

Abstract
Evolving evidence suggests that brain inflammation and the buildup of proinflammatory cytokine increases the risk for cognitive decline and cognitive dysfunction. Interleukin-1β (IL-1β), acting via poorly understood mechanisms, appears to be a key cytokine in causing these deleterious effects along with a presumably related loss of long-term potentiation (LTP)-type synaptic plasticity. We hypothesized that IL-1β disrupts brain-derived neurotrophic factor (BDNF) signaling cascades and thereby impairs the formation of filamentous actin (F-actin) in dendritic spines, an event that is essential for the stabilization of LTP. Actin polymerization in spines requires phosphorylation of the filament severing protein cofilin and is modulated by expression of the immediate early gene product Arc. Using rat organotypic hippocampal cultures, we found that IL-1β suppressed BDNF-dependent regulation of Arc and phosphorylation of cofilin and cAMP response element-binding protein (CREB), a transcription factor regulating Arc expression. IL-1β appears to act on BDNF signal transduction by impairing the phosphorylation of insulin receptor substrate 1, a protein that couples activation of the BDNF receptor TrkB to downstream signaling pathways regulating CREB, Arc, and cofilin. IL-1β upregulated p38 mitogen-activated protein kinase (MAPK), and inhibiting p38 MAPK prevented IL-1β from disrupting BDNF signaling. IL-1β also prevented the formation of F-actin in spines and impaired the consolidation, but not the induction, of BDNF-dependent LTP in acute hippocampal slices. The suppressive effect of IL-1β on F-actin and LTP was prevented by inhibiting p38 MAPK. These findings define a new mechanism for the action of IL-1β on LTP and point to a potential therapeutic target to restore synaptic plasticity.
Introduction
Interleukin-1β (IL-1β) is a key component of the inflammatory response of the brain and mediates the effect of inflammation on cognition and synaptic plasticity. In a normal healthy brain, the concentration of IL-1β is low but increases as a result of peripheral infection, surgery, brain injury, social isolation, or during neurodegenerative diseases, such as Alzheimer's disease (AD) (Rothwell and Luheshi, 2000). An accumulating body of evidence shows that elevated IL-1β causes cognitive decline, especially on hippocampal-dependent tasks (Rachal Pugh et al., 2001). For example, 2 weeks of IL-1β overexpression in an inducible transgenic mouse impaired long-term contextual and spatial memory but spared short-term and non-hippocampal memory (Hein et al., 2010). Moreover, Escherichia coli infection impairs contextual fear conditioning and increases IL-1β levels in the hippocampus, and the loss of memory is prevented by the specific IL-1β receptor antagonist IL-1ra (Barrientos et al., 2009; Frank et al., 2010).
Brain-derived neurotrophic factor (BDNF) signaling promotes both long-term potentiation (LTP) and memory formation (Minichiello et al., 1998; Liu et al., 2004). The neurotrophin directly facilitates LTP (Kramár et al., 2004) and regulates key pathways mediating activity-driven formation of filamentous actin (F-actin) in spine heads, a process required for spine structural changes and stabilization of the potentiation effect (Rex et al., 2007). Actin filament assembly is dependent on sustained translation of the immediate early gene (IEG) product Arc (Bramham, 2008; Shepherd and Bear, 2011) and on inactivation via phosphorylation of the actin severing protein cofilin. BDNF upregulates Arc (Yin et al., 2002) and phosphorylates cofilin, thereby facilitating F-actin formation and changes in spine morphology (Messaoudi et al., 2007; Rex et al., 2007). Therefore, conditions that interfere with BDNF signaling are expected to negatively affect a variety of downstream functions required for the production of stable LTP. Supporting this notion, hippocampal LTP is severely impaired in mice deficient in BDNF or its synaptic receptor TrkB (Minichiello et al., 1998; Liu et al., 2004) and is acutely blocked by scavengers of released BDNF (Rex et al., 2007; Chen et al., 2010).
We reported recently that IL-1β suppresses BDNF signal transduction and gene regulation in low-density cortical neuronal cultures (Tong et al., 2008). In the current study, we investigated the mechanism by which IL-1β regulates BDNF signaling in organotypic cultures and adult hippocampal slices. Importantly, these preparations represent more complex systems than low-density cultures, showing the features of the intact hippocampus but allowing rigorous control of external and internal conditions not readily feasible in vivo (Peña, 2010). First, we tested whether IL-1β affects BDNF-dependent mechanisms associated with actin polymerization and LTP, namely Arc expression and cofilin phosphorylation. Second, we focused on the molecular mechanism by which IL-1β is coupled to BDNF signaling. Third, we extended our biochemical findings from organotypic cultures to hippocampal slices and LTP. By combining electrophysiological recordings and biochemical and imaging techniques, we investigated the extent to which IL-1β regulates BDNF-dependent LTP and F-actin formation.
Materials and Methods
Slice culture.
Experiments were performed on cultured organotypic hippocampal slices prepared according to the method described previously with minor modifications (Stoppini et al., 1991). Briefly, rat pups (7–10 d old) were decapitated, and transverse hippocampal slices (350 μm) were obtained using a tissue chopper (McIlwain). After stabilization, slices were separated and transferred to 30 mm Millicell-CM 0.4-μm-thick sterile tissue culture plate inserts (Millipore). The inserts were placed on six-well tissue culture plates (Falcon; Becton Dickinson Labware), each containing 1 ml of culture medium. The slices were maintained for 7–10 d in horse serum-containing medium (50% Neurobasal medium, 25% HBSS, 0.5% GlutaMax, pH 7.2, and 25% horse serum), then for 2 d in Neurobasal B27-containing medium plus 10% serum, 2 d in Neurobasal B27-containing medium plus 3% serum, and finally in serum-free Neurobasal B27-containing medium with GlutaMax. Slices were cultured for 10–14 d at 36.5°C, 100% humidity, in 5% CO2 atmosphere and fed twice weekly by 50% medium exchange. No glutamate was added to the culture media throughout the culture preparation and maintenance periods. Viability of the slices was assessed by LDH release assay. Slices were examined periodically for viability, and any dark or abnormal slices were discarded.
Experimental treatment.
Recombinant IL-1β (Sigma) was dissolved in DMEM and used after one freeze–thaw cycle at 50 ng/ml, a concentration validated previously by dose–response experiments performed by our laboratory (Tong et al., 2008) and consistent with previous in vitro research (for review, see Pinteaux et al., 2009). Exposure to BDNF (100 ng/ml; Peprotech) or vehicle was usually for 60 min, unless mentioned otherwise, and then slices were processed for biochemical analysis. The actual concentrations at the synapse are at present unknown. In CSF, BDNF is ∼250 pg/ml in healthy brain, and is slightly lower in AD (Li et al., 2009), and in serum BDNF is ∼20 ng/ml (Erickson et al., 2010). IL-1β in CFS from AD patients is ∼150 pg/ml (Cacabelos et al., 1991). It has been argued that the concentration of neurotransmitters and neuromodulators at the synapse may be 100-fold above extracellular levels (Yao et al., 2008). Most investigators consider concentrations of IL-1β in picomoles per milliliters to be healthy, whereas concentrations in nanograms per milliliters simulate pathological conditions (Bellinger et al., 1993; Ross et al., 2003; Pinteaux et al., 2009). Pilot experiments on the time course of the effect of IL-1β on BDNF signaling indicated that significant interference is detectable after 24 h exposure that was the preincubation time routinely used in these studies. As described previously (Tong et al., 2008), the IL-1 receptor antagonist IL-1ra (R&D Systems) was used at 5 μg/ml, and, when applied, cells were preincubated for 30 min before the addition of IL-1β.
mRNA quantification.
The levels of mRNA were measured by RT-PCR as described previously (Adlard et al., 2004). In brief, slices were extracted in Trizol (Invitrogen) to allow for the collection of mRNA. Total RNA was extracted, and RNA quality was assessed on an Agilent bioanalyzer system with RNA chips. RT-PCR amplifications were performed by using OneStep RT-PCR kits (Qiagen) with 0.2–0.4 μg of total RNA in 10 μl reaction volumes in a Robocycler PCR machine (Stratagene) with hot top. Then 1 μl of each reaction was diluted with 4 μl of water and analyzed on a DNA-500 LabChip (Agilent Technologies). RT-PCR reactions included oligonucleotides for amplification of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for normalization. Oligonucleotide sequences used for amplification are as follows (5′ to 3′): Homer1a (Minami et al., 2003) forward, CAAACACTGTTTATGGACTG; Homer1a reverse, TGCTGAATTGAATGTGTACC; Arc forward, GTTAGCCCCTATGCCATCAC; Arc reverse, CTCCTCAGCGTCCACATACA; GAPDH forward, TCCATGACAACTTTGGCATCGTGG; and GAPDH reverse, GTTGCTGTTGAAGTCACAGGAGAC.
