

Abstract
Dysfunction of the hypothalamic–pituitary–adrenal (HPA) axis is believed to play a role in the pathophysiology of depression. To investigate mechanisms that may underlie this effect, we examined several indices of HPA axis function – specifically, diurnal cortisol slope, cortisol awakening response, and suppression of cortisol release following dexamethasone administration – in 26 pre-menopausal depressed women and 23 never depressed women who were matched for age and body mass index. Salivary cortisol samples were collected at waking, 30 min after waking, and at bedtime over three consecutive days. On the third day, immediately after the bedtime sample, participants ingested a 0.5 mg dexamethasone tablet; they then collected cortisol samples at waking and 30 min after waking the following morning. As predicted, depressed women exhibited flatter diurnal cortisol rhythms and more impaired suppression of cortisol following dexamethasone administration than non-depressed women over the three sampling days. In addition, flatter diurnal cortisol slopes were associated with reduced cortisol response to dexamethasone treatment, both for all women and for depressed women when considered separately. Finally, greater self-reported depression severity was associated with flatter diurnal cortisol slopes and with less dexamethasone-related cortisol suppression for depressed women. Depression in women thus appears to be characterized by altered HPA axis functioning, as indexed by flatter diurnal cortisol slopes and an associated impaired sensitivity of cortisol to dexamethasone. Given that altered HPA axis functioning has been implicated in several somatic conditions, the present findings may be relevant for understanding the pathophysiology of both depression and depression-related physical disease.
Keywords: Stress, Depression, Diurnal cortisol slope, Cortisol awakening response, Dexamethasone suppression test, Inflammation, Health, Disease
1. Introduction
Major depressive disorder (MDD) is among the most common and costly of all psychiatric disorders. Nearly 20% of individuals experience depression at some point in their lives (Kessler et al., 2010), and most individuals who experience one depressive episode experience at least one additional episode in their lifetime (Monroe and Harkness, 2011). The strongest predictor of an impending major depressive episode is psychosocial stress (Kendler et al., 1999), which has been related to both the severity and clinical presentation of depression (Monroe et al., 2007b; Muscatell et al., 2009). It has been proposed that dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis may mediate the effects of early and adulthood life stress on the development of depression (Harkness et al., 2011; Holsboer, 2000). Exactly how HPA axis dysregulation manifests in depression, however, and what mechanisms underlie its perpetuation, remains unclear.
Upregulation of the HPA axis is critical for helping individuals manage social–environmental challenges. The production and release of cortisol, the end product of HPA axis activation, is a tightly regulated process. Under basal conditions of little or no stress, cortisol secretion follows a diurnal pattern characterized by high concentrations at wakening, a morning peak shortly after waking up, and a steady decline over the day, which is collectively referred to as the “diurnal slope”. Deviations from this diurnal cycle can occur, though, and these altered dynamics have been implicated in several disorders including cardiovascular disease, breast cancer, and post-traumatic stress disorder, as well as in depression (Bauer, 2008; Hatzinger, 2000).
Studies investigating atypical diurnal HPA patterns in depressed individuals have produced mixed results. Some studies have demonstrated that depressed individuals exhibit lower morning cortisol and higher evening cortisol than non-depressed individuals, resulting in a flatter diurnal slope (Gartside et al., 2003; Weinrib et al., 2010). However, other studies have found evidence of chronically elevated cortisol levels (i.e., hypercortisolemia) throughout the diurnal rhythm in depressed individuals, but no difference in diurnal slope between depressed and non-depressed persons (Maes et al., 1998; Vreeburg et al., 2009), suggesting increased HPA activity in depressed individuals regardless of time of day (for a review, see Heaney et al., 2010). Proposed explanations for these divergent findings have focused on several factors including possible differences in time of cortisol sampling, severity of depression, type of depression (e.g., typical vs. atypical), presence of comorbid conditions including anxiety and depression, age of participants, use of medications including oral contraceptives, stage of the menstrual cycle in women, and methodological differences such as sample type (e.g., urinary vs. plasma vs. salivary; Heaney et al., 2010). Nonetheless, the precise manner in which diurnal cortisol production is altered in depression remains a topic of ongoing debate.
The mechanisms that underlie altered HPA axis functioning in depression also remain unclear. One line of research into this issue has focused on the glucocorticoid receptor in the paraventricular nucleus and pituitary gland. In order to down-regulate cortisol production, cortisol must interact effectively with its receptor. Cortisol typically exerts negative feedback via glucocorticoid receptors in the hippocampus and medial frontal lobe, as well as the hypothalamic and pituitary portions of the HPA axis (Akana et al., 2001; de Kloet et al., 1998; Diorio et al., 1993; Thomson and Craighead, 2008). Inhibited negative feedback at one or more of these sites, however, can cause a flatter diurnal slope (Bradbury et al., 1994; Miyanaga et al., 1990). To assess HPA axis negative feedback sensitivity in depression, researchers have commonly used the dexamethasone suppression test. Dexamethasone is a synthetic ligand that binds specifically to the glucocorticoid receptor (Anacker et al., 2011). Binding of dexamethasone to the glucocorticoid receptor exerts negative feedback similar to that exerted via binding of the receptor by cortisol. Following dexamethasone administration, less effective suppression of endogenous cortisol is believed to be caused by reduced glucocorticoid receptor sensitivity (for review, see Hatzinger, 2000). Reduced glucocorticoid receptor functioning – commonly referred to as “glucocorticoid resistance” – has been proposed as one possible mechanism underlying altered HPA axis regulation in depression (Anacker et al., 2011; Pace et al., 2007). Although the causal relationship between glucocorticoid resistance and depression is not fully understood, it is believed that HPA axis dysregulation and glucocorticoid resistance may play an important role in the pathophysiology of depression and depression-related physical disease (Chida and Hamer, 2008; Leonard and Myint, 2009; Pace et al., 2007). Direct simultaneous comparison of different aspects of HPA axis regulation – including diurnal cortisol rhythms, cortisol awakening responses, and glucocorticoid receptor sensitivity to dexamethasone – may further illuminate the link between HPA axis dysregulation and depression.
To characterize the specific type of HPA axis dysregulation that is associated with major depression and to examine whether such dysregulation is related to altered glucocorticoid receptor sensitivity, we recruited a sample of healthy pre-menopausal adult women with depression and a comparison group of non-depressed healthy women who were matched for age and body mass index (BMI). We restricted our sample to pre-menopausal women for several reasons. First, women are nearly twice as likely to experience depression as men (Marcus et al., 2005). Second, prevalence of depression appears to follow a bimodal pattern in women, characterized by an initial emergence in adolescence and a reemergence in mid- to late-adulthood, making pre-menopausal adult women particularly susceptible to depression (Hickie et al., 2012; Parker and Hadzi-Pavlovic, 2004). Third, although pre-menopausal women are at high risk for developing depression (American Psychiatric Association, 2000), they are also still relatively early in their cumulative disease process, permitting us to investigate hormonal processes that occur before onset of other confounding physical health conditions, such as sleep deprivation and cardiovascular disease. Finally, as we noted previously, the manner in which HPA axis activity and cortisol production is dysregulated in pre-menopausal, depressed women remains unclear.
Participants in the present study were followed for a week, during which time they provided three samples of salivary cortisol per day for four consecutive days. Prior to the last sampling day, they self-administered a 0.5 mg dose of dexamethasone. This study design allowed us to investigate cortisol rhythm over multiple days in closely matched depressed and non-depressed women, thus minimizing the possibility of confounding influences resulting from between group factors. By allowing women to collect their own salivary samples and administer the dexamethasone suppression test, we also maximized the likelihood that salivary cortisol levels reflected naturalistic concentrations. Based on prior research (Deuschle et al., 1997; Gartside et al., 2003), we hypothesized that depressed women would exhibit flatter diurnal cortisol slopes than non-depressed women, suggesting dysregulation in the form of impaired negative feedback response. Given the critical role that glucocorticoid receptors play in regulating HPA axis activity, we hypothesized further that flatter diurnal cortisol slopes would be associated with a weaker response to dexamethasone. Lastly, because depression symptom severity has been associated with degree of HPA axis dysregulation (Harkness et al., 2011; Hsiao et al., 2010; Knight et al., 2010), we hypothesized that among depressed women, greater depression severity would be associated with flatter diurnal cortisol slopes, blunted cortisol awakening response, and weaker dexamethasone-related suppression of cortisol.
