could play a critical role in the neuropathic pain-like state [63].
Some studies suggest the importance of the triad neuron-astrocyte-microglia in physiological and pathological inflammatory states [67].
3.3. Astrocyte-Endothelial Cell Interactions
Astroglial-endothelial signalling is altered under pathological conditions, such as infection, inflammation, stroke, or trauma, leading to BBB opening [6]. The coupling between the abluminal capillary cell membrane and the surrounding glial end-foot processes is reduced in pathological conditions [68, 69]. Stimulation of astrocytes, in coculture with brain endothelial cells, with 5-hydroxytryptamine (5-HT) generated a pronounced increase in intracellular Ca2+ release in the presence of inflammatory or pain-mediating activators, such as substance P, calcitonin gene-related peptide (CGRP), lipopolysaccharide (LPS), or leptin [70]. Mu-opioid agonists inhibit the enhanced intracellular Ca2+ responses in inflammatory-activated astrocytes cocultured with brain endothelial cells [70]. Overexpression of endothelin-1 in astrocytes, but not in endothelial cells, ameliorates inflammatory pain response after formalin injection [71]. The role played in chronic pain development by in vivo endothelial-astrocyte interaction at the barrier has not been investigated yet.
3.4. Pericyte-Endothelial Cell Interactions
In vivo studies in wild-type mice have shown that pericytes are more numerous in the brain than in the spinal cord [72]. Whereas brain regions such as the neocortex, hippocampus, and caudate nucleus show almost uniform presence of pericytes, the spinal cord shows significantly nonuniform distributions along the rostrocaudal extent, with the thoracic region being richer in pericytes, but with no more than 70% of brain levels. This reduced number of pericytes in the spinal cord correlates with (i) a higher BSCB permeability, as probed by fluorescent dextran and (ii) a diminished expression of tight junction proteins ZO-1, occluding, and claudin-5. Compared to wild-type mice, in Pdgfrβ F7/F7 pericyte-deficient mice, pericytes are reduced more in spinal cord capillaries, leading to BSCB disruption to serum proteins. ZO-1 and occludin are also reduced, and the accumulation in motor neurons of cytotoxic thrombin and fibrin leads to motor neuron loss [72]. In another pericyte-deficient model, the Pdgfrβ ret/ret mouse, an increase in BBB permeability to water and to a range of low- as well as high-molecular-mass tracers has been shown [73]. Pericytes express MRP1, MRP4, and MRP5 transporters, which might imply a role played by these cells in regulating xenobiotic transport through the BBB [74].
Abnormal interactions between pericytes and endothelial cells have been implicated in a number of human pathological conditions, including tumor angiogenesis, diabetic microangiopathy, ectopic tissue calcification, and stroke and dementia syndrome CADASIL [75]. In pathological conditions implying BBB damage, such as stroke, hypoxia, and traumatic brain injury, the pericytes migrate away from brain microvessels wall and it seems to have an important role in neurovascular unit repair [76–78].
Morphine potentiates endothelial-pericyte interaction via platelet-derived growth factor-BB (PDGF-BB)/PDGF receptor-β (PDGFR-β) signaling and promotes tumor angiogenesis, pericyte recruitment, and coverage of tumor vessels [79]. The role of pericyte-endothelial cell interaction in chronic pain development and the role of pericytes during BBB disruption are still open topics.
4. BBB and BSCB in Chronic Pain: “To Be or Not to Be” Permeabilized/Disrupted
4.1. Acute/Chronic Pain Induces Changes at the BBB and BSCB Level
Does chronic pain cause BBB/BSCB permeabilization or disruption? The variety and complexity of the clinical conditions that involve chronic pain make a simple answer impossible, and studies on animal models of acute/chronic pain provide controversial responses to this hypothesis.
In the literature, the terms BBB “opening,” “leakage,” and “breakdown” are often used interchangeably, but more caution should be paid, when choosing between them [80]. A distinction should be made between BBB “permeabilization” and BBB “disruption” in experimental animal models. For the purpose of this review, the term BBB permeabilization refers to leukocytic recruitment associated with increased endothelial permeability, with no tight junction opening or altered efflux transport. As BBB “opening” or “permeabilization” is a physiological phenomenon, it should be reserved to transient processes [80]. On the other hand, we consider the BBB “disrupted” if Evans blue (EB) or albumins are extravasated into brain or spinal cord parenchyma. BBB “disruption” or “breakdown” represents a long-term opening associated with often-irreversible phenomena [80–82].
4.1.1. BSCB Permeabilization in Neuropathic Pain and Disruption in Chronic Pain Animal Models
Peripheral nerve injury produced by either sciatic nerve constriction or selective transection (peroneal and tibial nerve branches, but not sural branch) causes a transient increase in BSCB permeability in the lumbar and thoracic spinal cord, peaking about 24–48 h. after injury and returning to normal levels after 7 days, as assessed by EB dye or horseradish peroxidase accumulation in the parenchyma [10]. BSCB permeability was also increased 24 hours after electrical stimulation of the sciatic nerve at intensity sufficient to activate C-fibers, but not A-fibers, or after capsaicin application on the sciatic nerve [10].
Partial sciatic nerve ligation in rats, a model of neuropathic pain, triggers an increase of BSCB permeability in the lumbar, but not in the thoracic, spinal cord to tracers of different size (e.g., EB, sodium fluorescein), which was prominent between day 3 and day 7, stayed significant for at least 4 weeks after injury, and returned to normal levels after 2 months [11]. Contrasting results on BSCB permeability in extralumbar spinal cord regions (e.g., thoracic) [10, 11] could likely be attributed to EB protocol differences.
Plasma proteins (IgG and fibronectin) immunopositive deposits in the ipsilateral side of the spinal parenchyma and downregulation of tight junction proteins (ZO-1, occludin-1, and caveolin-1) in isolated microvessels of the spinal cord were found 3 days after injury [11]. BSCB permeabilization occurs independently of the activation of resident microglial cells, EB extravasation being present while a microglial inhibitor minocycline is infused intrathecally from day 0 to day 7 [11]. Additionally, it was shown that the administration in rats of high doses of IL-1β (intravenous) impairs BSCB disruption, while TGF-β1 and IL-10 (intrathecal) shut down the openings in BSCB [11].
In a recent study of perispinal inflammation induced by applying the toll-like receptor (TLR)-2 agonist zymosan to the dorsal dural surface of the L1/L2 spinal cord, the lack of BSCB permeabilization was inferred from the lack of serum proteins in the spinal parenchyma 24 h after surgery [12]. No immunohistological evidence of T-cell or Mac-1-positive macrophages crossing into the parenchyma was found, but ATF-3 (a transcription factor that is also a sensitive indicator of neuronal injury) expression was observed in the dorsal horn of the same spinal cord segments after 1 day [12]. Thus, inflammatory signals are indeed transduced across the BSCB at the site of the inflammatory stimulus, within a 400–500 μm radius. Astrocyte activation and gliosis are significantly increased in the superficial dorsal horn 1–7 days after surgery, with a transient recovery after 14 days, while resident microglia cells show a steady increase in staining density within the superficial dorsal horn beginning 1 day after surgery [12].
Neuropathic pain induced by L4 spinal nerve lesions in animal models is accompanied by astrocyte activation and albumin leakage, revealing BSCB disruption more prominent in the gray matter of the lesioned side compared to the contralateral in both dorsal and ventral horns [83]. Inflammatory events and changes in astrocyte and microglia reactivity at the spinal level in response to injury or disease are important processes that can initiate pain hypersensitivity [84, 85]. Studies conducted in a T-cell-deficient Rag1-null adult mouse have shown that T-cell infiltration and activation in the dorsal horn of the spinal cord following peripheral nerve injury contribute to the evolution of neuropathic pain-like hypersensitivity [86]. Most likely, the T-cell infiltration into the spinal cord is higher than normal in the nerve-injured animals, a fact that may be correlated with an increase in BSCB permeability.
BSCB permeabilization is a delayed event with respect to the initial injury and has a transient character. Studies addressing the role of the endothelium in BSCB disruption have been carried out, but the inclusion of the NVU as a whole is needed [87]. While the activation of glia may be important for the development of chronic pain, it is still unclear if the activation is required for BSCB disruption or if the two phenomena are independent. Peripheral inflammation or nerve injury in animal models induces astrocytes and microglia activation in the spinal cord [52, 88–91], but in these studies evidence regarding BSCB permeabilization is not available. In this view, new approaches connecting glia activation to BSCB opening would be very useful.
4.1.2. BBB Permeabilization in Animal Models of Inflammatory Pain
Inflammation induced by an intraplantar injection of λ-carrageenan into the rat hindpaw causes increased brain uptake (in situ brain perfusion) of [14C]sucrose at 1, 3, 6 and 48 h after injection [92]. In the same study, Western blot analysis on isolated cerebral microvessels indicated a transitory increase in ZO-1 expression (increase after 1–6 h, returned to control after 12 h.) and a reduction in occludin expression (after 1, 3, 6, 12, and 48 h) [92]. These expression patterns indicate increased BBB permeability and suggest a link with the development of inflammatory pain. In another study devoted to inflammatory pain, [14C]sucrose in situ brain uptake, [3H] in situ cerebral flow, and Western blot analysis (occludin, ZO-1, CL-1, and actin expression) were performed 1 h after formalin injection, 3 h after λ-carrageenan injection and 3 days after complete Freund's adjuvant (CFA) injection, and BBB permeabilization was observed [93].
In a rat model of inflammatory pain (injection of CFA into the plantar hindpaw), significant edema formation and hyperalgesia were observed 72 h. after treatment, together with significant increases in brain sucrose uptake. Expression of the transmembrane TJ proteins occludin, claudin-3 and -5, and junction adhesion molecule-1 (JAM-1) significantly changed 24–72 h after CFA injection, as proved by Western blotting [9] and confocal microscopy [94].
The induction of peripheral inflammatory pain through the injection of λ-carrageenan was associated with increased BBB permeability in a study that showed, by means of SDS-PAGE/Western blot analysis, a significant change in the relative amounts of oligomeric, dimeric, and monomeric occludin isoforms in BBB endothelial cells, presumably promoted by the disruption of disulfide-bonded occludin oligomeric assemblies [95].
Expression of organic anion-transporting polypeptide 1a4 (Oatp1a4) is upregulated after 3 h exposure to λ-carrageenan; the upregulation is prevented by diclofenac, suggesting the implication of acute/chronic inflammatory pain [36]. This modulation of BBB permeability in inflammatory pain appears to be controlled by the TGF-beta/activin receptor-like kinase-5 (ALK5) signaling pathway [96]. λ-carrageenan-induced peripheral inflammatory pain generates increased [ 14C]sucrose and [ 3H]codeine in situ brain uptake, and rats pretreated (10 min before λ-carrageenan injection) with tempol, a pharmacological ROS scavenger, have an attenuated radiotracers uptake [97]. In the same study, other indirect pieces of evidence for BBB modulation have been presented consisting in increase of the nitrosylated proteins in isolated brain vessels extract.