Western blot analysis.
Slices were microdissected and lysed in a buffer containing 1% Triton X-100, 150 mm NaCl, 5 mm EDTA, 10 mm NaF, 2 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, 0.7 μg/ml pepstatin, 1 μg/ml leupeptin, and 20 mm Tris-HCl, pH 7.5. Lysates were centrifuged at 12,000 × g at 4°C for 30 min, and then the protein concentration was determined by the BCA protein assay (Pierce). Equivalent amounts of protein for each sample were electrophoresed on SDS-polyacrylamide gel (10% acrylamide gel). Proteins were then electrotransferred to PVDF membranes, blocked with 5% nonfat milk in Tris-buffered saline, and probed with various antibodies. The immunoreactivity was revealed using horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit IgG or goat anti-mouse IgG; Vector Laboratories) and enhanced chemiluminescence (ECL; GE Healthcare) according to the recommended conditions. Immunoreactivity was quantified using NIH Image. The following antibodies were used for the biochemical studies: from Millipore, phospho-specific cAMP response element-binding protein (P-CREB; Ser-133; 1:2000), total CREB (T-CREB, 1:2000), P-cofilin (Ser-3; 1:1000), cofilin (1:1000), and TrkB (1:1000); from Santa Cruz Biotechnology, anti-Arc (C7; 1:500); from Cell Signaling Technology, P-mitogen-activated protein kinase (P-MAPK) (Thr-202 and Tyr-204; 1:2000), T-MAPK (1:2000), P-Akt (Ser-473; 1:1000), T-Akt (1:1000), P-Trk (Tyr-490; 1:500), P-p38 MAPK (P-p38; Thr-180 and Tyr-182; 1:1000), T-p38 MAPK (T-p38; 1:1000), and T-insulin receptor substrate 1 (T-IRS-1; 1:1000). P-IRS-1 (Tyr-612; 1:1000) was from Abcam. Quantification of the data obtained from Western blots derived from cultures under the various experimental conditions was analyzed using one-way ANOVA, followed by the least significant difference (LSD) post hoc test as indicated in the legends.
Immunoprecipitation.
Slices were lysed in 500 μl of immunoprecipitation buffer (1% Triton X-100, 150 mm NaCl, 50 mm Tris, pH 8.0, 0.2 mm Na3VO4, 0.2 mm phenylmethylsulfonyl fluoride, 1 mg/ml pepstatin, 1 mg/ml leupeptin, and 1 mg/ml antipain). Lysates were centrifuged at 10,000 × g for 30 min, and protein concentration of the clarified lysates was determined using the Micro-BCA protein assay (Pierce). Proteins were immunoprecipitated with various antibodies at 4°C overnight. The immunoprecipitation was followed by addition of protein G-Sepharose, and the samples were rotated at 4°C for 1 h. The immune complexes were pelleted by centrifugation at 10,000 × g at 4°C for 1 min. The supernatant was decanted, and the pellet was washed with 1 ml of immunoprecipitation buffer. The wash steps were repeated three times, and finally the pellet was suspended in 60 μl of SDS-sample buffer (62.5 mm Tris, pH 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, and 0.01% bromphenol blue). Proteins of the suspended immunoprecipitate (30 μl) were separated on a 10% SDS-PAGE gel and analyzed by Western blotting with anti-phosphotyrosine 4G10 or anti-PLCγ (Millipore) antibodies, as indicated.
Cortical neuron transfection and luciferase assay.
Cortical neurons were cultured and transfected with plasmid pIII(170)Luc at 3 d in vitro (DIV) using a procedure described previously (Tong et al., 2001). Briefly, all transfections were conducted in six-well 35-mm dishes with LipofectAMINE (Invitrogen) according to the instructions of the manufacturer. One well was transfected with 1 μg of reporter plasmid and 0.25 μg of an internal control Renilla luciferase plasmid pGL4.74 [hRluc/TK] (Promega) to normalize pIII(170)Luc activity. After 48 h, cultures received treatments, plates were washed twice with cold PBS, and cells were lysed with 200 μl of lysis buffer (Promega). Cell extracts (20 μl) were used for a dual-luciferase reporter assay (Promega) according to the instructions of the manufacturer.
Immunocytochemistry.
For immunostaining, slices were fixed in 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB) for 4 h and cryoprotected in 20% sucrose/PB for 12–24 h at 4°C and 0.1 m glycine for 30 min. For Arc immunostaining, tissues were sectioned on a freezing microtome at 20 μm and then transferred to blocking/permeabilizing solution: PB containing 4% bovine serum albumin and 0.3% Triton X-100 (PBT). Sections were then incubated (2 h at room temperature) in PBT containing the primary antibody anti-Arc (C7; 1:200; Santa Cruz Biotechnology). Slides were then washed in PB (three times for 10 min), incubated (45 min at room temperature) with fluorescein-conjugated secondary antibody in PBT, rinsed in PB (three times for 10 min), mounted on Starfrost slides (Sigma), and coverslipped with Fluoromount-G (Southern Biotechnology Associates). Low-magnification images were taken by a confocal laser-scanning microscope (Bio-Rad), mostly with 10× and 20× objectives, and exported to NIH Image. For P-CREB and mitogen-activated protein-2 (MAP2) double immunostaining, tissues were stained with anti-P-CREB rabbit (1:500; Millipore) and anti-MAP2 (1:5000) antibodies. Double-label immunofluorescence images were taken by a confocal laser-scanning microscope (LSM 510) and exported to NIH Image. Appropriate controls lacking primary antibodies were performed for each antibody used.
Electrophysiology slice preparation and recording technique.
All studies used young adult (2–3 months old) male Sprague Dawley rats (Charles River). Slice preparation was performed as described previously (Kramár et al., 2004). Briefly, after deep anesthesia, animals were decapitated, and the brain was quickly removed and placed in ice-chilled oxygenated dissection medium containing the following (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 5 MgSO4, 26 NaHCO3, and 10 glucose. Transverse hippocampal slices (350 μm) through the middle third of the septo-temporal axis of hippocampus were prepared using a McIlwain tissue chopper and immediately transferred to an interface recording chamber containing preheated artificial CSF (aCSF) of the following composition (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 1.5 MgSO4, 2.5 CaCl2, 26 NaHCO3, and 10 glucose (maintained at 31 ± 1°C). Slices were continuously perfused with this solution at a rate of 2.0–2.5 ml/min while the surface of the slices was exposed to warm, humidified 95% O2/5% CO2. Recordings began after 2 h of incubation.
Field EPSPs (fEPSPs) were recorded from stratum radiatum of CA1b using a single glass pipette filled with 2 m NaCl (yielding a resistance of 2–3 MΩ) in response to antidromic and orthodromic stimulation (twisted nichrome wires, 65 μm) of the Schaffer collateral–commissural projections in CA1a and CA1c stratum radiatum, respectively. Alternating pulses were delivered to the stimulation electrodes at 0.03 Hz with current test intensity adjusted to obtain 50–60% of the maximum fEPSP. After establishing a 20 min stable baseline, test compounds (see below) were introduced into the infusion line by switching from control aCSF to drug-containing aCSF. LTP was induced by delivering 10 theta bursts in one of the stimulation pathways with each burst consisting of four pulses at 100 Hz and the bursts themselves separated by 200 ms [i.e., theta burst stimulation (TBS)]. The stimulation intensity was not increased during TBS. The second pathway was used to monitor the stability of the responses over time. In some experiments, a subthreshold level of LTP was induced by delivering two theta bursts, and the stimulation intensity was increased by 30% during TBS. Data were collected and digitized by NAC 2.0 Neurodata Acquisition System (Theta Burst Corp.) and stored on a disk.
To examine the effects of drug application on theta-induced LTP, the perfusion system consisted of chemically inert infusion lines (silicon tubing) connected to a 60 ml syringe. Flow was induced by gravity and rate controlled with an intravenous regulator. BDNF stock and IL-1β (Sigma) were prepared fresh on a daily basis in aCSF before being added to the infusion line. The p38 MAPK inhibitor SB202190 [4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole] (Sigma) was dissolved in 100% DMSO and diluted with aCSF to a working concentration of 1 μm (DMSO < 0.01%) before additional use.
In situ phalloidin labeling.