2. Method
2.1. Participants and procedures
Participants were 49 pre-menopausal healthy adult women (26 depressed women and 23 non-depressed women) between the ages of 21 and 40 (M = 30.0, SD = 6.11) recruited from a larger study examining the effects of stress on psychological and biological functioning in depression. To characterize how and why diurnal cortisol rhythms are altered in depression, the study was a case-matched, prospective study, where clinically depressed and non-depressed participants were matched for age (±3 years) and body mass index (±5 kg/m2). All but three depressed participants were matched with a non-depressed control participant at the time of this manuscript. Demographic and clinical characteristics of the sample are presented in Table 1.
Table 1.
Demographic and clinical characteristics of the sample by diagnostic group.
| Characteristic | Diagnostic group
|
Difference | |
|---|---|---|---|
| Depressed | Non-depressed | ||
| Age (years) | 29.8 ± 1.3 | 30.1 ± 1.1 | n.s. |
| Marital status | p < 0.0001 | ||
| Married | 8% | 25% | |
| Divorced | 12% | 4% | |
| Never married | 64% | 42% | |
| Separated | 4% | 0% | |
| Living together | 12% | 29% | |
| Ethnicity | n.s. | ||
| Caucasian | 50% | 54% | |
| African American | 11.5% | 4% | |
| Asian | 19% | 25% | |
| Latina | 11.5% | 4% | |
| Other | 8% | 12.5% | |
| Household income | $35,000–$39,999 | $50,000–$59,999 | n.s. |
| Education | p < 0.0001 | ||
| High school | 11.5% | 0% | |
| Some college | 4% | 0% | |
| Bachelors degree | 14% | 47% | |
| Advanced degree | 31% | 53% | |
| Lifetime MDEs | 2.74 ± 0.38 | n/a | n/a |
| IDS | 29.8 ± 1.5 | 5.7 ± 0.7 | p < 0.0001 |
| Cortisol (nmol/L) | |||
| Waking | 10.59 ± 0.48 | 12.37 ± 0.68 | p = 0.0354 |
| Morning peak | 16.44 ± 0.78 | 18.33 ± 1.01 | n.s. |
| Evening | 3.25 ± 0.36 | 2.74 ± 0.35 | n.s. |
Note: Income was measured on a 22-point scale where 1 represents less than $3000/year and 22 represents more than $200,000/year; a score of 14 represents $40,000–$49,999. Values expressed as M ± SE. MDE, major depressive episode and IDS, Inventory of Depressive Symptomatology.
Participants were recruited from the General Medicine Clinic at the University of California, San Francisco (UCSF), and from the greater San Francisco Bay area community using newspaper and online classified advertisements, a study website, and fliers posted around the UCSF campus. Individuals who passed an initial eligibility telephone screen were invited to complete a comprehensive diagnostic interview. All participants were examined by a physician or nurse practitioner. In addition to a medical history and physical exam, standard laboratory tests were performed to screen out participants who showed signs of physical illness, abnormal thyroid function, or abnormal blood glucose concentrations. To be included in the depressed group, participants had to meet Diagnostic and Statistical Manual of Mental Disorders (DSM-IV; American Psychiatric Association, 2000) criteria for current MDD or depressive disorder not otherwise specified, as assessed by the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID; First et al., 1995). To be included in the non-depressed group, participants must have been free from all current or past depressive symptoms meeting sub-threshold or threshold levels (i.e., 2 or 3) according to the SCID.
Regardless of diagnostic group, individuals were excluded if they had current posttraumatic stress disorder; a lifetime history of mania, hypomania, or primary psychotic symptoms; a current eating disorder; or a recent history (i.e., past 6 months) of alcohol or substance abuse or dependence. Participants were also excluded if they were unable to provide informed consent or were pregnant, post-menopausal, non-English speaking, were under 21 years old or over 40 years of age, had BMI scores below 17 or above 40 kg/m2, experiencing physical health problems, or taking medications that affect HPA axis or immune system functioning. Participants were allowed to take antidepressants (10 of 26 depressed women) and oral contraceptives (18 of total sample, equally distributed across diagnostic groups). As described below, however, both medications were examined for inclusion as potential covariates in the statistical models.
Participants who met all inclusion requirements were mailed a questionnaire packet and consent form, as well as a saliva collection log that contained instructions for the diurnal cortisol sampling and dexamethasone suppression test protocols (see below). Within one week of completing the saliva collection protocol, participants attended an in-person assessment session in which they completed several interviews and questionnaires assessing depression severity (see below). At the time of the in-person visit, height and weight were measured to calculate BMI scores, and hip and waist measurements were made to calculate waist-to-hip ratios. In addition, questions about current menstrual status were asked to determine the average length of each participant’s menstrual cycle and to calculate the menstrual phase during the saliva sampling protocol. For participating in the parent study on psychological and biological aspects of depression, non-depressed participants were paid $150 and depressed participants were paid $200, as they also completed two follow-up sessions (not reported here).
2.2. Salivary cortisol and dexamethasone suppression test
Saliva sampling is a minimally invasive method for investigating concentrations of the biologically active unbound form of cortisol (Kirschbaum and Hellhammer, 1989). To obtain these samples, participants passively drooled into saliva collection vials and immediately placed the sealed vials into a refrigerator. This was repeated for four consecutive days while participants were at home. Over the first three days, participants collected samples at three time points during the day: (1) at wakening, (2) 30 min after wakening (i.e., “morning peak”), and (3) before going to bed in the evening. Participants were instructed to collect all of the wakening samples while still in bed, and to not eat, drink, or brush their teeth for at least 20 min prior to any of the collection times. On average, actual collection times for wakening, morning peak, and evening were 7:31 AM, 8:04 AM, and 11:21 PM, respectively, and these times did not differ significantly across sampling days, or for depressed versus non-depressed women (all ps > 0.45).
To assess the effectiveness of HPA-axis negative feedback, a dexamethasone suppression test was conducted from the end of the third day to the beginning of the fourth consecutive day of sampling. Specifically, on the third day of sampling, immediately following the evening cortisol sampling, participants self-administered 0.5 mg of dexamethasone orally. Low-dose dexamethasone (0.5 mg) was selected because studies have demonstrated that this dose most effectively distinguishes functional and dysfunctional HPA activity (Poland et al., 1985). Saliva samples were collected the following (i.e., fourth) day at wakening and morning peak. Response to dexamethasone administration was indexed as the difference between pre- and post-dexamethasone cortisol levels at wakening and morning peak, averaged across sampling days 1–3.
Following collection, all saliva samples were delivered to the General Clinical Research Center at UCSF, where they were processed and frozen until time of assay. Prior to assay, samples were thawed and then centrifuged at 3000 rpm for 20 min to separate the aqueous component from mucins and other suspended particles. Salivary concentrations of cortisol were estimated in duplicate using commercial radioimmunoassay kits (Siemens Healthcare Diagnostics Inc., Los Angeles, CA). Assay procedures were modified to accommodate overall lower levels of cortisol in human saliva relative to plasma as follows: (1) standards were diluted to concentrations ranging from 2.76 to 317.4 nmol/L; (2) sample volume was increased to 200 μL; and (3) incubation times were extended to 3 h. Serial dilution of samples indicated that the modified assay displayed a linearity of 0.98 and a least detectable dose of 1.3854 nmol/L. Intra- and inter-assay coefficients of variation were 4.59% and 6.11%, respectively.
A cortisol collection logbook was provided to all participants. It contained instructions for the cortisol sampling, as well as questions about sample collection times and activities before the time of saliva collection. Questions focused on participants’ bedtime each previous night and the time of waking for each sample day, as well as their eating, drinking, teeth brushing, and vigorous activity behavior before each collection time point. In addition, at the end of each day, participants answered detailed questions about their recent tobacco, alcohol, caffeine, and medication use, as well as the amount of time they spent exercising. Participants also rated the quality of their workday [e.g., “Please check one of the following statements that best describes today for you: (a) Today I had a lower workload or felt less stressed than usual, (b) Today was typical in terms of my workload and stress level, or (c) Today I had a greater workload or felt more stressed than usual”].