In a λ-carrageenan inflammatory pain model, unidirectional permeability coefficients for several selected brain regions (hypothalamus, cerebellum, midbrain, cerebrum, hippocampus, brainstem, and thalamus) were calculated. Three hours after λ-carrageenan injection, the BBB resulted in an increased permeability in cerebrum and brainstem; diclofenac administration reversed this effect [98]. Western blot analysis of occludin expression in the same brain regions, however, did not reveal any significant changes [98]. In conclusion, correlating occludin expression changes with BBB “permeabilization” is problematic on the basis of the available data.
Administration of EB, which readily binds to serum albumins, is “classically” employed to assess BBB integrity, since in normal conditions the dye should not be found in the brain parenchyma [10]. However, in order to be revealed by EB, BBB disruption must be of a substantial degree (e.g., ischemic stroke [99]), while inflammatory pain per se does not constitute sufficient stimulus [100]. Inflammatory pain is more likely related to BBB permeabilization, as suggested by [14C]sucrose transport through the BBB using in situ brain perfusion [9].
Despite the valuable information contained in the above described studies, there are several experimental pitfalls to be considered. First, only indirect pieces of evidences are available in support of the idea of BBB permeabilization in inflammatory or chronic inflammatory pain. It is difficult to assess BBB permeabilization based on changes in TJ protein expression in an homogenate of isolated brain capillaries or to expand results from in situ brain perfusion with radioactive tracers to the BBB permeabilization. Another problem is that relatively short experimental durations (such as 24–72 h) are considered equivalent to a “chronic” pain state [9], while similar experiments on BSCB permeabilization were carried out over a significantly longer time scale (1 week–2 months) [10–12]. More consistent studies, based on in vivo brain uptake of Evans blue or [14C]sucrose should be done in order to prove BBB permeability changes. Alternative in vivo methods, such as intravital microscopy [101] or nuclear imaging of radioisotope-labeled leukocytes [72], are still unexplored in the field of chronic pain. A regional brain mapping of BBB permeabilization from the initial acute pain induction to the late chronic pain phase would be of significant use. In any case, clinical translation of the results obtained with experimental inflammatory pain models is still far from accomplished.
Possible changes in BBB and/or BSCB permeability as a result of acute and chronic pain are shown in Figure 1.
Figure 1.
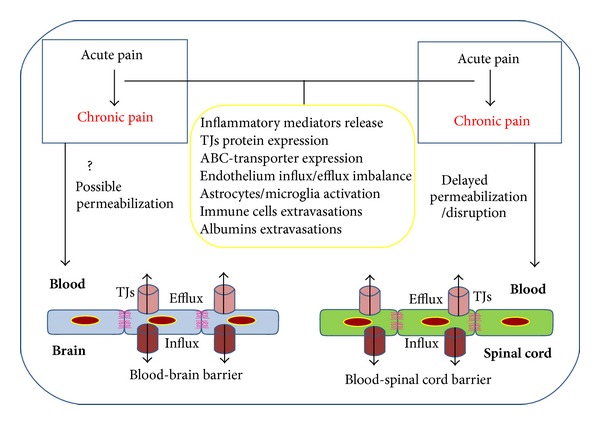
Acute pain occurs in a first step as a result of peripheral injury and/or inflammation. Chronic pain appears as a delayed event associated with permeabilization or brain/spinal cord capillary endothelium disruption. Different processes, such as inflammatory mediator release, changes in TJs protein and ABC transporters expression, activation of microglia and/or astrocytes, immune cells and albumin extravasation, may occur independently or in an “orchestrated” manner, and might contribute to the process of BBB/BSCB permeabilization or disruption.
4.2. Chronic Pain Treatments and NVU
Two major classes of analgesic drugs are currently in use for chronic pain treatment: opioids and nonsteroid anti-inflammatory drugs (NSAIDs). NSAIDs are used to treat chronic mild to moderate pain, while opioids are powerful analgesic agents used to treat moderate to severe chronic pain [102].
On the other hand, beside a wide range of adverse effects, long-term clinical administration of opioids (e.g., morphine) in chronic pain therapy is prevented by tolerance and dependence [102]. A classical dogma holds that agonist-induced μ-opioid receptor internalization contributes directly to functional receptor desensitization and opioid tolerance [103]. By contrast, other studies suggest that opioid receptor internalization can reduce opioid tolerance in vivo (reviewed by [103]). Beside neurons, other NVU players (e.g., glial cells, pericytes) have been considered to contribute to opioid tolerance development [62, 79, 104]. Endothelial cell lining represents the first “defence” to be crossed by opioids before interacting with CNS cells and therefore efflux alterations at this level are crucial in opioid tolerance.
In the λ-carrageenan rat model, acute inflammatory pain generates an increased functional expression and trafficking to membrane domains of endothelial efflux transporters (e.g., P-gp) in the BBB microvasculature [26, 37]. On the other hand, the same rats treated with morphine show reduced brain uptake of the drug due to increased P-gp activity [26]. Coadministration of cyclosporine A (P-gp inhibitor) with morphine in rats increased morphine transport through the BBB in a dose-dependent manner [26]. In the clinical practice, reducing tolerance to morphine by co-administration with cyclosporine A is unfeasible due to severe side effects (nephro- and neurotoxicity) [105, 106].
Chronic morphine treatment induced an increase in the expression of interleukin (IL)-18 by microglia, IL-18 receptor (IL-18R) by astrocytes, and protein kinase Cγ (PKCγ) by neurons in the spinal dorsal horn. The results were interpreted by the authors as signs of a complex glia-neuron dialogue in the process of developing tolerance to morphine [62]. Morphine also potentiates endothelial-pericyte interaction via PDGF-BB/PDGFR-β signaling [79]. Morphine upregulates sphingolipid ceramide (in spinal astrocytes and microglia, but not in neurons) and spinal sphingosine-1-phosphate [104]. In turn, sphingosine-1-phosphate modulates spinal glial function, increasing the production of glial-related proinflammatory cytokines, in particular TNF-α, IL-1β, and IL6 [104].
Another major line of chronic pain treatment is represented by nonopioid analgesics such as NSAIDs. These drugs have several side effects, the most important being the risk of serious upper gastrointestinal complications, including bleeding, ulcers, and perforation [102]. NSAIDs act on the descending pain control system, which includes the periaqueductal gray matter and rostral ventromedial region of the medulla, which are also targets for endogenous opioids. Therefore, repeated administration of NSAIDs (e.g., metamizol, lysine-acetylsalicylate, analgine, ketorolac, and xefocam) to rats induces tolerance to themselves and cross-tolerance to opioids [107, 108].
Studies suggest that NSAIDs interact in several different ways with the brain endothelium, either by reducing edema and BBB/BSCB permeabilization [109, 110] or by inhibiting endothelial ABC transporters (e.g., MRP1, MRP4) [42, 43]. Diclofenac attenuates edema and hyperalgesia induced by λ-carrageenan in the cerebral and brainstem regions [98]. Indomethacin, an inhibitor of cyclooxygenase (COX)-1 and COX-2, reduces BBB damage induced by intracerebral injection of TNF-α [109]. Pretreatment with p-chlorophenylalanine, indomethacin, ibuprofen, and nimodipine of rats with spinal cord injury, reduced edema formation, BSCB permeabilization, and blood flow [110]. Indomethacin was shown to be an inhibitor of MRP1 function [42] and indomethacin, indoprofen, ketoprofen, and flurbiprofen inhibit MRP4 [43]. Diclofenac is transported by BCRP, but not by P-gp [39].
5. Comorbidities of Chronic Pain with Neuroinflammatory and Neurodegenerative Diseases: Role of the Neurovascular Unit
Chronic pain has an extensive palette of comorbidities, but only neuroinflammatory and neurodegenerative diseases with known alterations of the NVU are here discussed (Figure 2). High prevalence of chronic pain can be observed in all these CNS pathologies. BBB/BSCB alterations in epilepsy, Alzheimer's disease, Parkinson's disease, multiple sclerosis, and amyotrophic lateral sclerosis will be briefly described, with regard to chronic pain syndromes. Understanding the exact role played by each pathology in permeabilizing/disrupting brain and spinal cord capillaries' endothelium is a crucial step in finding better therapeutic solutions.
Figure 2.
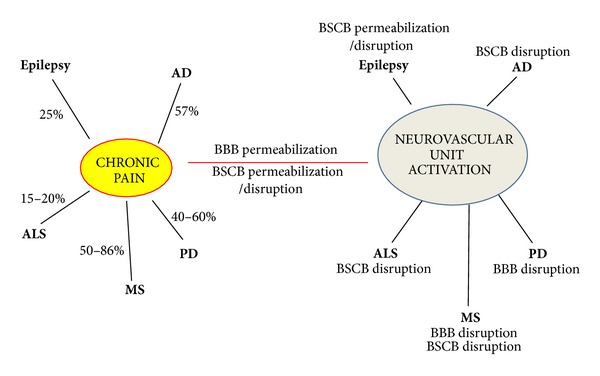
Similarities between chronic pain and NVU activation. Studies from the literature indicate that different NVU components are activated in a given pathology (e.g., epilepsy, Alzheimer's disease, Parkinson's disease, multiple sclerosis, amyotrophic lateral sclerosis, chronic pain) with a special focus on BBB/BSCB permeability alterations. Neuroinflammatory/neurodegenerative diseases are associated with chronic pain (see indicated percentages), but further studies are necessary to establish whether NVU activation may represent a “missing link” in the association. While the intrinsic mechanisms relating NVU activation, chronic pain, and neuroinflammatory/neurodegenerative disorders remain unclear, BBB/BSCB permeabilization appears to play a role.
5.1. Epilepsy and Chronic Pain
Epilepsy is a set of chronic neurological disorders characterized by abnormal, excessive, or hypersynchronous neuronal activity in the brain. The Epilepsy Comorbidities and Health (EPIC) Survey recently performed in the United States indicated that epilepsy is comorbid with several pain disorders, such as migraine, chronic pain, fibromyalgia, and neuropathic pain [111]. Additionally, the EPIC study indicated that chronic pain is prevalent in 25.4% of epileptic versus 17.7% of nonepileptic survey responders [111]. Chronic pain and fibromyalgia may be related to physical inactivity, which is more prevalent among adults with a history of epilepsy than among those without epilepsy [112].
The recent IASP taxonomy includes epilepsy in the list of generalized syndromes of chronic pain and includes chronic paroxysmal hemicrania—remitting form and hemicrania continua in the list of the chronic pain conditions [1]. Epilepsy and ictal epileptic headache share several pathophysiological mechanisms, such as (i) EEG abnormalities—lateralized or generalized, ipsilateral or contralateral, with focal theta activity or generalized spike-waves, and brief or longer-lasting episodes and (ii) headache and EEG anomalies resolve within minutes of i.v. antiepileptic medication administration [113]. The overlap between migraine and epilepsy may be partial or complete, not necessarily synchronous (preictal, ictal, or postictal), and in some cases the headache may represent the only ictal phenomenon [113]. In pediatrics studies, 3.1% of the patients suffered from idiopathic headache and idiopathic or cryptogenic epilepsy or unprovoked seizures [114]. The same study showed a strong association between migraine and epilepsy: in migraineurs the risk of epilepsy was 3.2 times higher when compared to tension-type headache, and children with epilepsy had a 4.5-fold increased risk of developing migraine than tension-type headache [114]. Postictal headache occurred in 41% of temporal lobe epilepsy patients, 40% of frontal lobe epilepsy patients, and 59% of occipital lobe epilepsy patients [115].