Methods were performed as described previously (Kramár et al., 2006). Alexa Fluor-568 (Tocris Bioscience)–phalloidin (6 μm) was applied topically from a micropipette every 5 min for 20 min either (1) at the end of the experiment in slices that received low-frequency stimulation (LFS) (0.03 Hz) or (2) 60 min after the delivery of theta bursts. Slices were then collected and prepared for microscopy. Labeling was examined using a Carl Zeiss Axioskop microscope (40×, Apochromat, numerical aperture 1.2) and an Axiocam camera. Identification and quantification of labeled spines was performed on a 35,700 μm2 sampling area within the zone of physiological recording as described previously (Lin et al., 2005; Kramár et al., 2006; Rex et al., 2007). For each field, five sequential images were collected at 2 μm focal steps totaling a thickness of 10 μm on the z-axis. Exposure times were adjusted for each experiment so that ∼6–12 spines were visualized in images from control slices. Images used for comparisons were collected with the same exposure parameters.
Results
IL-1β reduces induction of IEG Arc by BDNF
Several studies have shown that IL-1β disrupts the production of stable LTP in adult rodents (Bellinger et al., 1993; Ross et al., 2003). We hypothesized that this effect involves suppression of the positive contributions of BDNF to the induction and stabilization of LTP. We first assessed the effect of the cytokine on BDNF-induced gene expression of Arc in organotypic slices. Arc/Arg3.1 is a gatekeeper protein for LTP and learning (Bramham et al., 2010; Shepherd and Bear, 2011), a function likely involving its contributions to cytoskeletal reorganization shortly after induction of LTP (Messaoudi et al., 2007). BDNF (100 ng/ml) applied for 1 h caused an almost threefold increase in Arc mRNA levels in preparations obtained at postnatal days 7–10 and maintained in culture for 10–14 d before testing (Fig. 1A). A 24 h pretreatment with IL-1β (50 ng/ml) reduced BDNF-induced Arc expression by 60%, and this effect was completely blocked by the highly selective, competitive antagonist IL-1ra (Rothwell and Luheshi, 2000). IL-1β also caused a 70% reduction in BDNF-elicited expression of Homer1a mRNA, another IEG product that participates in spine remodeling (Sato et al., 2001; Sala et al., 2003) (Fig. 1B). Neurotoxic effects, as assessed with LDH assay, were not detected after the IL-1β treatments (10–50 ng/ml). At the highest concentration of IL-1β, cell survival was 92 ± 8% (n = 3) of control.
Figure 1.
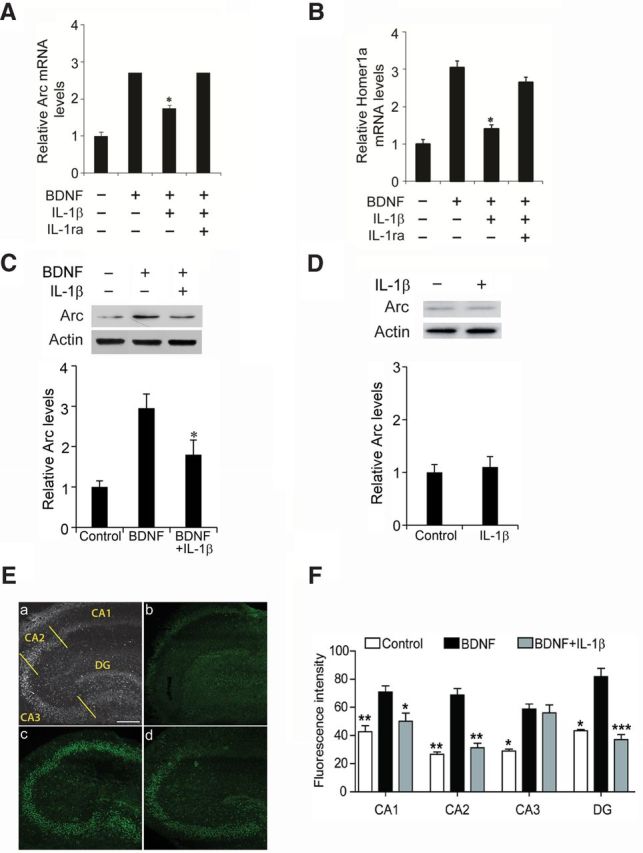
IL-1β pretreatment decreased BDNF-induced Arc and Homer1a expression. Arc (A) and Homer1a (B) mRNA levels were determined by RT-PCR. Slices were exposed to BDNF (100 ng/ml, 1 h), IL-1β (50 ng/ml, 24 h pretreatment), and IL-1ra (5 μg/ml, 30 min before IL-1β) as indicated. Data are the mean ± SEM (n = 3) expressed in terms of control cultures. *p < 0.05 for BDNF versus BDNF plus IL-1β, ANOVA followed by the LSD post hoc test. Notably, IL-1ra blocked the suppression by IL-1β of BDNF-induced expression of Arc and Homer1a. C, Representative Western blots showing that pretreatment with IL-1β suppressed the effect of BDNF (100 ng/ml, 2 h) on Arc expression in CA1. Bottom row shows the quantification of Arc protein levels. Unless otherwise indicated, here and in the other legends, the quantification data for Western blots are expressed relative to the means in the control cultures (n = 3). *p < 0.05 BDNF versus BDNF plus IL-1β, ANOVA followed by the LSD post hoc test. D, IL-1β alone (50 ng/ml, 24 h) did not affect the expression of Arc protein in CA1. E, BDNF-induced Arc expression was evaluated in CA1, CA2, CA3, and DG (as indicated in the left top, a) of 10 DIV hippocampal organotypic cultures. Scale bar, 300 μm. Figure shows representative slices from the different experimental conditions. Compared with control cultures (b), there was an increase in the fluorescence intensity when cultures were treated with BDNF (100 ng/ml; c). Notably, the BDNF-induced increase in staining was affected by a preincubation with IL-1β (100 ng/ml; d) in CA1, CA2, and DG but not in CA3. F, Quantification of the immunoreactivity of Arc (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 compared with BDNF (ANOVA followed by post hoc test).
We next evaluated Arc expression at the protein level. Because BDNF facilitates LTP (Figurov et al., 1996; Kramár et al., 2004) and LTP-induced structural plasticity (Tanaka et al., 2008) in hippocampal subfield CA1, we dissected this region from organotypic cultured slices and used Westerns blots to assess the effects of IL-1β and BDNF on Arc concentrations. BDNF increased Arc protein level in CA1, and, consistent with the mRNA results, this effect was markedly attenuated by pretreatment with IL-1β (Fig. 1C). Importantly, the cytokine alone (50 ng/ml, 24 h) did not affect the expression of Arc protein in CA1 (Fig. 1D). Possible regional differences in the interactions between IL-1, Arc, and BDNF were studied using immunohistochemistry. Arc was expressed at low basal levels across all hippocampal subfields [CA1, CA2, CA3, and dentate gyrus (DG)] in control organotypic cultures, and BDNF treatment significantly increased immunostaining in each of these areas. Notably, IL-1β reduced the BDNF-induced Arc labeling in CA1, CA2, and DG but not in CA3 (Fig. 1E,F).
We next tested whether the pattern of results obtained for the influence of BDNF and IL-1β on Arc expression also holds for phosphorylation of cofilin, a second downstream step closely related to actin polymerization and LTP stabilization. Immunoblots of dissected field CA1 from organotypic slices were used for these studies. BDNF increased cofilin phosphorylation at Ser-3 in accord with previous reports (Rex et al., 2007), and this effect was markedly attenuated by pretreatment with IL-1β. Interleukin alone did not reduce baseline levels of P-cofilin (Fig. 2A).
Figure 2.