2.3. Depression history and severity
Depression history and severity were assessed during the SCID session. Depression history (i.e., number of lifetime episodes of depression) was assessed using the SCID. Self-reported depression severity was assessed by the Inventory of Depressive Symptomatology (IDS). The IDS is a widely used, 30-item self-report measure of depression that assesses for the presence of cognitive, somatic, behavioral, and physiological symptoms that characterize the disorder (Rush et al., 1996). The measure has good internal consistency (Cronbach’s α = 0.85) and correlates strongly with clinician-rated instruments, such as the Hamilton Ratings Scale for Depression (Rush et al., 1986).
2.4. Statistical analyses
All variables were first evaluated by inspection of descriptive statistics. Any values that exceeded two standard deviations from the mean were excluded. This resulted in the exclusion of 5 of the original 546 cortisol data points. Next, data were divided according to clinical diagnostic group (i.e., depressed vs. non-depressed), and t-tests and χ2 analyses were used to evaluate whether the groups differed with respect to demographic or clinical characteristics. Correlational analyses were conducted to evaluate associations among the variables, and reported statistics have been adjusted for multiple comparisons (Bland and Altman, 1995). Cortisol values were greatly skewed and were thus log-transformed prior to statistical analyses.
Tests of our primary hypotheses were conducted using mixed models analyses (PROC MIXED in SAS v. 9.2, Cary, NC). The model utilized the sample collection day (1–3), sample collection time (wakening = 0, morning peak = 1, evening = 2), and diagnostic group (depressed = 0, non-depressed = 1) as predictors of log-transformed cortisol values. In addition, interaction effects between sample collection time and group were investigated. A second model testing the effects of the dexamethasone test used participants’ average wakening and morning peak samples from days 1 to 3 and compared them to their post-dexamethasone wakening and morning peak samples on day 4. This model permitted us to examine the effects of the dexamethasone test on participants’ wakening cortisol concentration while simultaneously exploring the effects of the dexamethasone test on participants’ cortisol awakening response. A third model utilized the previously described measures of depression history and severity (i.e., number of lifetime episodes of depression and IDS depression severity score) as predictors of log-transformed cortisol values. All statistical models controlled for the exact sampling times of individual cortisol samples, the exact time of awakening for each participant, the amount of time slept the previous night, medication use, menstrual cycle phase, education level, and marital status, all of which were included as covariates in the model. Post hoc analyses were conducted with t-tests for main effects and repeated measures analysis of variance for interactions.
3. Results
3.1. Preliminary analyses
As illustrated in Table 1, depressed and non-depressed participants differed with respect to marital status and level of education (ps < 0.0001). However, these factors were unrelated to the primary outcomes of interest – namely, participants’ diurnal cortisol concentrations and their dexamethasone suppression test response (ps > 0.34). Further, their inclusion in statistical models did not significantly alter the results. In addition, as expected, depressed and non-depressed participants differed on measures of depression, including depression history (as measured by the SCID-I; p < 0.0001) and depression severity (as measured by the IDS; p < 0.0001).
Cortisol data from all 49 depressed and non-depressed women were used to determine the overall diurnal pattern of cortisol in the sample. The diurnal cortisol rhythm showed the expected pattern of elevated morning cortisol upon wakening, followed by an immediate increase 30 min after wakening, followed by a decline to the lowest levels in the late evening (see Fig. 1). General linear models analysis revealed that time of day was a significant predictor of cortisol concentration (F2,276 = 376.44, p < 0.0001, η2 = 0.46). Post hoc analyses designed to examine this effect showed that, as expected, participants’ cortisol levels were significantly higher at morning peak than at wakening (t141 = −9.28, p < 0.0001, d = −1.56), and significantly lower in the evening than at wakening (t139 = 22.85, p < 0.0001, d = 3.88) or morning peak (t138 = 26.35, p < 0.0001, d = 4.49). Cortisol concentrations did not differ significantly across days 1–3 (F2,276 = 0.95, p = 0.35, η2 < 0.01), and there was no interaction between time of sampling and day of sampling (F4,270 = 0.14, p = 0.80, η2 < 0.01).
Fig. 1.

Diurnal cortisol patterns averaged across three days for depressed and non-depressed women. (A) When considered together, depressed and non-depressed women exhibited the expected diurnal cortisol rhythm, marked by higher cortisol concentrations at wakening and morning peak compared to in the evening, with the highest concentrations occurring at the morning peak. (B) When considered separately by diagnostic group, as predicted, depressed women exhibited a flatter diurnal cortisol slope than non-depressed women. *Significant differences by t-test, †significant interaction between diagnostic group and sample time (p < 0.05). W, wakening; MP, morning peak; and E, evening.
3.2. Group differences in cortisol awakening response and diurnal cortisol pattern
Participants’ cortisol awakening response was indexed as the difference between cortisol levels at wakening and at morning peak, relative to the amount of time that passed between the two samples (i.e., approximately 30 min). Analyses were conducted to test whether diagnostic groups differed according to cortisol awakening response across days 1–3. However, no overall difference was found for depressed versus non-depressed women with respect to this outcome (p = 0.80).
We also tested for possible group differences in cortisol concentrations at each of the sampling time points. Relative to non-depressed women, depressed women had significantly lower wakening cortisol levels (depressed: 0.95 ± 0.028; non-depressed: 1.03 ± 0.025; t147 = 2.07, p = 0.041, d = 0.34) and morning peak cortisol levels (depressed: 1.12 ± 0.034; non-depressed: 1.23 ± 0.023; t144 = 2.52, p = 0.013, d = 0.42), and marginally higher evening cortisol levels (depressed: 0.41 ± 0.036; non-depressed: 0.32 ± 0.029; t141 = 1.78, p = 0.077, d = 0.30).
Mixed models analyses were used to test for differences in log-transformed cortisol levels as a function of diagnostic group (depressed vs. non-depressed), day of sampling (days 1–3), and time of day of sample collection (wakening, morning peak, or evening) over the first three days of the saliva collection protocol (i.e., while excluding values from the dexamethasone suppression test on day 4). As expected, cortisol levels differed significantly by time of day (F2,47 = 385.94, p < 0.0001, η2 = 0.64), with cortisol values being higher (on average) at morning peak (M = 1.17 nmol/L, SD = 0.26) than at either wakening (M = 0.99 nmol/L, SD = 0.24) or evening (M = 0.37 nmol/L, SD = 0.28). Although there was no main effect of diagnostic group on overall cortisol levels averaged across all days and sampling time points (p > 0.45), the diurnal slope differed for depressed versus non-depressed participants, as indicated by a significant interaction between diagnostic group and time of sampling (F2,47 = 5.52, p = 0.0043, η2 = 0.01). As predicted, post hoc analysis with repeated measures analysis of variance revealed that declines in cortisol concentrations from wakening to evening were steeper for non-depressed women (0.71 ± 0.05 nmol/L) than for depressed women (0.56 ± 0.06 nmol/L; F1,47 = 5.55, p = 0.0042, η2 = 0.01), indicating a flatter diurnal cortisol slope for depressed women relative to non-depressed women. There were no differences in cortisol concentrations between the three days of sampling (p > 0.25), and no other interactions predicted log-transformed cortisol levels (all ps > 0.34). Covariates assessed in this model included the actual time that samples were collected (e.g., 7:05 AM), time of awakening, total time awake (i.e., time between waking and bedtime), duration of previous night’s sleep, BMI, medication use, menstrual cycle phase, level of education, and marital status. However, none of these covariates significantly predicted participants’ cortisol values (all ps > 0.23). In addition, removing these covariates from the model did not affect the significant diagnostic group by sampling time interaction (with covariates: F2,360 = 5.99, p = 0.0028; without covariates: F2,376 = 7.09, p = 0.0009).