Several mechanisms have been proposed to explain comorbidity of epilepsy and chronic pain (such as that characterizing migraine), such as (i) the essential role of glutamate as a mediator of the hyperexcitability in both focal seizures and migraine, considering that seizure generation and spread are mediated by synaptically released glutamate acting on AMPA receptors, while triggering of cortical spreading depression depends on NMDA receptors and spread does not require synaptic transmission; (ii) mutations in genes for the membrane ion transport proteins CACNA1A (P/Q-type voltage-gated calcium channel), ATP1A2 (Na+-K+ ATPase), and SCN1A (voltage-gated sodium channel) [116].
Another important mechanism implied in chronic pain comorbidity with epilepsy is NVU activation. In this respect, brain endothelium seems to play an important role.
BBB disruption induces epileptiform activity [117–122]. We have previously shown that BBB leakage is induced by acute seizure activity but prevented by blockade of leukocyte-vascular adhesion, either with blocking antibodies or by genetically interfering with P-selectin glycoprotein ligand-1 (PSGL-1) function in mice [123]. Endothelial proinflammatory chemokines induce complex signal transduction pathways leading to integrin activation and controlling leukocyte recruitment, and therefore play a critical role in epileptogenesis [124, 125].
ABC transporters in the BBB are also affected in epilepsy. Shortly after status epilepticus, MRP1, MRP2, and BCRP are upregulated in astrocytes within several limbic structures, including hippocampus [47]. In chronic epileptic rats, these proteins are overexpressed in the parahippocampal cortex, specifically in blood vessels and astrocytes surrounding these vessels [47].
Whether transient BBB opening occurs during migraine attacks is controversial. Some magnetic resonance imaging studies have reported negative results [126, 127] while others have found indications of BBB leakage [128]. In migraine, indirect evidence for BBB permeabilization is provided by increased circulating levels of matrix metalloproteinases (MMPs) 2 [129] and 9 [130] that have been attributed to MMPs release from the extracellular matrix of the neurovascular unit.
5.2. Alzheimer's Disease and Chronic Pain
Alzheimer's disease (AD) is the most common cause of dementia. It is a neurodegenerative disorder characterized by synaptic and neuronal loss, by the accumulation in the extracellular matrix of beta-amyloid deposits, and by the presence of abnormal aggregates of microtubule-associated proteins, the so-called neurofibrillary tangles, in neuronal cell bodies.
Prevalence of pain in AD was estimated at 57% of all patients [131], although such assessment is complicated by two factors. First, pain processing may be altered in dementias [132, 133] including AD [134]. Second, the primary method for pain assessment is patient reporting [135], but pain affects cognitive function [136, 137] and cognitive function in turn affects pain [133], which makes pain assessment in AD very difficult.
Astrocytes tend to localize around fibrillar amyloid plaques, suggesting that Aβ deposition is a potent trigger of astroglial activation in the AD brain [138]. Additionally, an increase in the number of IL-1 immunoreactive microglia associated with AD plaques has been shown [139]. A variety of biomarkers for microglial activation in AD have been proposed, such as chitotriosidase, CCL18 (pulmonary activation-regulated chemokine; PARC), YKL-40, CCL2 (monocyte chemoattractant protein 1; MCP-1), CD14, and neopterin [140].
Immunohistochemistry on postmortem human brains affected by AD or vascular dementia indicated an increased expression of CL-2, Cl-5, and CL-11 in neurons and of CL-2 and CL-11 in astrocytes and oligodendrocytes [141]. There is a strong relationship between neurodegeneration, cognitive decline, and BBB disruption in AD [142]. It was suggested that during neurodegeneration the receptor for advanced glycation end products (RAGE), which mediates transfer of amyloid-β to the brain through the endothelial cells, can be upregulated [143]. In AD transgenic mice, BBB alteration was proven to precede accumulation of senile plaques [144].
Neuroinflammation represents a crucial part in the pathogenesis of AD and other neurodegenerative diseases [145]. Inflammatory mediators, such as IL-1β, IL-6, TNF-α, IL-8, transforming growth factor-β (TGF-β), and macrophage inflammatory protein-1α (MIP-1α), are upregulated in AD [146].
5.3. Parkinson's Disease and Chronic Pain
Parkinson's disease (PD) is a degenerative disorder of the CNS, mainly characterized by loss of dopamine-generating cells in the substantia nigra. Prevalence of pain (musculoskeletal pain, neuritic or radicular pain, dystonia-associated pain, primary or central pain, and akathitic discomfort) in PD is estimated around 40–60% [147, 148]. See [149] for a comprehensive review of pain in PD.
Impairment of BBB function has been implicated in the pathogenesis of PD. Accumulation of verapamil (normally extruded from the brain by P-gp) in the brain of PD patients proves a dysfunction of BBB [150]. Injection of dopamine neurotoxin 6-hydroxydopamine (6OHDA, which produces Parkinson's-like dopaminergic neuron lesions) into the striatum of rats induced FITC-labeled albumin leakage in areas of the brain that are not protected by the BBB (e.g., the hypothalamus around the third ventricle and area postrema along the floor of the fourth ventricle) but no leakage in BBB-protected areas (e.g., ipsilateral parietal cortex or hippocampus, or into contralateral structures) [151]. The presence of neuroinflammatory markers, such as activated microglia or astrocytes, is also an important feature of PD [152]. Microglia activation and upregulation of inflammatory mediators can be induced by α-synuclein and contributes to PD pathogenesis [153]. On the other hand, astrocyte activation in PD is still under debate [152, 154–158]. In a recent study performed on aged c-rel−/− mice developing PD-like degeneration of substantia nigra pars compacta, we observed a marked microglia activation in the substantia nigra pars compacta and striatum, but no GFAP-positive astrocyte activation [159].
5.4. Multiple Sclerosis and Chronic Pain
Multiple sclerosis (MS) is a chronic inflammatory disease of the CNS, which leads to demyelination, neurodegeneration, perivascular edema, and inflammatory infiltrates [160]. Prevalence of pain in MS is estimated around 50–86% [161, 162]. A recent classification based on pathophysiological mechanisms and response to treatment identified nine types of MS-related pain: trigeminal neuralgia and Lhermitte's phenomenon (paroxysmal neuropathic pain due to ectopic impulse generation along primary afferents), ongoing extremity pain (deafferentation pain secondary to lesion in the spinothalamocortical pathways), painful tonic spasms and spasticity pain (mixed pains secondary to lesions in the central motor pathways but mediated by muscle nociceptors), pain associated with optic neuritis (nerve trunk pain originating from nervi nervorum), musculoskeletal pains (nociceptive pain arising from postural abnormalities secondary to motor disorders), migraine (nociceptive pain favored by predisposing factors or secondary to midbrain lesions), and treatment-induced pains [163].
BBB disruption is an early event in the progression of MS, as proved by magnetic resonance imaging studies [164, 165]. Diapedesis of monocytes and subsequent trafficking of monocyte-derived macrophages into the brain are key mediators of demyelination and axonal damage in MS. Endothelin 1 (ET-1), its type B receptor (ET(B)) and endothelin-converting enzyme-1 (ECE-1) are mediators for monocyte diapedesis through the human BBB and play a key role in demyelination and axonal damage in MS [166]. In experimental models of MS, such as experimental autoimmune encephalomyelitis (EAE), BBB disruption is induced by T-cells in conjunction with antigen-presenting dendritic cells [167, 168], and monocytes [169]. MS lesions are often found in proximity to blood vessels [170], associated with loss of occludin and ZO-1 in the microvasculature [171–173]. Leukocyte extravasation through BBB is mediated by cytokines: TNF-α, IL-1B, and interferon-γ [174]. Infiltration of inflammatory cells are localized perivascularly, but can also be located in the CNS parenchyma. In acute inflammatory lesions, CD4+ and CD8+ T cells and B cells infiltrate the lesion site. Lesions at later MS stages show an abundance of macrophages with internalized myelin degradation products and reactive proliferating astrocytes [175]. Sodium channels contribute to activation of microglia and macrophages in EAE [176].
MS is also characterized by significant changes in the composition and dynamics of the BSCB [177]. CD3-positive T-cells accumulate within the dorsal horn in mice with EAE, early in the disease course when cold and tactile allodynia are observed [178]. BSCB disruption is greatest at disease onset, followed by inflammation and demyelination, indicating that increased BSCB permeability precedes the destructive inflammatory process [177]. A recent study showed that autoreactive T cells access CNS via the fifth lumbar spinal cord in EAE mouse model [179].
In an EAE model, a recent study suggested a signalling role for Wnt (a family of secreted signaling proteins) in MS-associated chronic pain pathogenesis, although only neurons and glial cells were examined [180]. On the other hand, Wnt signaling contributes to brain angiogenesis, BBB formation, influences vascular sprouting, remodelling, and arteriovenous specification by modulating the Notch pathway [181]. Therefore, further studies on Wnt signalling in brain microvasculature could bring new insights in to MS-related pain syndromes.
5.5. Amyotrophic Lateral Sclerosis and Chronic Pain
Amyotrophic lateral sclerosis (ALS) is a chronic, progressive, and ultimately fatal neurodegenerative disease of motor neurons in the brain and spinal cord [182]. Prevalence of chronic pain (especially located at the arms level) in ALS is estimated around 15–20% [183, 184].
Increased permeability of the BSCB has been implicated in the pathogenesis of ALS [185]. Studies conducted in the ALS mouse model SOD1-G93A have shown BBB and BSCB disruption [186, 187], in areas of motor neuron degeneration (early and late ALS stages) [186] and capillary rupture in brainstem (early symptomatic ALS stage) [186]. Some studies indicate reduction in tight junction proteins (ZO-1, occluding, and claudin-5) before motor neuron loss, in presymptomatic ALS stages [188], while other data point out the reduction in tight junctions proteins (ZO-1 and occludin) and basement membrane protein agrin in symptomatic ALS stages [187]. Therefore, it is still controversial if the BBB/BSCB disruption is the cause or the consequence of ALS development. In SOD1-G93A mice, an increase was detected in mRNA and protein levels for P-gp and BCRP at the level of capillary endothelium in several regions, such as whole spinal cord, cerebral cortex, and cerebellum [189]. Additionally, the transport activity of P-gp and BCRP increased with ALS progression in spinal cord and cerebral cortex capillaries [189].
T lymphocytes are able to cross into the brain and spinal cord parenchyma, where they interact with resident microglia, inducing them to adopt either an M1 (cytotoxic) or M2 (protective) phenotype, depending on ALS stage [190]. Clinical studies evidenced perivascular and intraparenchymal CD4+ T-lymphocytes in the proximity of degenerating corticospinal tracts and ventral horns in two-thirds of ALS patients [191]. CD4+ T-lymphocytes slow disease progression, modify the microglial phenotypes, and extend survival [192, 193]. A potential mechanism behind the longer life expectancy may be mediated by the augmented secretion of IL-4 from mutant Cu2+/Zn2+ superoxide dismutase regulatory T lymphocytes that directly suppressed the toxic properties of microglia [193]. It was suggested that CD4+CD25HighFoxP3+ regulatory T lymphocytes (Tregs) are neuroprotective and slow ALS progression [194].