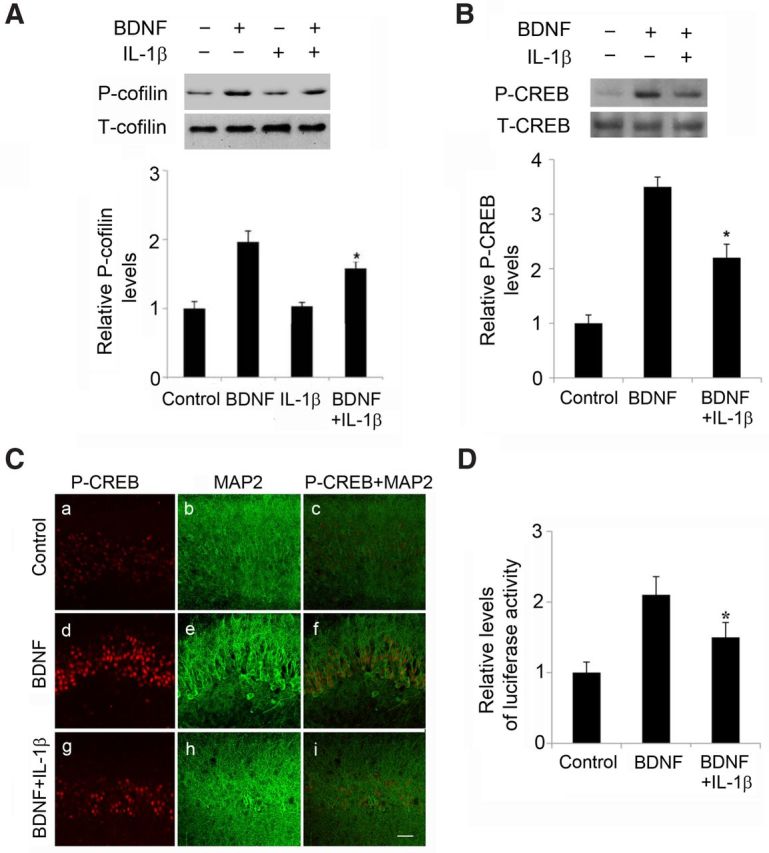
IL-1β pretreatment decreased BDNF-induced elevated levels of cofilin phosphorylation and CREB activation. A, Representative Western blots and quantification of P-cofilin levels showing that pretreatment with IL-1β suppressed the effect of BDNF (100 ng/ml, 1 h) on cofilin phosphorylation in CA1; *p < 0.05 BDNF versus BDNF plus IL-1β (n = 4). B, Representative Western blots and quantification of the detection of CREB phosphorylated at Ser133 (P-CREB). Exposure to BDNF (100 ng/ml, 1 h) increased the amount of P-CREB. Pretreatment with IL-1β suppressed the effect of BDNF on P-CREB but had no effect on T-CREB levels; *p < 0.05 BDNF versus BDNF plus IL-1β. C, Representative images showing the immunoreactivity of P-CREB in CA1. An antibody specific to P-CREB was used. Compared to control (a), P-CREB immunoreactivity increased after BDNF treatment (100 ng/ml; d). IL-1β decreased BDNF-induced immunoreactivity of P-CREB (g). MAP2 staining: control (b), BDNF-treated cultures (e), BDNF plus IL-1β treatment (h). Merged CREB and MAP2 staining: control (c), BDNF (f), BDNF plus IL-1β (i). Scale bar, 50 μm. D, CRE transcriptional activity. Cortical neurons at 3 DIV were transfected with plasmid pIII(170)Luc containing the promoter of BDNF exon III (Tao et al., 1998, numbering of promoter has been changed to exon IV according to Aid et al., 2007) comprising the CRE-like sequence and exon III fused to a luciferase reporter gene and an internal control Renilla luciferase plasmid. At 48 h after transfection, cultures were switched to fresh medium and incubated for 90 min in the presence or absence of 50 ng/ml IL-1β, before the addition of BDNF (50 ng/ml) or vehicle and additional incubation for 1 h. Transcription activity was assayed by measuring luciferase activity and expressed as the ratio of the luciferase activity in extracts from cells exposed BDNF to the luciferase activity in extracts from unstimulated cells. IL-1β treatment decreased BDNF-induced activation of pIII(170)Luc. Data are the mean ± SEM (n = 3) expressed in terms of the normalized pIII(170)Luc levels obtained in the control cultures. *p < 0.05 BDNF versus BDNF plus IL-1β, ANOVA followed by the LSD post hoc test.
IL-1β impairs BDNF-associated signal transduction
To characterize the upstream molecules involved in the above described inhibitory crosstalk between IL-1β and BDNF, we tested for an interleukin influence on BDNF-associated signal transduction. The biological functions of BDNF are mediated by the TrkB receptor, which activates the MAPK, phosphatidylinositol-3 kinase (PI3K)/Akt, and PLCγ pathways (Huang and Reichardt, 2003), along with small GTPase-initiated actin signaling cascade (Rex et al., 2007). Farther downstream, BDNF signaling leads to the activation of CREB, a transcription factor involved in the expression of Arc and Homer1 (Sato et al., 2001; Yin et al., 2002; Kawashima et al., 2009).
We used Western blots to test for an effect of IL-1β on BDNF-induced phosphorylation of CREB at Ser-133 (P-CREB). Pretreatment with IL-1β (24 h) did not measurably affect basal levels in field CA1 (data not shown) but significantly suppressed BDNF-evoked phosphorylation of CREB. T-CREB levels were not affected by the experimental manipulations (Fig. 2B). We next performed an immunohistochemical detection of P-CREB in organotypic slices after the above treatments. Consistent with the Western blot measurements, immunohistochemical staining showed that IL-1β pretreatment attenuates BDNF-induced phosphorylation of CREB in field CA1 neurons (Fig. 2C). cAMP response element (CRE)-driven gene expression was then evaluated to assess the consequences of IL-1β-mediated downregulation of CREB activity. Activation of BDNF exon IV promoter, which contains a CRE site (Tao et al., 1998), was monitored using a transient transcription activity assay with luciferase as a reporter gene. BDNF increased BDNF exon IV promoter activity, and this effect was significantly (30%) reduced by IL-1β pretreatment (50 ng/ml, 2 h) (Fig. 2D).
The PI3K/Akt pathway has a pivotal role in synaptic plasticity in the hippocampus (Sanna et al., 2002; Opazo et al., 2003) and also modulates CREB (Du and Montminy, 1998; Simão et al., 2012). Furthermore, in vitro data suggest that the PI3K/Akt/mammalian target of rapamycin (mTOR)/eukaryotic elongation factor 2 (eEF2) signaling cascade is involved in Arc expression induced by BDNF (Chen et al., 2009). We therefore tested whether IL-1β affects BDNF-induced activation of Akt by measuring Ser-473 phosphorylation. IL-1β pretreatment (50 ng/ml, 24 h) did not measurably affect basal levels in field CA1 (data not shown) but substantially reduced the levels of P-Akt response to BDNF in CA1 samples dissected from hippocampal cultures (Fig. 3A). Interestingly, IL-1β did not affect BDNF-dependent activation of PLCγ (Fig. 3B), the molecule linked to the TrkB/PLCγ/CaMK pathway (Huang and Reichardt, 2003).
Figure 3.

IL-1β pretreatment affected BDNF-induced Akt activation. A, Representative Western blots showing the detection of P-Akt. Quantification of P-Akt levels showed that exposure to BDNF (100 ng/ml, 1 h) increased the amount of P-Akt, whereas pretreatment with IL-1β suppressed the effect of BDNF on P-Akt but had no effect on T-Akt. *p < 0.05 BDNF versus BDNF plus IL-1β (n = 4). B, Western blots showing that BDNF-induced (100 ng/ml, 1 h) tyrosine phosphorylation of PLCγ is unaffected by IL-1β pretreatment (50 ng/ml, 24 h) in hippocampal slices. Tyrosine phosphorylation of PLCγ was examined by immunoprecipitation (IP) followed by Western blotting, as described in Materials and Methods. C, Representative Western blots and quantification of the levels of P-p42 and P-p44 isoforms of MAPK in hippocampal slices treated as indicated. Data are the mean ± SEM (n = 3) expressed in terms of P-MAPK levels obtained in the control cultures. *p < 0.05 for all treatment groups versus control, ANOVA followed by the LSD post hoc test.
MAPK signaling, along with the PI3K/Akt pathway, contributes to synaptic plasticity and long-term memory formation (Davis and Laroche, 2006), and we therefore evaluated it in hippocampal slices cultures after the treatments described above. All manipulations, BDNF alone, IL-1β alone, or the IL-1β/BDNF combination, significantly increased the levels of P-p42 and P-p44 isoforms of MAPK in CA1 area (Fig. 3C). The absence of a difference in P-MAPK isoforms in BDNF-treated and nontreated cultures in the presence of IL-1β could be attributable to the increases produced by IL-1β alone. IL-1β does not cause MAPK activation in low-density pure neuronal cultures (Tong et al., 2008), raising the possibility that its positive action in organotypic cultures reflects an action on microglia (Kim et al., 2004).