3.3. Group differences in responses to dexamethasone
As summarized in Table 2, initial analyses of group responses to dexamethasone administration were conducted by comparing depressed and non-depressed participants’ salivary cortisol concentrations at wakening and morning peak prior to dexamethasone administration, and at wakening and morning peak following dexamethasone administration. To evaluate differences in participants’ responses to dexamethasone administration, we conducted a 2 × 2 × 2 repeated measures ANOVA with diagnostic group (depressed vs. non-depressed) as a between-subjects factor, sampling days (pre-dexamethasone vs. post-dexamethasone administration) and sampling time point (wakening vs. morning peak) as within-subjects factors, and log-transformed cortisol concentrations as the dependent variable. These analyses revealed that dexamethasone effectively suppressed cortisol for all participants, as indicated by significantly lower cortisol concentrations following dexamethasone administration (F1,47 = 1030.52, p < 0.0001, η2 = 0.81; see Fig. 2). The expected cortisol awakening response was evidenced by elevated morning peak concentrations relative to wakening concentrations regardless of diagnostic group (F1,47 = 17.40, p < 0.0001, η2 = 0.01); however, in both diagnostic groups the magnitude of the cortisol awakening response was attenuated by dexamethasone administration (F1,47 = 4.18, p = 0.0428, η2 = 0.01), and post hoc analyses revealed that cortisol awakening response was significantly reduced following dexamethasone administration (t44 = 3.71, p = 0.0006, d = 1.12).
Table 2.
Cortisol levels following the dexamethasone suppression test by diagnostic group.
| Time point | Depressed | Non-depressed | Difference* |
|---|---|---|---|
| Pre-DST wakening levels | 0.95 ± 0.03 | 1.03 ± 0.02 | −0.08 (p = 0.04) |
| Pre-DST morning peak levels | 1.12 ± 0.03 | 1.23 ± 0.02 | −0.11 (p = 0.01) |
| Pre-DST evening levels | 0.41 ± 0.04 | 0.32 ± 0.03 | 0.08 (p = 0.08) |
| Post-DST wakening levels | 0.27 ± 0.04 | 0.20 ± 0.02 | 0.07 (p = 0.10) |
| Post-DST morning peak levels | 0.32 ± 0.05 | 0.24 ± 0.04 | 0.08 (p = 0.23) |
| Change in wakening levels from pre- to post-DST | 0.71 ± 0.06 | 0.86 ± 0.04 | 0.14 (p = 0.05) |
| Change in morning peak levels from pre- to post-DST | 0.83 ± 0.05 | 0.98 ± 0.08 | 0.15 (p = 0.08) |
Note: Levels of log-transformed cortisol expressed as M ± SE. DST, dexamethasone suppression test.
Values shown represent average difference between groups and (p-value).
Fig. 2.
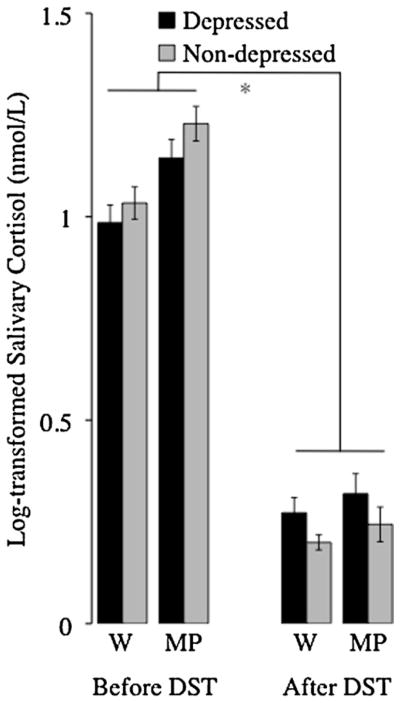
Dexamethasone effectively suppressed cortisol across the entire sample. However, as expected, depressed and non-depressed women differed with respect to how they responded to the dexamethasone suppression test. Specifically, dexamethasone suppression of cortisol, as indexed by the difference between morning cortisol levels before and after dexamethasone administration, was weaker for depressed women than for non-depressed women. *Significant main effect of sampling day, significant interaction between diagnostic group and sampling day, and significant interaction between sampling day and sampling time (p < 0.05). W, wakening; MP, morning peak; and DST, dexamethasone suppression test.
As predicted, depressed and non-depressed participants’ responses to the dexamethasone suppression test differed, such that the extent to which dexamethasone effectively suppressed cortisol was not equivalent for depressed and non-depressed women (F1,47 = 6.04, p = 0.0152, η2 = 0.01). Specifically, dexamethasone suppression of cortisol, as indexed by the difference between wakening cortisol before and after dexamethasone administration, was greater for non-depressed women (0.86 ± 0.04 nmol/L) than for depressed women (0.71 ± 0.0577 nmol/L; t42 = −2.32, p = 0.0519, d = 0.72). When examined by time of day, percent of cortisol suppression (i.e., the difference in pre- and post-dexamethasone cortisol divided by pre-dexamethasone cortisol) was greater for non-depressed women (80.8 ± 1.9%) than for depressed women at wakening (70.9 ± 3.9%; t43 = 2.20, p = 0.0333, d = 0.67), but not at morning peak (non-depressed: 80.3 ± 4.1%; depressed: 72.7 ± 3.7%; t45 = 1.38, p = 0.1739, d = 0.41). Considered together, these results indicate that compared to non-depressed women, depressed women exhibited (on average) more impaired suppression of endogenous cortisol production in response to an exogenous synthetic glucocorticoid (i.e., dexamethasone). No covariates were significant predictors of transformed cortisol values, or of the cortisol response to dexamethasone treatment (all ps > 0.10). Further, the strength of the interaction between diagnostic group and response to dexamethasone was not significantly altered when these covariates were removed from the model (with covariates: F1,47 = 9.81, p < 0.0001; without covariates: F1,47 = 7.38, p = 0.0074).
The dexamethasone suppression test was used to gain insight into a biological mechanism that might explain differences in diurnal cortisol slope for depressed versus non-depressed women. Therefore, we next investigated the association between dexamethasone-induced suppression of cortisol and diurnal cortisol slope using Spearman’s correlations. Diurnal cortisol slopes were quantified as the average differences between wakening and bedtime cortisol concentrations across the first three days of the study. Dexamethasone-induced suppression of cortisol, in turn, was calculated as the difference between wakening cortisol levels before and after dexamethasone administration. As expected, the degree to which dexamethasone suppressed both wakening and morning peak cortisol was positively correlated with diurnal cortisol slopes for the entire sample, with greater dexamethasone suppression predicting greater diurnal changes (wakening: r45 = 0.65, p < 0.0001; morning peak: r47 = 0.37, p = 0.0110; see Fig. 3). Put another way, less dexamethasone suppression of cortisol was related to flatter diurnal cortisol slopes. Importantly, these relations remained significant when depressed women were examined separately (depressed: wakening: r24 = 0.68, p = 0.0003; morning peak: r24 = 0.52, p = 0.0092; non-depressed: wakening: r21 = 0.48, p = 0.0277; morning peak: r23 = 0.09, p > 0.68). Consistent with the formulation that increased glucocorticoid resistance is a mechanism underlying the altered diurnal cortisol pattern observed in depression, 46% of the variability in depressed women’s diurnal cortisol slope was explained by their degree of glucocorticoid receptor sensitivity.
Fig. 3.
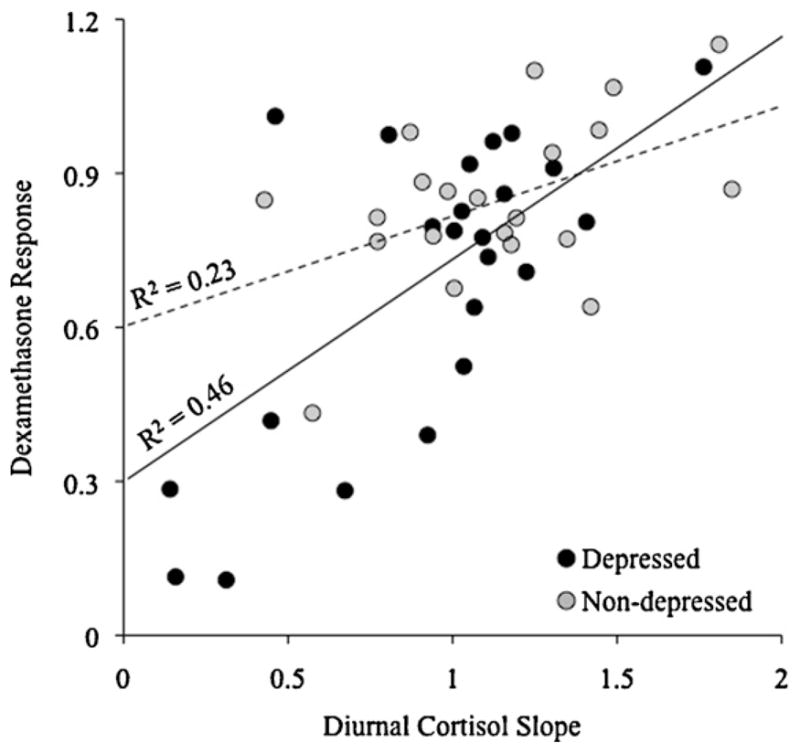
Response to dexamethasone suppression was strongly associated with diurnal cortisol slope. Specifically, when participants’ response to dexamethasone administration was indexed as the degree to which dexamethasone suppressed wakening cortisol (i.e., change in absolute cortisol values from before to after dexamethasone administration), and diurnal slope was indexed as the average change in absolute cortisol values from wakening to evening divided by the time between the two samples, women with less cortisol suppression following dexamethasone had (on average) flatter diurnal cortisol slopes over the first three consecutive days of the study (r45 = 0.65, p < 0.0001). As depicted, this association appears to be stronger for depressed women (R2 = 0.46) than for non-depressed women (R2 = 0.23), although the strength of these two associations is not statistically different (z = 1.59, p = 0.111).