6. Potential Strategies Targeting BBB or BSCB for Chronic Pain Relief
The molecular mechanisms of BBB/BSCB permeabilization due to chronic pain have yet to be clarified. Nevertheless, the barriers represent promising targets in designing new therapeutic strategies for chronic pain. Several approaches tested in preclinical and clinical studies, such as the use of Rho-kinase inhibitors, antiepileptic compounds, and statins, might turn out to be viable solutions in the future.
6.1. Rho-Kinase Inhibitor
Rho kinase (ROCK) is involved in various physiological functions, including cell motility, vasoconstriction, and neurite extension. ROCK inhibition reduces tissue-type plasminogen activator (t-PA)/plasminogen-mediated increase in permeability of in vitro models of the BBB [195]. Fasudil, a specific ROCK inhibitor, partly alleviates EAE-dependent damage by decreasing BBB and BSCB permeability [196]. In preclinical models of pain, fasudil (30 mg/kg) significantly attenuated mechanical allodynia in spinal-nerve ligation, chronic constriction injury, capsaicin-induced secondary mechanical hypersensitivity, sodium iodoacetate-induced pain, and capsaicin-induced acute flinching behaviors, but failed to attenuate or had only modest effects on inflammatory thermal hyperalgesia following carrageenan injection and mechanical allodynia following complete Freund's Adjuvant injection [197]. Fasudil also proved to be efficient in adjuvant-induced arthritis model (inflammatory arthritis model) and a monoiodoacetate-induced arthritis model (noninflammatory arthritis model) [198].
6.2. Antiepileptic Drugs
It is difficult to consider currently market-available antiepileptic drugs (AEDs) as an alternative for classical analgesics because of their side effects, potential drug interactions, and unsatisfactory efficacy (epilepsy resistance). Between 1990 and 2012, 16 new AEDs were approved, most of them developed using mechanism-unbiased anticonvulsant animal models [199]. In order to be attractive for the pharmaceutical industry, the future design of new AEDs must also include a potential in nonepileptic CNS disorders, such as bipolar disorder and neuropathic pain [199]. Resistance to AEDs is encountered in more than 40% of epileptic patients [25], probably due to upregulation of the efflux transporters in brain capillary endothelium [200].
Only three AEDs are currently approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of neuropathic pain: carbamazepine (CBZ), gabapentin (GBP), and pregabalin (PGB), all of them considered first-line treatment options for several neuropathic pain conditions (reviewed by [199]). Randomized clinical trials in spinal cord injury-related pain indicate gabapentin and pregabalin as powerful analgesics [201]. Cochrane Library reports based on extended clinical trials indicate GBP, PGB, and lacosamide, but not valproic acid, to be efficient against neuropathic pain or fibromyalgia [202–205].
Levetiracetam (LEV) may constitute a novel approach for BBB protection [206]. Clinical studies have evidenced the effects of levetiracetam (LEV) in various pain conditions, such as postmastectomy pain syndrome, trigeminal neuralgia, chronic general or central pain in MS, lumbar radiculopathy, chronic daily headache, polyneuropathy, and central poststroke pain [207–212]. In a rat model of hypothermia-induced cortical dysplasia, LEV and topiramate were found to protect the BBB [212]. However, a recent clinical trial failed to reveal significant effects of LEV against spinal cord injury-related pain [213]. Despite LEV's protective properties on the BBB, clinical efficacy against chronic pain is still controversial.
6.3. Statins
Beside the well-known efficacy of statins (inhibitors of HMGCoA (3-hydroxy-3-methyl-glutaryl-coenzyme A) reductase) in lowering plasma cholesterol levels, these compounds show a large palette of pleiotropic effects. Statins can improve endothelial function (thereby regulating the BBB permeability), decrease the oxidative stress and inflammation, and generally have a beneficial effect on the immune system, central nervous system, and bone [214]. Some of these effects point out statins as good candidates for chronic pain treatment. In vivo preclinical tests showed that Atorvastatin (a lipophilic statin) restored the BBB permeability in mice fed with saturated fatty acids (which compromised BBB integrity) [215]. In primary human skeletal muscle myoblast cells, atorvastatin and rosuvastatin proved to be substrates for MRP1, MRP4, and MRP5 transporters [216].
An analgesic effect was revealed by hot-plate test for some statins [217]. Preclinical tests have been performed to evaluate statin efficacy in neuropathic pain. Daily administration of statin for two weeks completely prevented the development of mechanical allodynia and thermal hyperalgesia in a nerve injury model [218]. Such approaches provide promising results for considering statins as a possible future generation of drugs against chronic pain, especially for patients with dislipidemy.
7. Future Perspectives
General mechanisms of chronic pain onset, development, and maintenance still await clarification, and the particular relationship between chronic pain and NVU function is an especially complex issue. Whether permeabilization/disruption of the endothelial barrier in brain or spinal cord could be a cause and/or a consequence of chronic pain is an open topic. Clearly, a better knowledge of the neurovascular unit contribution to chronic pain physiopathology would be highly beneficial in the clinical practice, especially in view of pharmacological targeting of the NVU.
The use of currently available analgesics (opioids and NSAIDs, in particular) in chronic pain is limited by their side effects and by the induction of tolerance and/or dependence. In this review, we have described some aspects of the neurobiological mechanisms of chronic pain, with particular emphasis on NVU players' interactions, also in view of present and future treatments. Future strategies against chronic pain should take into account the essential role played by the neurovascular unit in the efficacy of analgesics in an effort to overcome the already-known problems.
As many neurodegenerative/neuroinflammatory pathologies are comorbid with chronic pain in a significant number of patients, the identification of dual-target therapeutic strategies should be considered a priority.
With the NVU as an increasingly relevant target for the treatment of chronic pain, development of immunologically based strategies for preventing BBB and/or BSCB permeabilization or disruption would also represent an opportunity.
Authors' Contribution
Giuseppe Bertini and Paolo Francesco Fabene share the senior authorship position.
Acknowledgment
This work was supported by Fondazione Cariverona (Beatrice Mihaela Radu and Mihai Radu), Italian Ministry of Education and Research (MIUR) (Paolo Francesco Fabene), Fondazione San Paolo (Giuseppe Bertini and Paolo Francesco Fabene), Romanian Ministry of Education, Research, Youth and Sports (Grant no. PCE 117/2011-2014) (Maria-Luisa Flonta).
References
- 1. www.iasp-pain.org.
- 2.Ramirez-Maestre C, Esteve R. Disposition and adjustment to chronic pain. Current Pain and Headache Reports. 2013;17, article 312 doi: 10.1007/s11916-012-0312-9. [DOI] [PubMed] [Google Scholar]
- 3.Institute of Medicine (US) Committee on Advancing Pain Research. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. National Academies Press; 2011. [PubMed] [Google Scholar]
- 4.Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. European Journal of Pain. 2006;10(4):287–333. doi: 10.1016/j.ejpain.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 5.Leadley RM, Armstrong N, Lee YC, Allen A, Kleijnen J. Chronic diseases in the European Union: the prevalence and health cost implications of chronic pain. Journal of Pain and Palliative Care Pharmacotherapy. 2012;26(4):310–325. doi: 10.3109/15360288.2012.736933. [DOI] [PubMed] [Google Scholar]
- 6.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature Reviews Neuroscience. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 7.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacological Reviews. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 8.Strazielle N, Ghersi-Egea JF. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Molecular Pharmaceutics. 2013;10(5):1473–1491. doi: 10.1021/mp300518e. [DOI] [PubMed] [Google Scholar]
- 9.Brooks TA, Hawkins BT, Huber JD, Egleton RD, Davis TP. Chronic inflammatory pain leads to increased blood-brain barrier permeability and tight junction protein alterations. American Journal of Physiology. 2005;289(2):H738–H743. doi: 10.1152/ajpheart.01288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beggs S, Liu XJ, Kwan C, Salter MW. Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Molecular Pain. 2010;6, article 74 doi: 10.1186/1744-8069-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Echeverry S, Shi XQ, Rivest S, Zhang J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. Journal of Neuroscience. 2011;31(30):10819–10828. doi: 10.1523/JNEUROSCI.1642-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tenorio G, Kulkarni A, Kerr BJ. Resident glial cell activation in response to perispinal inflammation leads to acute changes in nociceptive sensitivity: implications for the generation of neuropathic pain. Pain. 2013;154(1):71–81. doi: 10.1016/j.pain.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Morita S, Miyata S. Different vascular permeability between the sensory and secretory circumventricular organs of adult mouse brain. Cell and Tissue Research. 2012;349(2):589–603. doi: 10.1007/s00441-012-1421-9. [DOI] [PubMed] [Google Scholar]
- 14.Nag S, Begley DJ. Blood-brain barrier, exchange of metabolites and gases. In: Kalimo H, editor. Pathology and Genetics. Cerebrovascular Diseases. Basel, Switzerland: ISN Neuropath Press; 2005. pp. 22–29. [Google Scholar]
- 15.Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. The blood-spinal cord barrier: morphology and clinical implications. Annals of Neurology. 2011;70(2):194–206. doi: 10.1002/ana.22421. [DOI] [PubMed] [Google Scholar]
- 16.Sharma HS. Pathophysiology of blood-spinal cord barrier in traumatic injury and repair. Current Pharmaceutical Design. 2005;11(11):1353–1389. doi: 10.2174/1381612053507837. [DOI] [PubMed] [Google Scholar]
- 17.Daniel PM, Lam DKC, Pratt OE. Changes in the effectiveness of the blood-brain and blood-spinal cord barriers in experimental allergic encephalomyelitis. Possible relevance to multiple sclerosis. Journal of the Neurological Sciences. 1981;52(2-3):211–219. doi: 10.1016/0022-510x(81)90006-x. [DOI] [PubMed] [Google Scholar]
- 18.Prockop LD, Naidu KA, Binard JE, Ransohoff J. Selective permeability of [3H]-D-mannitol and [14C]-carboxyl-inulin across the blood-brain barrier and blood-spinal cord barrier in the rabbit. The Journal of Spinal Cord Medicine. 1995;18(4):221–226. doi: 10.1080/10790268.1995.11719399. [DOI] [PubMed] [Google Scholar]
- 19.Pan W, Banks WA, Kastin AJ. Permeability of the blood-brain and blood-spinal cord barriers to interferons. Journal of Neuroimmunology. 1997;76(1-2):105–111. doi: 10.1016/s0165-5728(97)00034-9. [DOI] [PubMed] [Google Scholar]
- 20.Ge S, Pachter JS. Isolation and culture of microvascular endothelial cells from murine spinal cord. Journal of Neuroimmunology. 2006;177(1-2):209–214. doi: 10.1016/j.jneuroim.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 21.Tsai PS, Kaufhold JP, Blinder P, et al. Correlations of neuronal and microvascular densities in murine cortex revealed by direct counting and colocalization of nuclei and vessels. Journal of Neuroscience. 2009;29(46):14553–14570. doi: 10.1523/JNEUROSCI.3287-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiology of Disease. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 23.Wilhelm I, Fazakas C, Krizbai IA. In vitro models of the blood-brain barrier. Acta Neurobiologiae Experimentalis. 2011;71(1):113–128. doi: 10.55782/ane-2011-1828. [DOI] [PubMed] [Google Scholar]