The suppressive effect of IL-1β on BDNF-driven activation of Akt could occur at upstream steps of the signal transduction: initially, BDNF acts on TrkB at the cell surface, causing its autophosphorylation and leading to activation of the PI3K/Akt pathway via the protein IRS-1, which is activated when phosphorylated at Tyr-612 (Huang and Reichardt, 2003). IL-1β pretreatment (50 ng/ml, 24 h) substantially reduced the total tyrosine phosphorylation of IRS-1 in response to BDNF in CA1 but did not affect basal levels (Fig. 4A); specifically, it was found that IL-1β affected the BDNF-induced increase in P-IRS-1 Tyr-612 (Fig. 4B). Similar to our previous study in cultured cortical neurons, BDNF-driven TrkB phosphorylation was not affected by IL-1β treatment (Fig. 4C). These data indicate that IL-1β impairs the proximal signaling associated with TrkB activation at IRS-1. They are consistent with previous results obtained with low-density primary cortical neuronal cultures (Tong et al., 2008) but importantly extend the mechanism to CA1 area in organotypic cultures.
Figure 4.

IL-1β pretreatment affected BDNF-induced IRS-1 activation. Slices were treated with BDNF (100 ng/ml, 1 h) and IL-1β (50 ng/ml, 24 h pretreatment). Representative Western blots and quantification relative to control cultures of the levels of total P-tyrosine IRS (IRS-1 pY; A), IRS-1 phosphorylated at Tyr-612 (IRS-1 pY612; B), and P-TrkB (C) in CA1. *p < 0.05 BDNF versus BDNF plus IL-1β (n = 3). IP, Immunoprecipitation.
p38 MAPK may mediate the suppressive effect of IL-1β on BDNF signaling
Activation of p38 is an early event initiated by IL-1β stimulation in many cell types (Srinivasan et al., 2004) and has been implicated in the regulation of synaptic plasticity and the onset of neurodegeneration (Rush et al., 2002; Gallagher et al., 2004; Wang et al., 2004; Origlia et al., 2008). There is also evidence linking p38 to amyloid-β (Aβ)-induced synaptic depression (Hsieh et al., 2006) and to Aβ-mediated inhibition of LTP (Wang et al., 2004). Therefore, we examined the role of the kinase in IL-1β-mediated suppression of BDNF signaling in organotypic slice cultures. IL-1β activated p38 at 4 h, and this effect was maintained for 24 h (Fig. 5A). SB202190, a p38 inhibitor (Fig. 5A), blocked the inhibitory effect of IL-1β on BDNF-induced cofilin phosphorylation in CA1 (Fig. 5C). Similarly, the pharmacological inhibition of p38 blocked the inhibitory effect of IL-1β on BDNF-induced Akt phosphorylation (Fig. 5B) and the expression of Homer1a (Fig. 5D). Thus, p38 activation is one of the mechanisms whereby IL-1β disrupts BDNF signaling.
Figure 5.
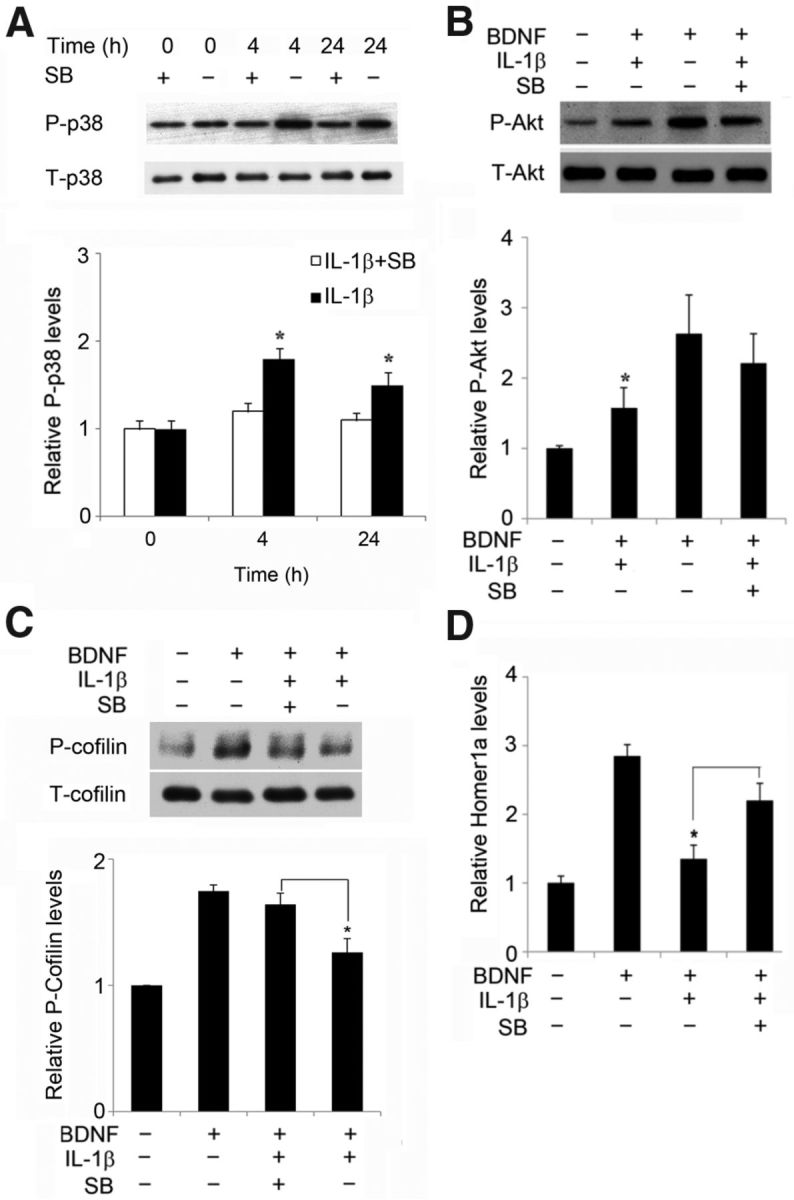
The p38 inhibitor decreased the impairing effect of IL-1β on BDNF signaling. A, IL-1β treatment (50 ng/ml) for 4 or 24 h increased the amount of phosphorylation of p38 at Thr-180/Tyr-182 (P-p38) but had no effect on T-p38 levels. In the presence of the p38 inhibitor SB202190 (2 μm; SB), the IL-1β-induced increase in P-p38 level was suppressed, as shown in the quantification; *p < 0.05. B, Treatment with the p38 inhibitor SB239063 (50 μm) attenuated the effect of IL-1β on BDNF-elicited P-Akt (Ser473); *p < 0.05 BDNF plus IL-1β versus BDNF plus IL-1β plus SB202190. C, Treatment with the p38 inhibitor SB202190 (2 μm) attenuated the effect of IL-1β on the BDNF-induced cofilin phosphorylation; *p < 0.05 BDNF plus IL-1β versus BDNF plus IL-1β plus SB202190. D, SB239063 prevented the suppression by IL-1β of BDNF-induced expression of Homer1a, as determined by RT-PCR. *p < 0.05 for BDNF plus IL-1β versus BDNF plus IL-1β plus SB239063, ANOVA followed by the LSD post hoc test. All data are the mean ± SEM (n = 3) expressed in terms of control cultures.
IL-1β impairs BDNF-dependent LTP by activation of p38
We next tested whether the mechanisms described in the preceding sections are involved in the IL-1β-induced suppression of LTP described in previous reports (Bellinger et al., 1993; Ross et al., 2003). The naturalistic TBS pattern (Larson et al., 1986), which produces a robust, extremely stable, and BDNF-dependent (Korte et al., 1995; Kramár et al., 2004) form of LTP, was used in these acute hippocampal slice experiments. Slices were treated with IL-1β (10–50 ng/ml, 1 h) before the delivery of TBS and compared with slices treated with aCSF alone. In contrast to previous reports (Bellinger et al., 1993; Ross et al., 2003), a 1 h pretreatment with the cytokine had no detectable effect on the slope or amplitude of fEPSPs recorded from the stratum radiatum of field CA1b. TBS caused a rapid increase in response size that was of equal magnitude in the IL-1β-treated and control slices (87 ± 8% of baseline at 90 s after TBS for controls and 110 ± 15% for treated, p > 0.20). The initial potentiation decayed steadily for 5–10 min and then stabilized in the control group but continued to decline in the IL-1β-treated slices; LTP at 60 min was 45 ± 4% above baseline for the aCSF-only cases and 16 ± 3% for the IL-1β slices (p < 0.001) (Fig. 6A,B). The decremental nature of potentiation without evident changes to the induction and expression of LTP strongly suggests that IL-1β selectively disrupted the actin filament assembly required for LTP consolidation. We tested this by topically applying Alexa Fluor-568–phalloidin at the conclusion of physiological testing and then using automated methods to count spine-sized structures containing high concentrations of F-actin (Kramár et al., 2006). Densely labeled spines were infrequent in slices given only LFS but were increased in number by approximately sixfold after TBS (Fig. 6C,D). This pronounced effect was completely eliminated by a 1 h pretreatment with IL-1β (p < 0.01 for the TBS groups with vs without IL-1β) (Fig. 6C,D).