3.4. Depression severity, HPA axis regulation, and response to dexamethasone
To follow up on these findings, we restricted our analyses to participants with depression and examined how HPA regulation (i.e., cortisol slope and response to dexamethasone) differed as a function of depression history and severity using two analytic approaches. First, we used correlation analyses to examine relations between HPA axis function, and depression history and severity. Second, we used mixed models to test whether depression history and severity predicted HPA axis function. Depression history was indexed as participants’ number of SCID-rated lifetime episodes of MDD, and depression severity was indexed as participants’ IDS score.
As seen in Table 3, correlation analyses revealed that number of lifetime depressive episodes was unrelated to diurnal cortisol slopes (p > 0.46). However, as predicted, greater depression severity was strongly associated with flatter diurnal slope (r25 = −0.44, p = 0.0262), suggesting more severe HPA axis disruption for women experiencing more severe depression. We also examined the relation of depression history and severity to depressed participants’ responses to the dexamethasone administration. Number of lifetime depressive episodes was unrelated to depressed participants’ responses to dexamethasone administration (p > 0.14). However, depressed participants’ depression severity scores were strongly and negatively related to their dexamethasone responses, but only at the morning peak time point (r25 = −0.45, p = 0.0230; other time points, ps > 0.20). Together, these data indicate that, as expected, women experiencing more severe episodes of depression exhibit flatter diurnal cortisol slopes and weaker dexamethasone-related suppression of cortisol.
Table 3.
Correlations between HPA axis functioning and clinical characteristics of depression for (A) all women, (B) depressed women, and (C) non-depressed women.
| Diurnal cortisol slope | DST change in W cortisol | DST change in MP cortisol | Depression history (MDEs) | Depression severity (IDS) | |
|---|---|---|---|---|---|
| (A) All women | |||||
| Diurnal cortisol slope | – | ||||
| DST change in W cortisol | 0.65* | – | |||
| DST change in MP cortisol | 0.37* | 0.69* | – | ||
| (B) Depressed women | |||||
| Diurnal cortisol slope | – | ||||
| DST change in W cortisol | 0.68* | – | |||
| DST change in MP cortisol | 0.52* | 0.70* | – | ||
| Depression history (MDEs) | −0.06 | 0.01 | 0.09 | – | |
| Depression severity (IDS) | −0.44* | −0.27 | −0.45* | 0.05 | – |
| (C) Non-depressed women | |||||
| Diurnal cortisol slope | – | ||||
| DST change in W cortisol | 0.48* | – | |||
| DST change in MP cortisol | 0.09 | 0.64* | – | ||
Note: DST, dexamethasone suppression test; W, wakening; MP, morning peak; IDS, Inventory of Depressive Symptomatology; MDE, major depressive episodes, as assessed by the SCID-I. Pearson product moment correlation is shown.
p < 0.05 after Bonferroni adjustment.
As described above, we next used mixed models to test whether our measures of depression history and severity (i.e., number of SCID-rated lifetime episodes of MDD and self-reported IDS scores) were significant predictors of depressed participants’ HPA axis functioning, as indexed by their (1) levels of cortisol at every time point, (2) cortisol awakening response, (3) diurnal cortisol slope, and (4) response to dexamethasone administration. We also examined associations between these measures of depression and all potential covariates. Diurnal cortisol slope was unrelated to participants’ number of lifetime depressive episodes (p > 0.47). However, consistent with the correlation analyses reported above, greater depression severity was strongly associated with flatter diurnal cortisol slopes (F1,12 = 13.90, p = 0.0029, η2 = 0.45). Lastly, depressed participants’ responses to dexamethasone were not predicted by their depression history or severity (ps > 0.16). No other analyses were significant. Together, these results are consistent with the correlation analyses above and indicate that depression severity is strongly associated with diurnal cortisol slope.
4. Discussion
The purpose of this study was to test the integrity of HPA axis functioning in a sample of well-characterized, pre-menopausal women with MDD relative to an age and BMI-matched sample of non-depressed women using three clinically relevant metrics: basal, diurnal cortisol patterns; cortisol awakening response; and cortisol production following administration of dexamethasone. Consistent with prior research (Balardin et al., 2011; Knight et al., 2010), we found that women with depression exhibited (on average) flatter diurnal cortisol slopes compared to non-depressed women. Moreover, greater self-reported depression severity was strongly related to flatter diurnal slopes for depressed participants. To examine a biological mechanism that may underlie these effects, we focused on participants’ responses to dexamethasone (0.5 mg). Although the dexamethasone administration effectively suppressed endogenous cortisol production for all participants, mean suppression of cortisol was much greater for non-depressed women than for depressed women. Given that dexamethasone is a steroid agonist selective to glucocorticoid receptors in the hypothalamus and pituitary, these findings highlight glucocorticoid receptor functioning as a potential mechanism underlying altered diurnal cortisol patterns in depression. We also found that, for depressed participants, greater depression severity was related to less dexamethasone-related suppression of cortisol. Finally, in the depressed group, flatter diurnal cortisol slopes were strongly associated with less suppression of cortisol following dexamethasone administration, suggesting that having a flatter diurnal cortisol pattern may be due in part to impaired glucocorticoid sensitivity at the level of the paraventricular nucleus in the hypothalamus and pituitary gland. To our knowledge, these data are the first to demonstrate that flatter diurnal cortisol rhythms in depression may be partially attributed to impaired glucocorticoid receptor functioning, and that these alterations are most pronounced for women experiencing more severe episodes of depression. These results thus extend previous research showing a flatter diurnal slope among individuals with major depression (Gartside et al., 2003; Weinrib et al., 2010).
In healthy individuals, cortisol levels are typically highest in the morning and lowest in the early evening. This diurnal pattern is tightly controlled primarily by glucocorticoid receptors in the morning and by mineralocorticoid receptors in the evening (de Kloet et al., 1998; Deuschle et al., 1998; Otte et al., 2003). When cortisol concentrations are at an appropriate level, cortisol exerts a feedback signal on glucocorticoid and mineralocorticoid receptors in the hippocampus, hypothalamus, and pituitary gland to prevent the additional release of cortisol. The relatively flatter diurnal cortisol rhythm detected for depressed women in the present study suggests a failure at some point in the limbic–hypothalamic negative feedback system – an effect commonly referred to as “glucocorticoid resistance”. The present data help localize this mechanistic problem by suggesting that the regulatory failure is characterized in part by increased insensitivity of the glucocorticoid receptors in the hypothalamus and/or pituitary. These findings may have implications for understanding how depression develops. In addition, because flatter diurnal cortisol slopes have been observed in a variety of diseases including Parkinson’s Disease (Hartmann et al., 1997), breast cancer (Abercrombie et al., 2004; Sephton et al., 2000), and heart disease (Bhattacharyya et al., 2008), these findings may also have implications for understanding why depression tends to co-occur with these and other physical disease conditions (Lavoie and Fleet, 2000; Menza et al., 1993; Rudisch and Nemeroff, 2003; Somerset et al., 2004; Zellweger et al., 2004). Indeed, insofar as dysregulation of the HPA axis is associated with both depression and several somatic conditions, it is possible that HPA axis dysregulation or phenomena related to these altered dynamics (e.g., elevated inflammation) represent a common biological process that underlies both depression and certain depression-related somatic pathologies. To test this hypothesis, though, additional research is necessary.