- 24.Löscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2(1):86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nature Reviews Neuroscience. 2005;6(8):591–602. doi: 10.1038/nrn1728. [DOI] [PubMed] [Google Scholar]
- 26.Seelbach MJ, Brooks TA, Egleton RD, Davis TP. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. Journal of Neurochemistry. 2007;102(5):1677–1690. doi: 10.1111/j.1471-4159.2007.04644.x. [DOI] [PubMed] [Google Scholar]
- 27.Ronaldson PT, Davis TP. Targeting blood-brain barrier changes during inflammatory pain: an opportunity for optimizing CNS drug delivery. Therapeutic Delivery. 2011;2(8):1015–1041. doi: 10.4155/tde.11.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tournier N, Decleves X, Saubamea B, Scherrmann JM, Cisternino S. Opioid transport by ATP-binding cassette transporters at the blood-brain barrier: implications for neuropsychopharmacology. Current Pharmaceutical Design. 2011;17(26):2829–2842. doi: 10.2174/138161211797440203. [DOI] [PubMed] [Google Scholar]
- 29.Cornford EM, Hyman S, Cornford ME, Landaw EM, Delgado-Escueta AV. Interictal seizure resections show two configurations of endothelial Glut1 glucose transporter in the human blood-brain barrier. Journal of Cerebral Blood Flow and Metabolism. 1998;18(1):26–42. doi: 10.1097/00004647-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell and Tissue Research. 2009;335(1):75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- 31.Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascular Pharmacology. 2002;38(6):323–337. doi: 10.1016/s1537-1891(02)00200-8. [DOI] [PubMed] [Google Scholar]
- 32.Mariano C, Sasaki H, Brites D, Brito MA. A look at tricellulin and its role in tight junction formation and maintenance. European Journal of Cell Biology. 2011;90(10):787–796. doi: 10.1016/j.ejcb.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Willis CL. Glia-induced reversible disruption of blood-brain barrier integrity and neuropathological response of the neurovascular unit. Toxicologic pathology. 2011;39(1):172–185. doi: 10.1177/0192623310385830. [DOI] [PubMed] [Google Scholar]
- 34.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. Journal of Lipid Research. 2001;42(7):1007–1017. [PubMed] [Google Scholar]
- 35.Campos CR, Schroter C, Wang X, Miller DS. ABC transporter function and regulation at the blood-spinal cord barrier. Journal of Cerebral Blood Flow & Metabolism. 2012;32(8):1559–1566. doi: 10.1038/jcbfm.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ronaldson PT, Finch JD, DeMarco KM, Quigley CE, Davis TP. Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. Journal of Pharmacology and Experimental Therapeutics. 2011;336(3):827–839. doi: 10.1124/jpet.110.174151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCaffrey G, Staatz WD, Sanchez-Covarrubias L, et al. P-glycoprotein trafficking at the blood-brain barrier altered by peripheral inflammatory hyperalgesia. Journal of Neurochemistry. 2012;122(5):962–975. doi: 10.1111/j.1471-4159.2012.07831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dagenais C, Graff CL, Pollack GM. Variable modulation of opioid brain uptake by P-glycoprotein in mice. Biochemical Pharmacology. 2004;67(2):269–276. doi: 10.1016/j.bcp.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 39.Lagas JS, Van Der Kruijssen CMM, Van De Wetering K, Beijnen JH, Schinkel AH. Transport of diclofenac by breast cancer resistance protein (ABCG2) and stimulation of multidrug resistance protein 2 (ABCC2)-mediated drug transport by diclofenac and benzbromarone. Drug Metabolism and Disposition. 2009;37(1):129–136. doi: 10.1124/dmd.108.023200. [DOI] [PubMed] [Google Scholar]
- 40.Cartwright TA, Campos CR, Cannon RE, Miller DS. Mrp1 is essential for sphingolipid signaling to p-glycoprotein in mouse blood-brain and blood-spinal cord barriers. Journal of Cerebral Blood Flow & Metabolism. 2013;33(3):381–388. doi: 10.1038/jcbfm.2012.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goadsby PJ. Trigeminal autonomic cephalalgias. Continuum. 2012;18(4):883–895. doi: 10.1212/01.CON.0000418649.54902.0b. [DOI] [PubMed] [Google Scholar]
- 42.De Groot DJA, Van Der Deen M, Le TKP, Regeling A, De Jong S, De Vries EGE. Indomethacin induces apoptosis via a MRP1-dependent mechanism in doxorubicin-resistant small-cell lung cancer cells overexpressing MRP1. British Journal of Cancer. 2007;97(8):1077–1083. doi: 10.1038/sj.bjc.6604010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reid G, Wielinga P, Zelcer N, et al. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal anti inflammatory drugs. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(16):9244–9249. doi: 10.1073/pnas.1033060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zelcer N, Van De Wetering K, Hillebrand M, et al. Mice lacking multidrug resistance protein 3 show altered morphine pharmacokinetics and morphine-6-glucuronide antinociception. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(20):7274–7279. doi: 10.1073/pnas.0502530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin ZP, Zhu YL, Johnson DR, et al. Disruption of cAMP and prostaglandin E2 transport by multidrug resistance protein 4 deficiency alters cAMP-mediated signaling and nociceptive response. Molecular Pharmacology. 2008;73(1):243–251. doi: 10.1124/mol.107.039594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beck K, Hayashi K, Nishiguchi B, Le Saux O, Hayashi M, Boyd CD. The distribution of Abcc6 in normal mouse tissues suggests multiple functions for this ABC transporter. Journal of Histochemistry and Cytochemistry. 2003;51(7):887–902. doi: 10.1177/002215540305100704. [DOI] [PubMed] [Google Scholar]
- 47.Van Vliet EA, Redeker S, Aronica E, Edelbroek PM, Gorter JA. Expression of multidrug transporters MRP1, MRP2, and BCRP shortly after status epilepticus, during the latent period, and in chronic epileptic rats. Epilepsia. 2005;46(10):1569–1580. doi: 10.1111/j.1528-1167.2005.00250.x. [DOI] [PubMed] [Google Scholar]
- 48.Pekcec A, Unkrüer B, Stein V, et al. Over-expression of P-glycoprotein in the canine brain following spontaneous status epilepticus. Epilepsy Research. 2009;83(2-3):144–151. doi: 10.1016/j.eplepsyres.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 49.Bartels AL. Blood-brain barrier P-glycoprotein function in neurodegenerative disease. Current Pharmaceutical Design. 2011;17(26):2771–2777. doi: 10.2174/138161211797440122. [DOI] [PubMed] [Google Scholar]
- 50.Von Wedel-Parlow M, Wölte P, Galla HJ. Regulation of major efflux transporters under inflammatory conditions at the blood-brain barrier in vitro. Journal of Neurochemistry. 2009;111(1):111–118. doi: 10.1111/j.1471-4159.2009.06305.x. [DOI] [PubMed] [Google Scholar]
- 51.Watkins LR, Maier SF. Immune regulation of central nervous system functions: from sickness responses to pathological pain. Journal of Internal Medicine. 2005;257(2):139–155. doi: 10.1111/j.1365-2796.2004.01443.x. [DOI] [PubMed] [Google Scholar]
- 52.Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. Journal of Neuroscience. 2006;26(16):4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee HL, Lee KM, Son SJ, Hwang SH, Cho HJ. Temporal expression of cytokines and their receptors mRNAs in a neuropathic pain model. NeuroReport. 2004;15(18):2807–2811. [PubMed] [Google Scholar]
- 54.Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. European Journal of Neuroscience. 2004;20(2):467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- 55.McMahon SB, Cafferty WBJ, Marchand F. Immune and glial cell factors as pain mediators and modulators. Experimental Neurology. 2005;192(2):444–462. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 56.Guo W, Wang H, Watanabe M, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. Journal of Neuroscience. 2007;27(22):6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srinivasan D, Yen JH, Joseph DJ, Friedman W. Cell type-specific interleukin-1beta signaling in the CNS. Journal of Neuroscience. 2004;24(29):6482–6488. doi: 10.1523/JNEUROSCI.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansson E. Could chronic pain and spread of pain sensation be induced and maintained by glial activation? Acta Physiologica. 2006;187(1-2):321–327. doi: 10.1111/j.1748-1716.2006.01568.x. [DOI] [PubMed] [Google Scholar]
- 59.Ledeboer A, Sloane EM, Milligan ED, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115(1-2):71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 60.Owolabi SA, Saab CY. Fractalkine and minocycline alter neuronal activity in the spinal cord dorsal horn. FEBS Letters. 2006;580(18):4306–4310. doi: 10.1016/j.febslet.2006.06.087. [DOI] [PubMed] [Google Scholar]
- 61.Mika J, Osikowicz M, Makuch W, Przewlocka B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. European Journal of Pharmacology. 2007;560(2-3):142–149. doi: 10.1016/j.ejphar.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 62.Chen ML, Cao H, Chu YX, et al. Role of P2X7 receptor-mediated IL-18/IL-18R signaling in morphine tolerance: multiple glial-neuronal dialogues in the rat spinal cord. Journal of Pain. 2012;13(10):945–958. doi: 10.1016/j.jpain.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 63.Imai S, Ikegami D, Yamashita A, et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain. 2013;136(part 3):828–843. doi: 10.1093/brain/aws330. [DOI] [PubMed] [Google Scholar]
- 64.DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10(1):40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- 65.Kuzumaki N, Narita M, Narita M, et al. Chronic pain-induced astrocyte activation in the cingulate cortex with no change in neural or glial differentiation from neural stem cells in mice. Neuroscience Letters. 2007;415(1):22–27. doi: 10.1016/j.neulet.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 66.Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. Journal of Neuroscience. 2008;28(48):12775–12787. doi: 10.1523/JNEUROSCI.3512-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cerbai F, Lana D, Nosi D, et al. The neuron-astrocyte-microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS One. 2012;7, article e45250 doi: 10.1371/journal.pone.0045250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liwnicz BH, Leach JL, Yeh HS, Privitera M, Roberts DW. Pericyte degeneration and thickening of basement membranes of cerebral microvessels in complex partial seizures: electron microscopic study of surgically removed tissue. Neurosurgery. 1990;26(3):409–420. doi: 10.1097/00006123-199003000-00006. [DOI] [PubMed] [Google Scholar]
- 69.Hellstrom M, Gerhardt H, Kalen M, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. The Journal of Cell Biology. 2001;153(3):543–553. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hansson E, Westerlund A, Björklund U, Olsson T. μ-Opioid agonists inhibit the enhanced intracellular Ca2+ responses in inflammatory activated astrocytes co-cultured with brain endothelial cells. Neuroscience. 2008;155(4):1237–1249. doi: 10.1016/j.neuroscience.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 71.Hung VK, Chen SM, Tai LW, Chen AY, Chung SK, Cheung CW. Over-expression of endothelin-1 in astrocytes, but not endothelial cells, ameliorates inflammatory pain response after formalin injection. Life Sciences. 2012;91(13-14):618–622. doi: 10.1016/j.lfs.2012.06.038. [DOI] [PubMed] [Google Scholar]
- 72.Winkler EA, Sengillo JD, Bell RD, Wang J, Zlokovic BV. Blood-spinal cord barrier pericyte reductions contribute to increased capillary permeability. Journal of Cerebral Blood Flow & Metabolism. 2012;32(10):1841–1852. doi: 10.1038/jcbfm.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Armulik A, Genove G, Mae M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 74.Berezowski V, Landry C, Dehouck MP, Cecchelli R, Fenart L. Contribution of glial cells and pericytes to the mRNA profiles of P-glycoprotein and multidrug resistance-associated proteins in an in vitro model of the blood-brain barrier. Brain Research. 2004;1018(1):1–9. doi: 10.1016/j.brainres.2004.05.092. [DOI] [PubMed] [Google Scholar]