Figure 6.

IL-1β impairs theta burst-induced LTP in hippocampal slices. The slope of the fEPSP (mean ± SEM) was normalized to the mean of the last 10 min of baseline recording collected in the presence and absence of IL-1β (10–50 ng/ml) before application of TBS (upward arrow). A, A train of 10 theta bursts produced a significant degree of impairment in the level of potentiation 1 h after induction in IL-1β-treated slices relative to control aCSF-treated slices. B, Representative traces collected from control (i) and IL-1β-treated (ii) slices during baseline (black line), 60 min after start of drug infusion (red line), and 60 min after TBS (blue line). Calibration: 0.5 mV, 5 ms. C, Rhodamine-labeled phalloidin was applied to slices at the end of the experiment and then processed for epifluorescence microscopy. Survey micrographs show a marked increase in the number of phalloidin-positive spines (arrow) in control slices receiving TBS relative to slices receiving LFS (3 test pulses per minute). The TBS-induced increase in phalloidin staining was markedly reduced to baseline levels in slices treated with IL-1β. Scale bar, 200 μm. Inset, High magnification of phalloidin-labeled spine heads as indicated by arrow. Scale bar, 2 μm. D, IL-1β reduces LTP-dependent actin polymerization. The mean ± SEM number of dense phalloidin labeling in spine heads as assessed after LFS (n = 13) and 1 h after delivery of a 10-burst theta train in the presence (n = 14) and absence (n = 13) of IL-1β (**p < 0.001).
TBS activates synaptic TrkB receptors in a manner that is dependent on endogenous, presumably released, BDNF (Chen et al., 2010), and this effect is necessary for induced actin polymerization and the production of stable LTP. It is therefore possible that the disruptive effects of IL-1β on LTP are attributable to the above described negative influence of the cytokine on downstream responses to the TrkB activation. We tested this idea using the same p38 inhibitor, SB202190, that reduced the inhibitory actions of IL-1β in cultured slices. Slices were cotreated with SB202190 and IL-1β for 1 h before TBS was applied; LTP obtained under these conditions was not measurably different from that found in slices treated with SB202190 alone (Fig. 7A). Similarly, IL-1β did not block TBS-induced increases in the number of spines with high levels of F-actin in slices pretreated with the p38 antagonist (Fig. 7B,C).
Figure 7.

p38 inhibitor prevents IL-1β-induced impairment of LTP. A, Left, After a 20 min stable baseline recording, 1 μm SB202190 (SB; n = 3) infusions began for 1 h before delivering 10 theta bursts and continued throughout the remaining recording period. The level of potentiation 1 h after induction was no different from that measured in control slices (see Fig. 6A). Infusions of IL-1β (50 ng/ml) in the presence of 1 μm SB202190 was tested in a separate set of slices. Infusions with SB202190 plus IL-1β (n = 3) produced a stable level of potentiation similar to slices treated with SB202190 alone. Right, Representative traces collected from slices treated with SB202190 and SB202190 plus IL-1β before (black line) and 1 h after the start of drug infusion (red line) and 60 min after TBS (blue line). Calibration: 1 mV, 5 ms. B, The p38 inhibitor SB202190 prevents IL-1β-induced decrease in TBS-dependent actin polymerization. Survey photomicrographs showing that, relative to slices receiving baseline low-frequency test pulses (left), theta burst-induced actin polymerization is not reduced in SB202190-treated slices (middle) nor in slices infused with the combination of SB202190 plus IL-1β (right). Scale bar, 5 μm. C, The mean ± SEM number of dense phalloidin labeling in spine heads as assessed after LFS in control slices (n = 10) and 1 h after delivery of a 10-burst theta train in slices pretreated with SB202190 alone (n = 10) and SB202190 plus IL-1β (n = 11) (**p < 0.001).
To verify that IL-1β suppresses LTP by inhibiting BDNF signaling, we tested the effects of the cytokine under conditions in which exogenous neurotrophin is required for the production of lasting potentiation. Past studies showed that brief infusions of BDNF lower the number of theta bursts needed to generate lasting potentiation; we therefore asked whether, as predicted by the preceding results, this direct effect of BDNF is eliminated by cotreatment with IL-1β. Slices were infused with a low concentration of BDNF (2 ng/ml) either by itself or in the presence of IL-1β for 60 min and compared with aCSF-treated slices and then given two, instead of the normal 10, theta bursts. As in previous work (Kramár et al., 2004), the short trains failed to induce stable LTP in control slices but elicited robust and stable potentiation in the presence of BDNF. In contrast, the two burst trains were ineffective in cases treated with both BDNF and IL-1β (Fig. 8), and, consistent with above findings, the degree of LTP in slices bathed for 1 h in the presence of p38 inhibitor plus BDNF plus IL-1β was not different from that found in slices treated for the same period with BDNF alone. Thus, the facilitatory effects of BDNF on LTP signaling are blocked by IL-1β, as expected from the organotypic culture experiments.
Figure 8.

BDNF-dependent LTP is eliminated by IL-1β via p38. A 1 hour BDNF treatment (2 nm) before the delivery of two theta bursts (a degree of stimulation that is subthreshold for generating LTP) results in a measurable degree of LTP 60 min after induction with respect to control aCSF-treated slices. A 1 h infusion of IL-1β (50 ng/ml) in combination with 2 nm BDNF produced a similar decay in LTP as in control slices after the delivery of two theta bursts. The level of potentiation in slices infused for 1 h in the presence of BDNF, IL-1β, and 1 μm SB202190 were no different from slices treated with BDNF alone. Control pathway indicates that responses were stable throughout the recording period.
Discussion
A now substantial body of work indicates that the buildup of proinflammatory cytokines, and IL-1β in particular, contributes to age-related declines in cognitive functioning. The present results describe a detailed mechanism that is likely to be central to these effects. Specifically, we propose that the contributions of BDNF to the synaptic modifications and gene regulation produced by patterned afferent activity are blocked by IL-1β via a p38-mediated mechanism. Elevated levels of IL-1β occurring during brain aging would therefore interfere with events needed for memory encoding and the maintenance of neuronal viability.
The current studies examined the interactions of IL-1β with several key steps in the signaling pathways linking BDNF to the regulation of plasticity-related genes. Past work using cultured cortical neurons showed that BDNF increases the expression of Arc, a protein that modulates the subsynaptic cytoskeleton and LTP stabilization, via activation of the PI3K/Akt/mTOR/eEF2 signaling pathway (Chen et al., 2009). We established that the neurotrophin stimulates both Akt and the expression of Arc in cultured hippocampal slices and then found that both effects are substantially reduced by IL-1β. Importantly, the suppressive effects of the interleukin were regionally differentiated, being prominent in fields CA1, CA2, and the DG, but absent in the large pyramidal neurons of field CA3. Differential responses among the hippocampal areas to inflammatory stimuli have been described by others (Chang et al., 2009; Chapman et al., 2011), and it has been suggested that field CA1 is particularly vulnerable to neuroinflammation in aged animals (Chapman et al., 2010, 2011). Also of interest in this regard, basal and fear-conditioning-induced expression of BDNF were reduced in CA1, but not in CA3, during infection (intraperitoneal injection of E. coli) (Chapman et al., 2011). IL-1β also decreased BDNF-induced phosphorylation of CREB, a transcription factor involved in Arc (Kawashima et al., 2009) and BDNF exon IV expression in cultured slices. As predicted, we found that BDNF exon IV expression was suppressed by IL-1β. Consistent with these data, peripheral E. coli infection, which elevates brain IL-1β, suppresses the induction of BDNF exon IV after fear conditioning (Chapman et al., 2011).
In addition to the above described mechanisms, IL-1β could interfere with BDNF-dependent learning in neurodegenerative diseases and other conditions that raise IL-1β levels (e.g., peripheral infection) by reducing the expression of BDNF (for review, see Tapia-Arancibia et al., 2008). The presented data suggest that IL-1β acts rapidly on downstream mechanisms initiated by direct applications of the neurotrophin and thus are not dependent on BDNF mRNA expression; this strongly implies that the site of action involves BDNF signal transduction, a point supported by the results discussed immediately below.