This study has several strengths. First, the simultaneous examination of multiple indices of endogenous HPA axis function –namely, diurnal cortisol patterns and cortisol awakening response, in combination with a pharmacologic test of glucocorticoid receptor resistance to an exogenous glucocorticoid – provides a preliminary phenotype of HPA axis functioning in pre-menopausal women with depression. In addition, collecting cortisol samples across several consecutive days in a naturalistic setting (as opposed to in a novel, busy hospital setting) increases the likelihood that the observed cortisol concentrations reflect naturalistic levels. Moreover, by conducting the dexamethasone suppression test on the final consecutive day of sampling, we were able to examine whether altered glucocorticoid receptor sensitivity in the HPA axis could be a potential mechanism underlying the dysregulated diurnal cortisol patterns observed in our sample of depressed women. Another strength of the study was the ability to control for several factors known to influence HPA axis regulation. Specifically, depressed and non-depressed participants were matched for sex, age, and BMI, meaning these factors are unlikely to explain any between-group differences that were observed. Further, women reported the date of their last menstrual period, which allowed us to control for variability in menstrual cycle phase. Indeed, when these factors were included in the statistical models, they explained very little variability. Finally, participants reported the exact times that they collected their saliva samples, in addition to their waking time, which allowed us to accurately calculate diurnal slope values for each individual and to include these times as covariates in our statistical models.
Several limitations of the present study should also be noted. First, the sample included only pre-menopausal women. We focused on these women because they are at disproportionately high risk for depression, and because altered cortisol patterns may presage an increase in risk for late-life depression and the development of physical disease, as these women grow older and the cumulative impact of life experiences and biological aging emerges (McEwen and Seeman, 1999; Pace and Miller, 2009). Additional research is necessary to these hypotheses, however, as well as to determine whether the present findings generalize to men or to women in different reproductive stages. Second, because it is difficult to recruit pre-menopausal women who are not taking oral contraceptives and depressed women not taking antidepressant medication, several participants in the present study were taking oral contraceptives (37%) or antidepressant medication (38% of depressed sample). Flattened diurnal cortisol patterns (e.g., due to elevated evening cortisol levels) and decreased cortisol suppression following dexamethasone administration have been observed in depressed patients taking selective serotonin re-uptake inhibitors (SSRIs; Manthey et al., 2011; Klok et al., 2011). In addition, oral contraceptive use is often associated with lower cortisol levels and blunted cortisol responses to stress (Kirschbaum et al., 1995, 1999). We accounted for these effects by including participants’ oral contraceptive and SSRI use status as covariates in our statistical models; however, neither oral contraceptive use nor SSRI use were related to diurnal cortisol patterns or to cortisol levels following dexamethasone administration. Finally, given the sampling procedure and limited sample size of the current study, the present findings should be replicated with a larger sample of un-medicated men and women with depression. Indeed, although participants in our study showed no indication of chronic hypercortisolemia as observed elsewhere (Maes et al., 1998; Vreeburg et al., 2009), we cannot rule out the possibility that this was not due to some characteristic of the women sampled.
Because social–environmental factors such as chronic stress, social isolation, and social rejection have been related to altered HPA axis and glucocorticoid receptor functioning (Cohen et al., 2012; Miller et al., 2008), in addition to being strongly associated with depression (Monroe et al., 2007a; Slavich et al., 2009), future research is needed to examine how social stressors alter HPA axis-related mechanisms that increase risk for depression (Murphy et al., 2013). A particularly fruitful avenue for research may involve adopting a developmental approach that examines how early life stress shapes regulatory mechanisms of the HPA axis that persist over the lifespan and lead to an increasingly pro-inflammatory phenotype that elevates risk for depression (Miller et al., 2009, 2011; Pace and Miller, 2009). Additional research is also needed to identify neurocognitive processes involved in regulating stress-related HPA axis and inflammatory responding (Slavich et al., 2010a,b). In addition to elucidating pathways that link the external social environment with altered physiological and neuroendocrine functioning, this work can highlight new potential targets for treating and preventing depression (Haroon et al., 2012; Li et al., 2011). These include neurocognitive perceptions of social threat that are known to alter HPA-axis and inflammatory responsivity (O’Donovan et al., 2013; Sawyer et al., 2012; Slavich et al., 2010b).
In sum, the present data demonstrate that depressed women exhibit a flatter diurnal cortisol slope than non-depressed women, and that this effect may be explained in part by reduced glucocorticoid receptor sensitivity in the HPA axis. The present data also indicate that women with more severe episodes of depression are at greater risk for experiencing reduced glucocorticoid sensitivity and associated HPA axis dysregulation. Although this study cannot establish a causal link between glucocorticoid sensitivity and depression severity, antidepressant administration has been found to normalize cortisol levels in addition to alleviating depressive symptoms (Hinkelmann et al., 2012), and these effects are believed to be mediated by restoration of glucocorticoid negative feedback (Inder et al., 2001). Additional research is needed to examine the generalizability of our findings, to identify the health implications of HPA axis dysregulation, to determine the role stress plays in altering biological mechanisms implicated in depression, and to elucidate neurocognitive processes that may represent modifiable risk factors for depression and depression-related physical disease.
Acknowledgments
Funding
This study was supported by a Career Development Award from the National Institute of Mental Health (5K08MH75813-3), a Young Investigator Award from the Brain and Behavior Research Fund (formerly NARSAD), and a Clinical Research Pilot Award from the Clinical and Translational Science Institute (CTSI) at UCSF to Heather Burke, and by an institutional CTSI grant to UCSF (NIH/NCRR UL1 RR024131). Preparation of the present manuscript was supported by Ruth L. Kirschstein National Research Service Award T32 MH19925 to Michael Jarcho, and by a Society in Science: Branco Weiss Fellowship to George Slavich. These organizations had no role in designing the study; in collecting, analyzing, or interpreting the data; in writing this report; or in deciding to submit this report for publication.
We thank the National Institutes of Health, the Brain and Behavior Research Fund (formerly NARSAD), and the Clinical and Translational Science Institute at UCSF for supporting this research. We also acknowledge the Stress and Depression Study research team, including Kirstin Aschbacher, Erin Wipff, Laura Balch, Bryan Low, Vidita Chopra, Melissa Latham, and the other dedicated volunteer research assistants on the project. We also thank Sally Mendoza and Lisa Laughlin at the Endocrine Core Laboratory at the California National Primate Research Center at the University of California, Davis, for performing the salivary cortisol assays. Finally, and most importantly, we thank the women who participated in the Stress and Depression Study.
Footnotes
Conflict of interest
Owen Wolkowitz has served on Speakers’ Bureaus for Forest Labs and Merck Pharmaceuticals, and is on the Scientific Advisory Board of Telome Health Inc. These organizations were not involved in the study.
Contributors
GMS, HMB, and OMW designed the study and wrote the protocol. GMS, HT-S, and HMB ran the study (recruitment of subjects, diagnostic group assignment, collecting saliva samples). MRJ and GMS conducted all statistical analyses. MRJ and GMS wrote the initial drafts of the manuscript, and all authors contributed to and have approved the final manuscript.