- 75.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circulation Research. 2005;97(6):512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 76.Dore-Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, Rafols JA. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvascular Research. 2000;60(1):55–69. doi: 10.1006/mvre.2000.2244. [DOI] [PubMed] [Google Scholar]
- 77.Gonul E, Duz B, Kahraman S, Kayali H, Kubar A, Timurkaynak E. Early pericyte response to brain hypoxia in cats: an ultrastructural study. Microvascular Research. 2002;64(1):116–119. doi: 10.1006/mvre.2002.2413. [DOI] [PubMed] [Google Scholar]
- 78.Liu S, Agalliu D, Yu C, Fisher M. The role of pericytes in blood-brain barrier function and stroke. Current Pharmaceutical Design. 2012;18(25):3653–3662. doi: 10.2174/138161212802002706. [DOI] [PubMed] [Google Scholar]
- 79.Luk K, Boatman S, Johnson KN, et al. Influence of morphine on pericyte-endothelial interaction: implications for antiangiogenic therapy. Journal of Oncology. 2012;2012:10 pages. doi: 10.1155/2012/458385.458385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma HS, Sharma A. Breakdown of the blood-brain barrier in stress alters cognitive dysfunction and induces brain pathology: new perspectivesfor neuroprotective strategies. In: Ritsner MS, editor. Brain Protection in Schizophrenia, Mood and Cognitive Disorders. Dordrecht, The Netherlands: Springer; 2010. pp. 243–304. [Google Scholar]
- 81.Sharma HS, Sharma A. Blood-brain and spinal cord barriers in stress. In: Sharma HS, Westman J, editors. Blood-Spinal Cord and Brain Barriers in Health and Disease. San Diego, Calif, USA: Elsevier Academic Press; 2004. pp. 231–298. [Google Scholar]
- 82.Sharma HS. Blood-central nervous system barriers. A gateway to neurodegeneration, neuroprotection and neuroregeneration. In: Lajtha A, Naren Banik N, Ray SK, editors. Handbook of Neurochemistry and Molecular Neurobiology. San Diego, Calif, USA: Elsevier Academic Press; 2009. pp. 363–457. [Google Scholar]
- 83.Gordh T, Chu H, Sharma HS. Spinal nerve lesion alters blood-spinal cord barrier function and activates astrocytes in the rat. Pain. 2006;124(1-2):211–221. doi: 10.1016/j.pain.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 84.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nature Neuroscience. 2007;10(11):1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 85.Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annual Review of Neuroscience. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Costigan M, Moss A, Latremoliere A, et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. Journal of Neuroscience. 2009;29(46):14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rodrigues MCO, Hernandez-Ontiveros DG, Louis MK, et al. Neurovascular aspects of amyotrophic lateral sclerosis. New Perspectives of Central Nervous System Injury and Neuroprotection. 2012;102:91–106. doi: 10.1016/B978-0-12-386986-9.00004-1. [DOI] [PubMed] [Google Scholar]
- 88.Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Research. 1991;565(1):1–7. doi: 10.1016/0006-8993(91)91729-k. [DOI] [PubMed] [Google Scholar]
- 89.Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33(11):1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 90.Tsuda M, Shigemoto-Mogami Y, Koizumi S, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424(6950):778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 91.Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(16):5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huber JD, Hau VS, Borg L, Campos CR, Egleton RD, Davis TP. Blood-brain barrier tight junctions are altered during a 72-h exposure to λ-carrageenan-induced inflammatory pain. American Journal of Physiology - Heart and Circulatory Physiology. 2002;283(4):H1531–H1537. doi: 10.1152/ajpheart.00027.2002. [DOI] [PubMed] [Google Scholar]
- 93.Huber JD, Witt KA, Hom S, Egleton RD, Mark KS, Davis TP. Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. American Journal of Physiology - Heart and Circulatory Physiology. 2001;280(3):H1241–H1248. doi: 10.1152/ajpheart.2001.280.3.H1241. [DOI] [PubMed] [Google Scholar]
- 94.Brooks TA, Ocheltree SM, Seelbach MJ, et al. Biphasic cytoarchitecture and functional changes in the BBB induced by chronic inflammatory pain. Brain Research. 2006;1120(1):172–182. doi: 10.1016/j.brainres.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McCaffrey G, Seelbach MJ, Staatz WD, et al. Occludin oligomeric assembly at tight junctions of the blood-brain barrier is disrupted by peripheral inflammatory hyperalgesia. Journal of Neurochemistry. 2008;106(6):2395–2409. doi: 10.1111/j.1471-4159.2008.05582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-Β signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. Journal of Cerebral Blood Flow and Metabolism. 2009;29(6):1084–1098. doi: 10.1038/jcbfm.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lochhead JJ, McCaffrey G, Sanchez-Covarrubias L, et al. Tempol modulates changes in xenobiotic permeability and occludin oligomeric assemblies at the blood-brain barrier during inflammatory pain. American Journal of Physiology. 2012;302(3):H582–H593. doi: 10.1152/ajpheart.00889.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brooks TA, Nametz N, Charles R, Davis TP. Diclofenac attenuates the regional effect of λ-Carrageenan on blood-brain barrier function and cytoarchitecture. Journal of Pharmacology and Experimental Therapeutics. 2008;325(2):665–673. doi: 10.1124/jpet.107.135632. [DOI] [PubMed] [Google Scholar]
- 99.Nagaraja TN, Keenan KA, Fenstermacher JD, Knight RA. Acute leakage patterns of fluorescent plasma flow markers after transient focal cerebral ischemia suggest large openings in blood-brain barrier. Microcirculation. 2008;15(1):1–14. doi: 10.1080/10739680701409811. [DOI] [PubMed] [Google Scholar]
- 100.Lu P, Gonzales C, Chen Y, et al. CNS penetration of small molecules following local inflammation, widespread systemic inflammation or direct injury to the nervous system. Life Sciences. 2009;85(11-12):450–456. doi: 10.1016/j.lfs.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 101.Fabene PF, Navarro MG, Martinello M, et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nature Medicine. 2008;14(12):1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Labianca R, Sarzi-Puttini P, Zuccaro SM, Cherubino P, Vellucci R, Fornasari D. Adverse effects associated with non-opioid and opioid treatment in patients with chronic pain. Clinical Drug Investigation. 2012;32:53–63. doi: 10.2165/11630080-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 103.Koch T, Hollt V. Role of receptor internalization in opioid tolerance and dependence. Pharmacology & Therapeutics. 2008;117(2):199–206. doi: 10.1016/j.pharmthera.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 104.Muscoli C, Doyle T, Dagostino C, et al. Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. Journal of Neuroscience. 2010;30(46):15400–15408. doi: 10.1523/JNEUROSCI.2391-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gottschalk S, Cummins CL, Leibfritz D, Christians U, Benet LZ, Serkova NJ. Age and sex differences in the effects of the immunosuppressants cyclosporine, sirolimus and everolimus on rat brain metabolism. NeuroToxicology. 2011;32(1):50–57. doi: 10.1016/j.neuro.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 106.Zheng S, Tasnif Y, Hebert MF, et al. CYP3A5 gene variation influences cyclosporine a metabolite formation and renal cyclosporine disposition. Transplantation. 2013;95(6):821–827. doi: 10.1097/TP.0b013e31827e6ad9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vanegas H, Vazquez E, Tortorici V. NSAIDS, opioids, cannabinoids and the control of pain by the central nervous system. Pharmaceuticals. 2010;3(5):1335–1347. doi: 10.3390/ph3051335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tsiklauri N, Viatchenko-Karpinski V, Voitenko N, Tsagareli MG. Non-opioid tolerance in juvenile and adult rats. European Journal of Pharmacology. 2010;629(1–3):68–72. doi: 10.1016/j.ejphar.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 109.Candelario-Jalil E, Taheri S, Yang Y, et al. Cyclooxygenase inhibition limits blood-brain barrier disruption following intracerebral injection of tumor necrosis factor-α in the rat. Journal of Pharmacology and Experimental Therapeutics. 2007;323(2):488–498. doi: 10.1124/jpet.107.127035. [DOI] [PubMed] [Google Scholar]
- 110.Nandedkar SD. Assessment of spinal cord pathology following trauma using early changes in the spinal cord evoked potentials: a pharmacological and morphological study in the rat. Muscle and Nerve. 2002;25(supplement 11):S83–S91. doi: 10.1002/mus.10152. [DOI] [PubMed] [Google Scholar]
- 111.Ottman R, Lipton RB, Ettinger AB, et al. Comorbidities of epilepsy: results from the Epilepsy Comorbidities and Health (EPIC) survey. Epilepsia. 2011;52(2):308–315. doi: 10.1111/j.1528-1167.2010.02927.x. [DOI] [PubMed] [Google Scholar]
- 112.Kobau R, Zahran H, Thurman DJ, et al. Epilepsy surveillance among adults—19 States, Behavioral Risk Factor Surveillance System, 2005. Morbidity and Mortality Weekly Report Surveillance Summaries. 2008;57(6):1–20. [PubMed] [Google Scholar]
- 113.Parisi P, Striano P, Verrotti A, Villa MP, Belcastro V. What have we learned about ictal epileptic headache? A review of well-documented cases. Seizure. 2013;22(4):253–258. doi: 10.1016/j.seizure.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 114.Toldo I, Perissinotto E, Menegazzo F, et al. Comorbidity between headache and epilepsy in a pediatric headache center. The Journal of Headache and Pain. 2010;11(3):235–240. doi: 10.1007/s10194-010-0191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ito M, Adachi N, Nakamura F, et al. Multi-center study on post-ictal headache in patients with localization-related epilepsy. Psychiatry and Clinical Neurosciences. 2003;57(4):385–389. doi: 10.1046/j.1440-1819.2003.01136.x. [DOI] [PubMed] [Google Scholar]
- 116.Rogawski MA. Migraine and epilepsy—shared mechanisms within the family of episodic disorders. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. Oxford University Press; 2012. pp. 930–944. [Google Scholar]
- 117.Fabene PF, Marzola P, Sbarbati A, Bentivoglio M. Magnetic resonance imaging of changes elicited by status epilepticus in the rat brain: diffusion-weighted and T2-weighted images, regional blood volume maps, and direct correlation with tissue and cell damage. NeuroImage. 2003;18(2):375–389. doi: 10.1016/s1053-8119(02)00025-3. [DOI] [PubMed] [Google Scholar]
- 118.Seiffert E, Dreier JP, Ivens S, et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. Journal of Neuroscience. 2004;24(36):7829–7836. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Oby E, Janigro D. The blood-brain barrier and epilepsy. Epilepsia. 2006;47(11):1761–1774. doi: 10.1111/j.1528-1167.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- 120.Ivens S, Kaufer D, Flores LP, et al. TGF-β receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130(2):535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- 121.Van Vliet EA, Araújo SDC, Redeker S, Van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130(2):521–534. doi: 10.1093/brain/awl318. [DOI] [PubMed] [Google Scholar]
- 122.Marchi N, Oby E, Batra A, et al. In vivo and in vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48(10):1934–1946. doi: 10.1111/j.1528-1167.2007.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fabene PF, Butcker EC, Constantin G. Anti-leukocyte recruitment therapy for the treatment of seizures and epilepsy. US Patent, no: US, 2008/0025992 A1, 2008.