How does IL-1β disrupt BDNF-mediated signaling to events essential for enduring synaptic plasticity? The present studies included a first direct test of the possibility that cytokine blocks autophosphorylation (activation) of the TrkB receptors of BDNF: the results were negative. However, IL-1β did substantially reduce phosphorylation of IRS-1, a protein factor known from previous studies (Tong et al., 2008) to couple TrkB to the PI3K pathway. Consonant with these arguments, we observed that IL-1β increased the phosphorylation of p38 but decreased BDNF-stimulated IRS-1 phosphorylation in cultured slices. Moreover, a pharmacological inhibitor of p38 removed the inhibitory effects of IL-1β on BDNF-driven Akt phosphorylation, expression of IEGs, and LTP. These results describe a specific signaling pathway for mediating the negative effects of the interleukin on BDNF signaling. It is presently unknown how p38 may inactivate IRS-1, but one possibility is that it acts by way of the protein tyrosine phosphatase PTP1B, as suggested recently (García-San Frutos et al., 2012); this and other options remain to be tested.
It should be emphasized that IL-1β can affect signal transduction pathways associated with hippocampal synaptic plasticity and memory in addition to those engaged by BDNF/TrkB. For instance, it has been reported that IL-1β modulates synaptic functions by activating JNK, nuclear factor-κB, and caspase-1 (Vereker et al., 2000; Curran et al., 2003) and can regulate NMDA receptors via Src family kinases (Viviani et al., 2003). It is also possible that calcium plays a role in the IL-1β occlusion of BDNF effects on synaptic plasticity. In particular, Ca2+ influx via L-type channels contributes to IL-1β activation of JNK, extracellular signal-regulated kinase, and p38 (Størling et al., 2005). Therefore, the elevated postsynaptic intracellular [Ca2+] and L-type calcium channel activity in aging neurons (Thibault et al., 2001) may increase vulnerability for the p38-dependent impairments induced by IL-1β.
The demonstration that IL-1β blocks TrkB signaling in cultured slices provides a plausible explanation for the negative effects of interleukin on BDNF-dependent stabilization of LTP in adult hippocampus (Bellinger et al., 1993; Ross et al., 2003; Chapman et al., 2011). However, questions arise concerning the likelihood that the effects of IL-1β on expression, as described here, occur quickly enough to account for the negative influence of the cytokine on stabilization events developing in the first 5–10 min after TBS. This rapid, as opposed to protein synthesis dependent, phase of consolidation involves TBS-driven TrkB activation and stimulation of pathways leading to cofilin phosphorylation and actin filament assembly (Rex et al., 2007). The present studies confirmed previous reports that the negative effects of IL-1β on LTP are evident within 10 min after TBS and further demonstrated that the cytokine blocks the rapid facilitatory actions of exogenous BDNF on the LTP induced by short, subthreshold TBS trains. The latter result provides direct evidence that the interleukin impairs the BDNF-dependent step in LTP induction. IL-1β also markedly attenuated both BDNF-initiated cofilin phosphorylation in cultured slices and TBS-driven polymerization of spine actin in adult hippocampus. Moreover, a p38 inhibitor that prevented the suppressive effects of interleukin on BDNF signaling in organotypic cultures also rescued F-actin formation along with LTP stabilization in adult slices. We therefore conclude that the negative, p38-mediated action of IL-1β on the IRS-1 response to released BDNF disrupts the link between activated synaptic TrkB receptors and the machinery underlying the cytoskeletal modifications needed for rapid consolidation.
We have referred previously to the impairment of BDNF signaling as neurotrophin resistance (Tong et al., 2008), analogous to the development of insulin resistance (Böni-Schnetzler and Donath, 2011). In both cases, cells show decreased sensitivity to ligand activation in the presence of proinflammatory cytokines, which play prominent roles in diabetes and neurodegenerative diseases (Rothwell and Luheshi, 2000; Marette, 2002), through similar mechanisms, e.g., by negatively regulating key signaling molecules such as IRS-1 and Akt. The activation of docking proteins, including IRS-1 (Zick, 2001), can be suppressed by stress inflammatory MAPKs such as p38 in the insulin-resistance paradigm (Hemi et al., 2011). Similarly, activation of p38 by IL-1β leads to the suppression of the IRS-1 response to BDNF. Thus, there is a common pathway that can compromise multiple readouts of BDNF and insulin signaling. However, the parallels may not extend to the receptor desensitization induced by elevated ligand levels, as observed in some types of insulin resistance. At present, there is no evidence for compensatory upregulation of BDNF during chronic brain inflammation; on the contrary, the elevated IL-1β levels found in the aged brain are associated with decreased BDNF concentrations (Peng et al., 2005). In all, then, the IL-1β effects presented here resemble that form of inflammatory-induced insulin resistance reviewed by Marette (2002) and Olefsky and Glass (2010). It remains for future research to explicitly compare the mechanisms underlying insulin and neurotrophin resistance in the context of inflammation.
In conclusion, we found that IL-1β impaired BDNF-induced expression of molecules critical for activity-dependent synaptic plasticity in organotypic hippocampal slices, leading to the inhibition of BDNF-dependent LTP. The action of IL-1β was found to be associated with the reduction of TrkB-mediated BDNF signaling, CREB, Arc, cofilin regulation, and the activation of p38. The involvement of p38 in the IL-1β-induced inhibition of synaptic plasticity suggests that p38 is a key molecule linking multiple signal transduction pathways underlying inflammation-dependent memory loss. These results suggest that treatments that preserve neurotrophic signaling in brain may help to overcome aging-dependent memory decline and AD.
Footnotes
This work was supported by National Institutes of Health Grant PO1-AG000538 and R01-AG34667 (C.W.C.) and National Institute of Mental Health Grant MH083346 and Office of Naval Research Multidisciplinary University Research Initiative Grant N00014-10-1-10072 (G.L.). We thank Dr. Robert Balazs for helpful discussions on the design and conduct of this study.
The authors declare no competing financial interests.