References
- Abercrombie HC, Giese-Davis J, Sephton S, Epel ES, Turner-Cobb JM, Spiegel D. Flattened cortisol rhythms in metastatic breast cancer patients. Psychoneuroendocrinology. 2004;29:1082–1092. doi: 10.1016/j.psyneuen.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Akana SF, Chu A, Soriano L, Dallman MF. Corticosterone exerts site-specific and state-dependent effects in prefrontal cortex and amygdala on regulation of adrenocorticotropic hormone, insulin and fat depots. Journal of Neuroendocrinology. 2001;13:625–637. doi: 10.1046/j.1365-2826.2001.00676.x. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4. American Psychiatric Association; Washington, DC: 2000. Text Revision. [Google Scholar]
- Anacker C, Zunszain PA, Carvalho LA, Pariante CM. The glucocorticoid receptor: pivot of depression and of antidepressant treatment? Psychoneuroendocrinology. 2011;36:415–425. doi: 10.1016/j.psyneuen.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balardin JB, Vedana G, Luz C, Bromberg E. Subjective mild depressive symptoms are associated with abnormal diurnal cycle of salivary cortisol in older adults. Journal of Geriatric Psychiatry and Neurology. 2011;24:19–22. doi: 10.1177/0891988710373599. [DOI] [PubMed] [Google Scholar]
- Bauer ME. Chronic stress and immunosenescence: a review. Neuroimmunomodulation. 2008;15:241–250. doi: 10.1159/000156467. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya MR, Molloy GJ, Steptoe A. Depression is associated with flatter cortisol rhythms in patients with coronary artery disease. Journal of Psychosomatic Research. 2008;65:107–113. doi: 10.1016/j.jpsychores.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Bland JM, Altman DG. Multiple significance tests: the Bonferroni method. British Medical Journal. 1995;310:170. doi: 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury MJ, Akana SF, Dallman MF. Roles of type I and II corticosteroid receptors in regulation of basal activity in the hypothalamo-pituitary-adrenal axis during the diurnal trough and the peak: evidence for a nonadditive effect of combined receptor occupation. Endocrinology. 1994;134:1286–1296. doi: 10.1210/endo.134.3.8119168. [DOI] [PubMed] [Google Scholar]
- Chida Y, Hamer M. Chronic psychosocial factors and acute physiological responses to laboratory-induced stress in healthy populations: a quantitative review of 30 years of investigations. Psychological Bulletin. 2008;134:829–885. doi: 10.1037/a0013342. [DOI] [PubMed] [Google Scholar]
- Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, Turner RB. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5995–5999. doi: 10.1073/pnas.1118355109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocrine Reviews. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- Deuschle M, Gotthardt U, Schweiger U, Weber B, Körner A, Schmider J, Standhardt H, Lammers CH, Heuser I. With aging in humans the activity of the hypothalamus–pituitary–adrenal system increases and its diurnal amplitude flattens. Life Sciences. 1997;61:2239–2246. doi: 10.1016/s0024-3205(97)00926-0. [DOI] [PubMed] [Google Scholar]
- Deuschle M, Weber B, Colla M, Muller M, Kniest A, Heuser I. Mineralocorticoid receptor also modulates basal activity of hypothalamus–pituitary–adrenocortical system in humans. Neuroendocrinology. 1998;68:355–360. doi: 10.1159/000054384. [DOI] [PubMed] [Google Scholar]
- Diorio D, Viau V, Meaney MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic–pituitary–adrenal responses to stress. Journal of Neuroscience. 1993;13:3839–3847. doi: 10.1523/JNEUROSCI.13-09-03839.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders. New York Psychiatric Institute, Biomedical Research Division; New York: 1995. [Google Scholar]
- Gartside SE, Leitch MM, McQuade R, Swarbrick DJ. Flattening the glucocorticoid rhythm causes changes in hippocampal expression of messenger RNAs coding structural and functional proteins: Implications for aging and depression. Neuropsychopharmacology. 2003;28:821–829. doi: 10.1038/sj.npp.1300104. [DOI] [PubMed] [Google Scholar]
- Harkness KL, Stewart JG, Wynne-Edwards KE. Cortisol reactivity to social stress in adolescents: role of depression severity and child maltreatment. Psychoneuroendocrinology. 2011;36:173–181. doi: 10.1016/j.psyneuen.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: translational implications of the impact of inflammation on behavior. Neuropsychopharmacology. 2012;37:137–162. doi: 10.1038/npp.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Veldhuis JD, Deuschle M, Standhardt H, Heuser I. Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: Ultradian secretory pulsatility and diurnal variation. Neurobiology of Aging. 1997;18:285–289. doi: 10.1016/s0197-4580(97)80309-0. [DOI] [PubMed] [Google Scholar]
- Hatzinger M. Neuropeptides and the hypothalamic–pituitary–adrenocortical (HPA) system: review of recent research strategies in depression. World Journal of Biological Psychiatry. 2000;1:105–111. doi: 10.3109/15622970009150573. [DOI] [PubMed] [Google Scholar]
- Heaney JL, Phillips AC, Carroll D. Ageing, depression, anxiety, social support and the diurnal rhythm and awakening response of salivary cortisol. International Journal of Psychophysiology. 2010;78:201–208. doi: 10.1016/j.ijpsycho.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Hickie IB, Scott EM, Hermens DF, Naismith SL, Guastella AJ, Kaur M, Sidis A, Whitwell B, Glozier N, Davenport T, Pantelis C, Wood SJ, McGorry PD. Applying clinical staging to young people who present for mental health care. Early Intervention Psychiatry. 2012:31–43. doi: 10.1111/j.1751-7893.2012.00366.x. [DOI] [PubMed] [Google Scholar]
- Hinkelmann K, Moritz S, Botzenhardt J, Muhtz C, Wiedemann K, Kellner M, Otte C. Changes in cortisol secretion during antidepressive treatment and cognitive improvement in patients with major depression: a longitudinal study. Psychoneuroendocrinology. 2012;37:685–692. doi: 10.1016/j.psyneuen.2011.08.012. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Hsiao FH, Yang TT, Ho RTH, Jow GM, Ng SM, Chan CLW, Lai YM, Chen YT, Wang KC. The self-perceived symptom distress and health-related conditions associated with morning to evening diurnal cortisol patterns in outpatients with major depressive disorder. Psychoneuroendocrinology. 2010;35:503–515. doi: 10.1016/j.psyneuen.2009.08.019. [DOI] [PubMed] [Google Scholar]
- Inder WJ, Prickett TC, Mulder RT, Donald RA, Joyce PR. Reduction in basal afternoon plasma ACTH during early treatment of depression with fluoxetine. Psychopharmacology. 2001;156:73–78. doi: 10.1007/s002130100737. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. American Journal of Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Birnbaum H, Bromet E, Hwang I, Sampson N, Shahly V. Age differences in major depression: results from the National Comorbidity Survey Replication (NCS-R) Psychological Medicine. 2010;40:225–237. doi: 10.1017/S0033291709990213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschbaum C, Hellhammer DH. Salivary cortisol in psychobiological research: an overview. Neuropsychobiology. 1989;22:150–169. doi: 10.1159/000118611. [DOI] [PubMed] [Google Scholar]
- Kirschbaum C, Kudielka BM, Gaab J, Schommer NC, Hellhammer DH. Impact of gender, menstrual cycle phase, and oral contraceptives on the activity of the hypothalamus–pituitary–adrenal axis. Psychosomatic Medicine. 1999;61:154–162. doi: 10.1097/00006842-199903000-00006. [DOI] [PubMed] [Google Scholar]
- Kirschbaum C, Pirke KM, Hellhammer DH. Preliminary evidence for reduced cortisol responsivity to psychological stress in women using oral contraceptive medication. Psychoneuroendocrinology. 1995;20:509–514. doi: 10.1016/0306-4530(94)00078-o. [DOI] [PubMed] [Google Scholar]
- Klok MD, Vreeburg SA, Penninx BW, Zitman FG, de Kloet ER, DeRijk RH. Common functional mineralocorticoid receptor polymorphisms modulate the cortisol awakening response: interaction with SSRIs. Psychoneuroendocrinology. 2011;36:484–494. doi: 10.1016/j.psyneuen.2010.07.024. [DOI] [PubMed] [Google Scholar]
- Knight JM, Avery EF, Janssen I, Powell LH. Cortisol and depressive symptoms in a population-based cohort of midlife women. Psychosomatic Medicine. 2010;72:855–861. doi: 10.1097/PSY.0b013e3181f4ab87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie KL, Fleet RP. The impact of depression on the course and outcome of coronary artery disease: review for cardiologists. Canadian Journal of Cardiology. 2000;16:653–662. [PubMed] [Google Scholar]
- Leonard BE, Myint A. The psychoneuroimmunology of depression. Human Psychopharmacology. 2009;24:165–175. doi: 10.1002/hup.1011. [DOI] [PubMed] [Google Scholar]