- 124.Fabene PF, Bramanti P, Constantin G. The emerging role for chemokines in epilepsy. Journal of Neuroimmunology. 2010;224(1-2):22–27. doi: 10.1016/j.jneuroim.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 125.Fabene PF, Laudanna C, Constantin G. Leukocyte trafficking mechanisms in epilepsy. Molecular Immunology. 2013;55(1):100–104. doi: 10.1016/j.molimm.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 126.Cutrer FM, Sorensen AG, Weisskoff RM, et al. Perfusion-weighted imaging defects during spontaneous migrainous aura. Annals of Neurology. 1998;43(1):25–31. doi: 10.1002/ana.410430108. [DOI] [PubMed] [Google Scholar]
- 127.Jäger HR, Giffin NJ, Goadsby PJ. Diffusion- and perfusion-weighted MR imaging in persistent migrainous visual disturbances. Cephalalgia. 2005;25(5):323–332. doi: 10.1111/j.1468-2982.2004.00858.x. [DOI] [PubMed] [Google Scholar]
- 128.Dreier JP, Jurkat-Rott K, Petzold GG, et al. Opening of the blood-brain barrier preceding cortical edema in a severe attack of FHM type II. Neurology. 2005;64(12):2145–2147. doi: 10.1212/01.WNL.0000176298.63840.99. [DOI] [PubMed] [Google Scholar]
- 129.Gonçalves FM, Martins-Oliveira A, Lacchini R, et al. Matrix metalloproteinase (MMP)-2 gene polymorphisms affect circulating MMP-2 levels in patients with migraine with aura. Gene. 2013;512(1):35–40. doi: 10.1016/j.gene.2012.09.109. [DOI] [PubMed] [Google Scholar]
- 130.Martins-Oliveira A, Gonçalves FM, Speciali JG, et al. Specific matrix metalloproteinase 9 (MMP-9) haplotype affect the circulating MMP-9 levels in women with migraine. Journal of Neuroimmunology. 2012;252(1-2):89–94. doi: 10.1016/j.jneuroim.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 131.Pautex S, Michon A, Guedira M, et al. Pain in severe dementia: self-assessment or observational scales? Journal of the American Geriatrics Society. 2006;54(7):1040–1045. doi: 10.1111/j.1532-5415.2006.00766.x. [DOI] [PubMed] [Google Scholar]
- 132.Scherder EJ, Sergeant JA, Swaab DF. Pain processing in dementia and its relation to neuropathology. Lancet Neurology. 2003;2(11):677–686. doi: 10.1016/s1474-4422(03)00556-8. [DOI] [PubMed] [Google Scholar]
- 133.Schmidt R, Bach M, Dal-Bianco P, et al. Dementia and pain. Neuropsychiatry. 2010;24(1):1–13. [PubMed] [Google Scholar]
- 134.Pickering G, Eschalier A, Dubray C. Pain and Alzheimer’s disease. Gerontology. 2000;46(5):235–241. doi: 10.1159/000022166. [DOI] [PubMed] [Google Scholar]
- 135.Licht E, Siegler EL, Reid MC. Can the cognitively impaired safely use patient-controlled analgesia? Journal of Opioid Management. 2009;5(5):307–312. doi: 10.5055/jom.2009.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee DM, Pendleton N, Tajar A, et al. Chronic widespread pain is associated with slower cognitive processing speed in middle-aged and older European men. Pain. 2010;151(1):30–36. doi: 10.1016/j.pain.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 137.Moriarty O, McGuire BE, Finn DP. The effect of pain on cognitive function: a review of clinical and preclinical research. Progress in Neurobiology. 2011;93(3):385–404. doi: 10.1016/j.pneurobio.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 138.Carrero I, Gonzalo MR, Martin B, Sanz-Anquela JM, Arevalo-Serrano J, Gonzalo-Ruiz A. Oligomers of beta-amyloid protein (Abeta1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Experimental Neurology. 2012;236(2):215–227. doi: 10.1016/j.expneurol.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 139.Griffin WST, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. Journal of Neuropathology and Experimental Neurology. 1995;54(2):276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 140.Lautner R, Mattsson N, Scholl M, et al. Biomarkers for microglial activation in Alzheimer's disease. International Journal of Alzheimer'S DiSeaSe. 2011;2011:5 pages. doi: 10.4061/2011/939426.939426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Romanitan MO, Popescu BO, Spulber Ş, et al. Altered expression of claudin family proteins in Alzheimer’s disease and vascular dementia brains. Journal of Cellular and Molecular Medicine. 2010;14(5):1088–1100. doi: 10.1111/j.1582-4934.2009.00999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Popescu BO, Toescu EC, Popescu LM, et al. Blood-brain barrier alterations in ageing and dementia. Journal of the Neurological Sciences. 2009;283(1-2):99–106. doi: 10.1016/j.jns.2009.02.321. [DOI] [PubMed] [Google Scholar]
- 143.Zlokovic BV. Clearing amyloid through the blood-brain barrier. Journal of Neurochemistry. 2004;89(4):807–811. doi: 10.1111/j.1471-4159.2004.02385.x. [DOI] [PubMed] [Google Scholar]
- 144.Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003;10(6):463–470. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- 145.Esiri MM. The interplay between inflammation and neurodegeneration in CNS disease. Journal of Neuroimmunology. 2007;184(1-2):4–16. doi: 10.1016/j.jneuroim.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 146.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiology of Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Simuni T, Sethi K. Nonmotor manifestations of Parkinson’s disease. Annals of Neurology. 2008;64(2):S65–S80. doi: 10.1002/ana.21472. [DOI] [PubMed] [Google Scholar]
- 148.Ford B. Pain in Parkinson's Disease. Movement Disorders. 2010;25(3):S98–S103. doi: 10.1002/mds.22716. [DOI] [PubMed] [Google Scholar]
- 149.Borsook D. Neurological diseases and pain. Brain. 2012;135(part 2):320–344. doi: 10.1093/brain/awr271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kortekaas R, Leenders KL, Van Oostrom JCH, et al. Blood-brain barrier dysfunction in Parkinsonian midbrain in vivo . Annals of Neurology. 2005;57(2):176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 151.Carvey PM, Zhao CH, Hendey B, et al. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. European Journal of Neuroscience. 2005;22(5):1158–1168. doi: 10.1111/j.1460-9568.2005.04281.x. [DOI] [PubMed] [Google Scholar]
- 152.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? The Lancet Neurology. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 153.Zhang W, Wang T, Pei Z, et al. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. The FASEB Journal. 2005;19(6):533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 154.Forno LS, DeLanney LE, Irwin I, Di Monti D, Langston JW. Astrocytes and Parkinson’s disease. Progress in Brain Research. 1992;94:429–436. doi: 10.1016/s0079-6123(08)61770-7. [DOI] [PubMed] [Google Scholar]
- 155.Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience. 1993;52(1):1–6. doi: 10.1016/0306-4522(93)90175-f. [DOI] [PubMed] [Google Scholar]
- 156.Mirza B, Hadberg H, Thomsen P, Moos T. The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience. 1999;95(2):425–432. doi: 10.1016/s0306-4522(99)00455-8. [DOI] [PubMed] [Google Scholar]
- 157.Song YJC, Halliday GM, Holton JL, et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. Journal of Neuropathology and Experimental Neurology. 2009;68(10):1073–1083. doi: 10.1097/NEN.0b013e3181b66f1b. [DOI] [PubMed] [Google Scholar]
- 158.Chung YC, Ko HW, Bok E, et al. The role of neuroinflammation on the pathogenesis of Parkinson’s disease. BMB Reports. 2010;43(4):225–232. doi: 10.5483/bmbrep.2010.43.4.225. [DOI] [PubMed] [Google Scholar]
- 159.Baiguera C, Alghisi M, Pinna A, et al. Late-onset Parkinsonism in NFkappaB/c-Rel-deficient mice. Brain. 2012;135(Part 9):2750–2765. doi: 10.1093/brain/aws193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cayrol R, Saikali P, Vincent T. Effector functions of antiaquaporin-4 autoantibodies in neuromyelitis optica. Annals of the New York Academy of Sciences. 2009;1173:478–486. doi: 10.1111/j.1749-6632.2009.04871.x. [DOI] [PubMed] [Google Scholar]
- 161.O’Connor AB, Schwid SR, Herrmann DN, Markman JD, Dworkin RH. Pain associated with multiple sclerosis: systematic review and proposed classification. Pain. 2008;137(1):96–111. doi: 10.1016/j.pain.2007.08.024. [DOI] [PubMed] [Google Scholar]
- 162.Bermejo PE, Oreja-Guevara C, Díez-Tejedor E. Pain in multiple sclerosis: prevalence, mechanisms, types and treatment. Revista de Neurologia. 2010;50(2):101–108. [PubMed] [Google Scholar]
- 163.Truini A, Barbanti P, Pozzilli C, Cruccu G. A mechanism-based classification of pain in multiple sclerosis. Journal of Neurology. 2013;226(2):351–367. doi: 10.1007/s00415-012-6579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.de Vries HE, Dijkstra DD. Mononuclear phagocytes at the blood-brain barrier in multiple sclerosis. In: Sharma HS, Westman J, editors. Blood-Spinal Cord and Brain Barriers in Health and Disease. San Diego, Calif, USA: Elsevier; 2004. pp. 409–417. [Google Scholar]
- 165.de Vries HE, Kooij G, Frenkel D, Georgopoulos S, Monsonego A, Janigro D. Inflammatory events at blood-brain barrier in neuroinflammatory and neurodegenerative disorders: implications for clinical disease. Epilepsia. 2012;53(supplement 6):45–52. doi: 10.1111/j.1528-1167.2012.03702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Reijerkerk A, Lakeman KA, Drexhage JA, et al. Brain endothelial barrier passage by monocytes is controlled by the endothelin system. Journal of Neurochemistry. 2012;121(5):730–737. doi: 10.1111/j.1471-4159.2011.07393.x. [DOI] [PubMed] [Google Scholar]
- 167.Seeldrayers PA, Syha J, Morrissey SP, et al. Magnetic resonance imaging investigation of blood-brain barrier damage in adoptive transfer experimental autoimmune encephalomyelitis. Journal of Neuroimmunology. 1993;46(1-2):199–206. doi: 10.1016/0165-5728(93)90250-3. [DOI] [PubMed] [Google Scholar]
- 168.Kooij G, Backer R, Koning JJ, et al. P-glycoprotein acts as an immunomodulator during neuroinflammation. PloS one. 2009;4(12):p. e8212. doi: 10.1371/journal.pone.0008212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Morrissey SP, Stodal H, Zettl U, et al. In vivo MRI and its histological correlates in acute adoptive transfer experimental allergic encephalomyelitis: quantification of inflammation and oedema. Brain. 1996;119(1):239–248. doi: 10.1093/brain/119.1.239. [DOI] [PubMed] [Google Scholar]
- 170.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. The New England Journal of Medicine. 2000;343(13):938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 171.Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo . Neuroscience. 1998;86(4):1245–1257. doi: 10.1016/s0306-4522(98)00058-x. [DOI] [PubMed] [Google Scholar]