References
- Adlard PA, Perreau VM, Engesser-Cesar C, Cotman CW. The timecourse of induction of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus following voluntary exercise. Neurosci Lett. 2004;363:43–48. doi: 10.1016/j.neulet.2004.03.058. [DOI] [PubMed] [Google Scholar]
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF. Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun. 2009;23:46–54. doi: 10.1016/j.bbi.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- Böni-Schnetzler M, Donath MY. Increased IL-1beta activation, the culprit not only for defective insulin secretion but also for insulin resistance? Cell Res. 2011;21:995–997. doi: 10.1038/cr.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR. Local protein synthesis, actin dynamics, and LTP consolidation. Curr Opin Neurobiol. 2008;18:524–531. doi: 10.1016/j.conb.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Alme MN, Bittins M, Kuipers SD, Nair RR, Pai B, Panja D, Schubert M, Soule J, Tiron A, Wibrand K. The Arc of synaptic memory. Exp Brain Res. 2010;200:125–140. doi: 10.1007/s00221-009-1959-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacabelos R, Barquero M, García P, Alvarez XA, Varela de Seijas E. Cerebrospinal fluid interleukin-1 beta (IL-1 beta) in Alzheimer's disease and neurological disorders. Methods Find Exp Clin Pharmacol. 1991;13:455–458. [PubMed] [Google Scholar]
- Chang FY, Lee CC, Huang CC, Hsu KS. Unconjugated bilirubin exposure impairs hippocampal long-term synaptic plasticity. PLoS One. 2009;4:e5876. doi: 10.1371/journal.pone.0005876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman TR, Barrientos RM, Ahrendsen JT, Maier SF, Patterson SL. Synaptic correlates of increased cognitive vulnerability with aging: peripheral immune challenge and aging interact to disrupt theta-burst late-phase long-term potentiation in hippocampal area CA1. J Neurosci. 2010;30:7598–7603. doi: 10.1523/JNEUROSCI.5172-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman TR, Barrientos RM, Ahrendsen JT, Hoover JM, Maier SF, Patterson SL. Aging and infection reduce expression of specific brain-derived neurotrophic factor mRNAs in hippocampus. Neurobiol Aging. 2011;33:832.e1–e14. doi: 10.1016/j.neurobiolaging.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Sanaiha Y, Lynch G, Gall CM. Learning induces neurotrophin signaling at hippocampal synapses. Proc Natl Acad Sci U S A. 2010;107:7030–7035. doi: 10.1073/pnas.0912973107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TJ, Wang DC, Chen SS. Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-induced Arc expression in rat cortical neurons. J Neurosci Res. 2009;87:2297–2307. doi: 10.1002/jnr.22057. [DOI] [PubMed] [Google Scholar]
- Curran BP, Murray HJ, O'Connor JJ. A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience. 2003;118:347–357. doi: 10.1016/s0306-4522(02)00941-7. [DOI] [PubMed] [Google Scholar]
- Davis S, Laroche S. Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: a review. Genes Brain Behav. 2006;5(Suppl 2):61–72. doi: 10.1111/j.1601-183X.2006.00230.x. [DOI] [PubMed] [Google Scholar]
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- Erickson KI, Prakash RS, Voss MW, Chaddock L, Heo S, McLaren M, Pence BD, Martin SA, Vieira VJ, Woods JA, McAuley E, Kramer AF. Brain-derived neurotrophic factor is associated with age-related decline in hippocampal volume. J Neurosci. 2010;30:5368–5375. doi: 10.1523/JNEUROSCI.6251-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Hein AM, Biedenkapp JC, Watkins LR, Maier SF. IL-1RA blocks E. coli-induced suppression of Arc and long-term memory in aged F344xBN F1 rats. Brain Behav Immun. 2010;24:254–262. doi: 10.1016/j.bbi.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24:4859–4864. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-San Frutos M, Fernández-Agulló T, Carrascosa JM, Horrillo D, Barrús MT, Oliveros E, Sierra J, Ros M. Involvement of protein tyrosine phosphatases and inflammation in hypothalamic insulin resistance associated with ageing: effect of caloric restriction. Mech Ageing Dev. 2012;133:489–497. doi: 10.1016/j.mad.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Hein AM, Stasko MR, Matousek SB, Scott-McKean JJ, Maier SF, Olschowka JA, Costa AC, O'Banion MK. Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 2010;24:243–253. doi: 10.1016/j.bbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemi R, Yochananov Y, Barhod E, Kasher-Meron M, Karasik A, Tirosh A, Kanety H. p38 mitogen-activated protein kinase-dependent transactivation of ErbB receptor family: a novel common mechanism for stress-induced IRS-1 serine phosphorylation and insulin resistance. Diabetes. 2011;60:1134–1145. doi: 10.2337/db09-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Kawashima T, Okuno H, Nonaka M, Adachi-Morishima A, Kyo N, Okamura M, Takemoto-Kimura S, Worley PF, Bito H. Synaptic activity-responsive element in the Arc/Arg3.1 promoter essential for synapse-to-nucleus signaling in activated neurons. Proc Natl Acad Sci U S A. 2009;106:316–321. doi: 10.1073/pnas.0806518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Smith CJ, Van Eldik LJ. Importance of MAPK pathways for microglial pro-inflammatory cytokine IL-1 beta production. Neurobiol Aging. 2004;25:431–439. doi: 10.1016/S0197-4580(03)00126-X. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain- derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci. 2004;24:5151–5161. doi: 10.1523/JNEUROSCI.0800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci U S A. 2006;103:5579–5584. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368:347–350. doi: 10.1016/0006-8993(86)90579-2. [DOI] [PubMed] [Google Scholar]
- Li G, Peskind ER, Millard SP, Chi P, Sokal I, Yu CE, Bekris LM, Raskind MA, Galasko DR, Montine TJ. Cerebrospinal fluid concentration of brain-derived neurotrophic factor and cognitive function in non-demented subjects. PLoS One. 2009;4:e5424. doi: 10.1371/journal.pone.0005424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Kramár EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu IY, Lyons WE, Mamounas LA, Thompson RF. Brain-derived neurotrophic factor plays a critical role in contextual fear conditioning. J Neurosci. 2004;24:7958–7963. doi: 10.1523/JNEUROSCI.1948-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marette A. Mediators of cytokine-induced insulin resistance in obesity and other inflammatory settings. Curr Opin Clin Nutr Metab Care. 2002;5:377–383. doi: 10.1097/00075197-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Messaoudi E, Kanhema T, Soul é J, Tiron A, Dagyte G, da Silva B, Bramham CR. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci. 2007;27:10445–10455. doi: 10.1523/JNEUROSCI.2883-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami I, Kengaku M, Smitt PS, Shigemoto R, Hirano T. Long-term potentiation of mGluR1 activity by depolarization-induced Homer1a in mouse cerebellar Purkinje neurons. Eur J Neurosci. 2003;17:1023–1032. doi: 10.1046/j.1460-9568.2003.02499.x. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R. Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–345. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O'Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction. J Neurosci. 2008;28:3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña F. Organotypic cultures as tool to test long-term effects of chemicals on the nervous system. Curr Med Chem. 2010;17:987–1001. doi: 10.2174/092986710790820679. [DOI] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem. 2005;93:1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- Pinteaux E, Trotter P, Simi A. Cell-specific and concentration-dependent actions of interleukin-1 in acute brain inflammation. Cytokine. 2009;45:1–7. doi: 10.1016/j.cyto.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Rachal Pugh C, Fleshner M, Watkins LR, Maier SF, Rudy JW. The immune system and memory consolidation: a role for the cytokine IL-1beta. Neurosci Biobehav Rev. 2001;25:29–41. doi: 10.1016/s0149-7634(00)00048-8. [DOI] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramár EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FM, Allan SM, Rothwell NJ, Verkhratsky A. A dual role for interleukin-1 in LTP in mouse hippocampal slices. J Neuroimmunol. 2003;144:61–67. doi: 10.1016/j.jneuroim.2003.08.030. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Luheshi GN. Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 2000;23:618–625. doi: 10.1016/s0166-2236(00)01661-1. [DOI] [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R. Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro. J Neurosci. 2002;22:6121–6128. doi: 10.1523/JNEUROSCI.22-14-06121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Futai K, Yamamoto K, Worley PF, Hayashi Y, Sheng M. Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J Neurosci. 2003;23:6327–6337. doi: 10.1523/JNEUROSCI.23-15-06327.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suzuki K, Nakanishi S. NMDA receptor stimulation and brain-derived neurotrophic factor upregulate homer 1a mRNA via the mitogen-activated protein kinase cascade in cultured cerebellar granule cells. J Neurosci. 2001;21:3797–3805. doi: 10.1523/JNEUROSCI.21-11-03797.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat Neurosci. 2011;14:279–284. doi: 10.1038/nn.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão F, Matté A, Pagnussat AS, Netto CA, Salbego CG. Resveratrol prevents CA1 neurons against ischemic injury by parallel modulation of both GSK-3β and CREB through PI3-K/Akt pathways. Eur J Neurosci. 2012;36:2899–2905. doi: 10.1111/j.1460-9568.2012.08229.x. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Yen JH, Joseph DJ, Friedman W. Cell type-specific interleukin-1β signaling in the CNS. J Neurosci. 2004;24:6482–6488. doi: 10.1523/JNEUROSCI.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Størling J, Zaitsev SV, Kapelioukh IL, Karlsen AE, Billestrup N, Berggren PO, Mandrup-Poulsen T. Calcium has a permissive role in interleukin-1beta-induced c-jun N-terminal kinase activation in insulin-secreting cells. Endocrinology. 2005;146:3026–3036. doi: 10.1210/en.2005-0036. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GC, Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319:1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [Erratum (1998) 20:1297] [DOI] [PubMed] [Google Scholar]
- Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev. 2008;59:201–220. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–9756. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW. Beta-amyloid-(1–42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival is not compromised. J Biol Chem. 2001;276:17301–17306. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- Tong L, Balazs R, Soiampornkul R, Thangnipon W, Cotman CW. Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging. 2008;29:1380–1393. doi: 10.1016/j.neurobiolaging.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereker E, Campbell V, Roche E, McEntee E, Lynch MA. Lipopolysaccharide inhibits long term potentiation in the rat dentate gyrus by activating caspase-1. J Biol Chem. 2000;275:26252–26258. doi: 10.1074/jbc.M002226200. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao WD, Spealman RD, Zhang J. Dopaminergic signaling in dendritic spines. Biochem Pharmacol. 2008;75:2055–2069. doi: 10.1016/j.bcp.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Edelman GM, Vanderklish PW. The brain-derived neurotrophic factor enhances synthesis of Arc in synaptoneurosomes. Proc Natl Acad Sci U S A. 2002;99:2368–2373. doi: 10.1073/pnas.042693699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zick Y. Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol. 2001;11:437–441. doi: 10.1016/s0962-8924(01)02129-8. [Erratum (2002) 12:56] [DOI] [PubMed] [Google Scholar]