- Li M, Soczynska JK, Kennedy SH. Inflammatory biomarkers in depression: an opportunity for novel therapeutic interventions. Current Psychiatry Reports. 2011;13:316–320. doi: 10.1007/s11920-011-0210-6. [DOI] [PubMed] [Google Scholar]
- Maes M, Lin A, Bonaccorso S, van Hunsel F, Van Gastel A, Delmeire L, Biondi M, Bosmans E, Kenis G, Scharpe S. Increased 24-hour urinary cortisol excretion in patients with post-traumatic stress disorder and patients with major depression, but not in patients with fibromyalgia. Acta Psychiatry Scandanavia. 1998;98:328–335. doi: 10.1111/j.1600-0447.1998.tb10092.x. [DOI] [PubMed] [Google Scholar]
- Manthey L, Leeds C, Giltay EJ, van Veen T, Vreeburg SA, Penninx BW, Zitman FG. Antidepressant use and salivary cortisol in depressive and anxiety disorders. European Neuropsychopharmacology. 2011;21:691–699. doi: 10.1016/j.euroneuro.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Marcus SM, Young EA, Kerber KB, Kornstein S, Farabaugh AH, Mitchell J, Wisniewski SR, Balasubramani GK, Trivedi MH, Rush AJ. Gender differences in depression: findings from the STAR*D study. Journal of Affective Disorders. 2005;87:141–150. doi: 10.1016/j.jad.2004.09.008. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Seeman T. Protective and damaging effects of mediators of stress. Elaborating and testing the concepts of allostasis and allostatic load. Annals of the New York Academy of Science. 1999;896:30–47. doi: 10.1111/j.1749-6632.1999.tb08103.x. [DOI] [PubMed] [Google Scholar]
- Menza MA, Robertson-Hoffman DE, Bonapace AS. Parkinson’s disease and anxiety: comorbidity with depression. Biological Psychiatry. 1993;34:465–470. doi: 10.1016/0006-3223(93)90237-8. [DOI] [PubMed] [Google Scholar]
- Miller GE, Chen E, Fok AK, Walker H, Lim A, Nicholls EF, Cole S, Kobor MS. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14716–14721. doi: 10.1073/pnas.0902971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Parker KJ. Psychological stress in childhood and susceptibility to the chronic diseases of aging: moving toward a model of behavioral and biological mechanisms. Psychological Bulletin. 2011;137:959–997. doi: 10.1037/a0024768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, Ma R, Cole SW. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biological Psychiatry. 2008;64:266–272. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanaga K, Miyabo S, Ooya E. Negative feedback by corticosterone is the major mechanism responsible for corticotropin-releasing factor-induced desensitization. Journal of Neuroendocrinology. 1990;2:839–843. doi: 10.1111/j.1365-2826.1990.tb00649.x. [DOI] [PubMed] [Google Scholar]
- Monroe SM, Harkness KL. Recurrence in major depression: a conceptual analysis. Psychological Review. 2011;118:655–674. doi: 10.1037/a0025190. [DOI] [PubMed] [Google Scholar]
- Monroe SM, Slavich GM, Torres LD, Gotlib IH. Major life events and major chronic difficulties are differentially associated with history of major depressive episodes. Journal of Abnormal Psychology. 2007a;116:116–124. doi: 10.1037/0021-843X.116.1.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe SM, Slavich GM, Torres LD, Gotlib IH. Severe life events predict specific patterns of change in cognitive biases in major depression. Psycholological Medicine. 2007b;37:863–871. doi: 10.1017/S0033291707000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy ML, Slavich GM, Rohleder N, Miller GE. Targeted rejection triggers differential pro- and anti-inflammatory gene expression in adolescents as a function of social status. Clinical Psychological Science. 2013;1:30–40. doi: 10.1177/2167702612455743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscatell KA, Slavich GM, Monroe SM, Gotlib IH. Stressful life events, chronic difficulties, and the symptoms of clinical depression. Journal of Nervous and Mental Diseases. 2009;197:154–160. doi: 10.1097/NMD.0b013e318199f77b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan A, Slavich GM, Epel ES, Neylan TC. Exaggerated neurobiological sensitivity to threat as a mechanism linking anxiety with increased risk for diseases of aging. Neuroscience and Biobehavioral Reviews. 2013;37:96–108. doi: 10.1016/j.neubiorev.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte C, Yassouridis A, Jahn H, Maass P, Stober N, Wiedemann K, Kellner M. Mineralocorticoid receptor-mediated inhibition of the hypothalamic–pituitary–adrenal axis in aged humans. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2003;58:B900–B905. doi: 10.1093/gerona/58.10.b900. [DOI] [PubMed] [Google Scholar]
- Pace TW, Miller AH. Cytokines and glucocorticoid receptor signaling: relevance to major depression. Annals of the New York Academy of Science. 2009;1179:86–105. doi: 10.1111/j.1749-6632.2009.04984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TWW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain, Behavior, and Immunity. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker G, Hadzi-Pavlovic D. Is the female preponderance in major depression secondary to a gender difference in specific anxiety disorders? Psychological Medicine. 2004;34:461–470. doi: 10.1017/s0033291703001181. [DOI] [PubMed] [Google Scholar]
- Poland RE, Rubin RT, Lane LA, Martin DJ, Rose DE, Lesser IM. A modified dexamethasone suppression test for endogenous depression. Psychiatry Research. 1985;15:293–299. doi: 10.1016/0165-1781(85)90066-6. [DOI] [PubMed] [Google Scholar]
- Rudisch B, Nemeroff CB. Epidemiology of comorbid coronary artery disease and depression. Biological Psychiatry. 2003;54:227–240. doi: 10.1016/s0006-3223(03)00587-0. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Giles DE, Schlesser MA, Fulton CL, Weissenburger J, Burns C. The Inventory for Depressive Symptomatology (IDS): preliminary findings. Psychiatry Research. 1986;18:65–87. doi: 10.1016/0165-1781(86)90060-0. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH. The Inventory of Depressive Symptomatology (IDS): psychometric properties. Psychological Medicine. 1996;26:477–486. doi: 10.1017/s0033291700035558. [DOI] [PubMed] [Google Scholar]
- Sawyer PJ, Major B, Casad BJ, Townsend SS, Mendes WB. Discrimination and the stress response: psychological and physiological consequences of anticipating prejudice in interethnic interactions. American Journal of Public Health. 2012;102:1020–1026. doi: 10.2105/AJPH.2011.300620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton SE, Sapolsky RM, Kraemer HC, Spiegel D. Diurnal cortisol rhythm as a predictor of breast cancer survival. Journal of the National Cancer Institute. 2000;92:994–1000. doi: 10.1093/jnci/92.12.994. [DOI] [PubMed] [Google Scholar]
- Slavich GM, O’Donovan A, Epel ES, Kemeny ME. Black sheep get the blues: a psychobiological model of social rejection and depression. Neuroscience and Biobehavioral Review. 2010a;35:39–45. doi: 10.1016/j.neubiorev.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavich GM, Thornton T, Torres LD, Monroe SM, Gotlib IH. Targeted rejection predicts hastened onset of major depression. Journal of Socical and Clinical Psychology. 2009;28:223–243. doi: 10.1521/jscp.2009.28.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavich GM, Way BM, Eisenberger NI, Taylor SE. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proceedings of the National Academy of Sciences of the United States of America. 2010b;107:14817–14822. doi: 10.1073/pnas.1009164107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerset W, Stout SC, Miller AH, Musselman D. Breast cancer and depression. Oncology. 2004;18:1021–1034. [PubMed] [Google Scholar]
- Thomson F, Craighead M. Innovative approaches for the treatment of depression: targeting the HPA axis. Neurochemical Research. 2008;33:691–707. doi: 10.1007/s11064-007-9518-3. [DOI] [PubMed] [Google Scholar]
- Vreeburg SA, Hoogendijk WJ, van Pelt J, Derijk RH, Verhagen JC, van Dyck R, Smit JH, Zitman FG, Penninx BW. Major depressive disorder and hypothalamic–pituitary–adrenal axis activity: results from a large cohort study. Archives of General Psychiatry. 2009;66:617–626. doi: 10.1001/archgenpsychiatry.2009.50. [DOI] [PubMed] [Google Scholar]
- Weinrib AZ, Sephton SE, Degeest K, Penedo F, Bender D, Zimmerman B, Kirschbaum C, Sood AK, Lubaroff DM, Lutgendorf SK. Diurnal cortisol dysregulation, functional disability, and depression in women with ovarian cancer. Cancer. 2010;116:4410–4419. doi: 10.1002/cncr.25299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger MJ, Osterwalder RH, Langewitz W, Pfisterer ME. Coronary artery disease and depression. European Heart Journal. 2004;25:3–9. doi: 10.1016/j.ehj.2003.09.009. [DOI] [PubMed] [Google Scholar]