- 172.Plumb J, McQuaid S, Mirakhur M, Kirk J. Abnormal endothelial tight junctions in active lesions and normal-appearing white matter in multiple sclerosis. Brain Pathology. 2002;12(2):154–169. doi: 10.1111/j.1750-3639.2002.tb00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. Journal of Pathology. 2003;201(2):319–327. doi: 10.1002/path.1434. [DOI] [PubMed] [Google Scholar]
- 174.Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Multiple Sclerosis. 2003;9(6):540–549. doi: 10.1191/1352458503ms965oa. [DOI] [PubMed] [Google Scholar]
- 175.Hafler DA, Slavik JM, Anderson DE, O’Connor KC, De Jager P, Baecher-Allan C. Multiple sclerosis. Immunological Reviews. 2005;204:208–231. doi: 10.1111/j.0105-2896.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- 176.Craner MJ, Damarjian TG, Liu S, et al. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. GLIA. 2005;49(2):220–229. doi: 10.1002/glia.20112. [DOI] [PubMed] [Google Scholar]
- 177.Schellenberg AE, Buist R, Yong VW, Del Bigio MR, Peeling J. Magnetic resonance imaging of blood-spinal cord barrier disruption in mice with experimental autoimmune encephalomyelitis. Magnetic Resonance in Medicine. 2007;58(2):298–305. doi: 10.1002/mrm.21289. [DOI] [PubMed] [Google Scholar]
- 178.Olechowski CJ, Truong JJ, Kerr BJ. Neuropathic pain behaviours in a chronic-relapsing model of experimental autoimmune encephalomyelitis (EAE) Pain. 2009;141(1-2):156–164. doi: 10.1016/j.pain.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 179.Arima Y, Harada M, Kamimura D, et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell. 2012;148(3):447–457. doi: 10.1016/j.cell.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 180.Yuan SB, Shi YQ, Tang SJ. Wnt signaling in the pathogenesis of multiple sclerosis-associated chronic pain. Journal of Neuroimmune Pharmacology. 2012;7(4):904–913. doi: 10.1007/s11481-012-9370-3. [DOI] [PubMed] [Google Scholar]
- 181.Reis M, Liebner S. Wnt signaling in the vasculature. Experimental Cell Research. 2013;319(9):1317–1323. doi: 10.1016/j.yexcr.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 182.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227–1233. doi: 10.1212/WNL.0b013e3181bc01a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Franca JMC, D’Abreu A, Friedman JH, Nucci A, Lopes-Cendes I. Chronic pain in Machado-Joseph disease: a frequent and disabling symptom. Archives of Neurology. 2007;64(12):1767–1770. doi: 10.1001/archneur.64.12.1767. [DOI] [PubMed] [Google Scholar]
- 184.De Castro-Costa CM, Oriá RB, Machado-Filho JA, et al. Amyotrophic lateral sclerosis: clinical analysis of 78 cases from Fortaleza (Northeastern Brazil) Arquivos de Neuro-Psiquiatria. 1999;57(3B):761–774. doi: 10.1590/s0004-282x1999000500006. [DOI] [PubMed] [Google Scholar]
- 185.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 186.Garbuzova-Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, Sanberg PR. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Research. 2007;1157(1):126–137. doi: 10.1016/j.brainres.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 187.Nicaise C, Mitrecic D, Demetter P, et al. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Research. 2009;1301:152–162. doi: 10.1016/j.brainres.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 188.Zhong Z, Deane R, Ali Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nature Neuroscience. 2008;11(4):420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jablonski MR, Jacob DA, Campos C, et al. Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiology of Disease. 2012;47(2):194–200. doi: 10.1016/j.nbd.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Evans MC, Couch Y, Sibson N, Turner MR. Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Molecular and Cellular Neuroscience. 2013;53:34–41. doi: 10.1016/j.mcn.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 191.Engelhardt JI, Tajti J, Appel SH. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Archives of Neurology. 1993;50(1):30–36. doi: 10.1001/archneur.1993.00540010026013. [DOI] [PubMed] [Google Scholar]
- 192.Chiu IM, Chen A, Zheng Y, et al. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(46):17913–17918. doi: 10.1073/pnas.0804610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Beers DR, Henkel JS, Zhao W, et al. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134(5):1293–1314. doi: 10.1093/brain/awr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Henkel JS, Beers DR, Wen S, et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Molecular Medicine. 2013;5(1):64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Niego B, Freeman R, Puschmann TB, Turnley AM, Medcalf RL. t-PA-specific modulation of a human blood-brain barrier model involves plasmin-mediated activation of the Rho kinase pathway in astrocytes. Blood. 2012;119(20):4752–4761. doi: 10.1182/blood-2011-07-369512. [DOI] [PubMed] [Google Scholar]
- 196.Huang XN, Fu J, Wang WZ. The effects of fasudil on the permeability of the rat blood-brain barrier and blood-spinal cord barrier following experimental autoimmune encephalomyelitis. Journal of Neuroimmunology. 2011;239(1-2):61–67. doi: 10.1016/j.jneuroim.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 197.Boyce-Rustay JM, Simler GH, McGaraughty S, et al. Characterization of Fasudil in preclinical models of pain. Journal of Pain. 2010;11(10):941–949. doi: 10.1016/j.jpain.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 198.Yoshimi E, Kumakura F, Hatori C, et al. Antinociceptive effects of AS1892802, a novel rho kinase inhibitor, in rat models of inflammatory and noninflammatory arthritis. Journal of Pharmacology and Experimental Therapeutics. 2010;334(3):955–963. doi: 10.1124/jpet.110.167924. [DOI] [PubMed] [Google Scholar]
- 199.Bialer M. Why are antiepileptic drugs used for nonepileptic conditions? Epilepsia. 2012;53(supplement 7):26–33. doi: 10.1111/j.1528-1167.2012.03712.x. [DOI] [PubMed] [Google Scholar]
- 200.Bauer B, Hartz AMS, Pekcec A, Toellner K, Miller DS, Potschka H. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Molecular Pharmacology. 2008;73(5):1444–1453. doi: 10.1124/mol.107.041210. [DOI] [PubMed] [Google Scholar]
- 201.Finnerup NB, Baastrup C. Spinal cord injury pain: mechanisms and management. Current Pain and Headache Reports. 2012;16(3):207–216. doi: 10.1007/s11916-012-0259-x. [DOI] [PubMed] [Google Scholar]
- 202.Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews. 2009;(3):p. CD007076. doi: 10.1002/14651858.CD007076.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Moore RA, Wiffen PJ, Derry S, McQuay HJ. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews. 2011;3:p. CD007938. doi: 10.1002/14651858.CD007938.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gill D, Derry S, Wiffen PJ, Moore RA. Valproic acid and sodium valproate for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews. 2011;10 doi: 10.1002/14651858.CD009183.pub2.CD009183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hearn L, Derry S, Moore RA. Lacosamide for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews. 2012;2 doi: 10.1002/14651858.CD009318.pub2.CD009318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ahishali B, Kaya M, Orhan N, et al. Effects of levetiracetam on blood-brain barrier disturbances following hyperthermia-induced seizures in rats with cortical dysplasia. Life Sciences. 2010;87(19–22):609–619. doi: 10.1016/j.lfs.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 207.Vilholm OJ, Cold S, Rasmussen L, Sindrup SH. Effect of levetiracetam on the postmastectomy pain syndrome. European Journal of Neurology. 2008;15(8):851–857. doi: 10.1111/j.1468-1331.2008.02206.x. [DOI] [PubMed] [Google Scholar]
- 208.Jorns TP, Johnston A, Zakrzewska JM. Pilot study to evaluate the efficacy and tolerability of levetiracetam (Keppra®) in treatment of patients with trigeminal neuralgia. European Journal of Neurology. 2009;16(6):740–744. doi: 10.1111/j.1468-1331.2009.02585.x. [DOI] [PubMed] [Google Scholar]
- 209.Rossi S, Mataluni G, Codecà C, et al. Effects of levetiracetam on chronic pain in multiple sclerosis: results of a pilot, randomized, placebo-controlled study. European Journal of Neurology. 2009;16(3):360–366. doi: 10.1111/j.1468-1331.2008.02496.x. [DOI] [PubMed] [Google Scholar]
- 210.Holbech JV, Otto M, Bach FW, Jensen TS, Sindrup SH. The anticonvulsant levetiracetam for the treatment of pain in polyneuropathy: a randomized, placebo-controlled, cross-over trial. European Journal of Pain. 2011;15(6):608–614. doi: 10.1016/j.ejpain.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 211.Beran RG, Spira PJ. Levetiracetam in chronic daily headache: a double-blind, randomised placebo-controlled study. (The Australian KEPPRA Headache Trial [AUS-KHT]) Cephalalgia. 2011;31(5):530–536. doi: 10.1177/0333102410384886. [DOI] [PubMed] [Google Scholar]
- 212.Kaya M, Becker AJ, Gurses C. Blood-brain barrier, epileptogenesis, and treatment strategies in cortical dysplasia. Epilepsia. 2012;53(supplement 6):31–36. doi: 10.1111/j.1528-1167.2012.03700.x. [DOI] [PubMed] [Google Scholar]
- 213.Finnerup NB, Grydehøj J, Bing J, et al. Levetiracetam in spinal cord injury pain: a randomized controlled trial. Spinal Cord. 2009;47(12):861–867. doi: 10.1038/sc.2009.55. [DOI] [PubMed] [Google Scholar]
- 214.Liao JK, Laufs U. Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Pallebage-Gamarallage M, Lam V, Takechi R, Galloway S, Clark K, Mamo J. Restoration of dietary-fat induced blood-brain barrier dysfunction by anti-inflammatory lipid-modulating agents. Lipids in Health and Disease. 2012;11(117) doi: 10.1186/1476-511X-11-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Knauer MJ, Urquhart BL, Meyer Zu Schwabedissen HE, et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circulation Research. 2010;106(2):297–306. doi: 10.1161/CIRCRESAHA.109.203596. [DOI] [PubMed] [Google Scholar]
- 217.Kesim M, Kadioglu M, Okuyan M, et al. The evaluation of analgesic effects of simvastatin, pravastatin and atorvastatin in hot plate test. European Review for Medical and Pharmacological Sciences. 2012;16(6):789–796. [PubMed] [Google Scholar]
- 218.Shi XQ, Lim TKY, Lee S, Zhao YQ, Zhang J. Statins alleviate experimental nerve injury-induced neuropathic pain. Pain. 2011;152(5):1033–1043. doi: 10.1016/j.pain.2011.01.006. [DOI] [PubMed] [Google Scholar]