

Abstract
Human anthrax infection caused by exposure to Bacillus anthracis cannot always be treated by antibiotics. This is mostly because of the effect of the remaining anthrax toxin in the body. Lethal factor (LF) is a component of lethal toxin (LeTx), which is the major virulence of anthrax toxin. A murine IgG monoclonal antibody (mAb) against LF with blocking activity (coded LF8) was produced in a previous study. In this report, a human/murine chimeric Fab mAb (coded LF8-Fab) was developed from LF8 by inserting murine variable regions into human constant regions using antibody engineering to reduce the incompatibility of the murine antibody for human use. The LF8-Fab expressed in Escherichia coli could specifically identify LF with an affinity of 3.46 × 107 L/mol and could neutralize LeTx with an EC50 of 85 μg/mL. Even after LeTx challenge at various time points, the LF8-Fab demonstrated protection of J774A.1 cells in vitro. The results suggest that the LF8-Fab might be further characterized and potentially be used for clinical applications against anthrax infection.
1. Introduction
Anthrax infection is caused by Bacillus anthracis which primarily affects livestock but can spread to humans [1]. It is known that anthrax spores have the potential use as a weapon of bioterrorism. The anthrax attacks of 2001 heightened awareness concerning the treatment of anthrax exposure [2]. One of the current clinical treatments for anthrax is to use antibiotics which are effective but limited [3]. This is mainly because of the effect of the remaining anthrax toxin in the body, which cannot be eliminated by antibiotics. Anthrax toxin consists of three protein components: protective antigen (PA), lethal factor (LF), and edema factor (EF). PA combining with LF or EF constitutes lethal toxin (LeTx) or edema toxin (EdTx), respectively [4]. The 83 kDa form of PA (PA83) binds either of two known receptors on the surface of mammalian cells: anthrax toxin receptor 1 (ATXR1)/tumor endothelial marker 8 (TEM8) or anthrax toxin receptor 2 (ATXR2)/capillary morphogenesis protein 2 (CMG2) [5]. Then, PA83 is cleaved by a furin-like protease, producing PA20 and PA63. The latter oligomerizes to a heptamer and forms a pre-pore to bind LF and/or EF. The complex is internalized into cells by receptor-mediated endocytosis, and LF and/or EF are released to cytosol under acid conditions [6]. LF is the major virulent factor which is responsible for shock and death. LF is a zinc-dependent protease which can cleave several members of mitogen-activated protein kinase kinase (MAPKK) family causing lysis of macrophages [7]. In addition, LF offers Bacillus anthracis an efficient mechanism to evade the host immune responses by inhibiting interferon regulatory factor 3 (IRF3) activation by lipopolysaccharide and subsequent cytokine production through bacterial membrane components [8]. EF is a calcium-calmodulin-dependent adenylate cyclase which causes local edema [9].
Recent studies of antitoxin treatments have focused on three aspects: vaccines [10], monoclonal antibodies (mAbs), and other inhibitors, such as dominant-negative mutants of PA [11], soluble receptors [12], and noncatalytic domains of LF and EF [13]. Many neutralizing mAbs against PA have been developed and utilized in clinical trials [14], as PA shares the common part of LeTx and EdTx. However, the neutralization effect may become invalid against mutant strains of Bacillus anthracis [15]. Hence, EF and LF mAbs are alternative options to be used alone or in combination with PA mAb [16]. Murine mAbs may have some limitations to be used in humans directly because of the human anti-mouse antibody (HAMA) response [17]. It is necessary to develop mAbs with low immunogenicity including human, humanized, and chimeric mAbs. Human mAbs are generated by technologies of phage display library, transgenic mouse, EBV immortalized human B cell, and human-human hybridoma [18]. Humanized and chimeric mAbs, produced by genetic engineering, have the original target specificity of the murine precursor. Compared to the time-consuming and laborious mutations in development of humanized mAb, chimeric mAb is prepared by recombining of whole murine variable regions, not only CDRs, with human constant regions. Furthermore, in contrast to the repeating administration of the mAb against tumor, the dosage of the anti-infective mAb is not so frequent. Sometimes only a single dose is necessary before or after the exposure to the microorganism [19]. In this situation, chimeric mAb may have as fewer side effects as humanized and human mAbs.
In a previous study, we reported the production of a neutralizing murine mAb (coded LF8) against LF that blocks LeTx formation [20]. In this study, we develop a human/murine chimeric Fab mAb (coded LF8-Fab) which was generated by antibody engineering using LF8 variable regions combined with human constant regions. The LF8-Fab could bind LF specifically and protect J774A.1 cells against LeTx challenge in vitro under prophylactic and postexposure conditions. Our results suggest that this chimeric LF8-Fab mAb might be further characterized and potentially be used for clinical treatment of anthrax infection.
2. Materials and Methods
2.1. Murine LF8 and LeTx
Murine mAb against anthrax lethal factor (LF8) was developed and purified in our lab, as described previously [20]. Briefly, BALB/c mice were immunized with purified LF protein, and spleen cells were fused with P3X63AF8/653 myeloma cells using standard protocol. The LF8 was screened by ELISA, immune precipitation, Western blotting, and gel mobility-shifting assay. This murine mAb could inhibit LeTx both in vitro and in vivo. Purified LeTx (PA and LF) was produced in Dr. Nick Duesbery's lab at Van Andel Research Institute [21].
2.2. Construction of Expression Vector
Vectors pComb3XSS and pComb3XTT were kindly provided by the Barbas laboratory (Scripps Research Institute, USA). Total RNA was extracted from the LF8 hybridoma cells by the TRIzol reagent, and cDNA was synthesized using SuperScript II Reverse Transcriptase according to the manufacturer's protocols. The prokaryotic vector was constructed by cloning LF8-Fab into pComb3XSS following three rounds of PCR amplification as described previously [22]. First, murine variable regions of the heavy chain (VH) and the light chain (VL) were amplified by PCR using cDNA of LF8 as template. GAPDH severed as the internal control of RT-PCR using the primers (Forward: 5′-CCCTTCATTGACCTCAAC-3′ and Backward: TTCACACCCATCACAAAC) [23]. Human constant regions of both the heavy chain domain 1 (CH1) and the light chain (CL) were amplified by PCR using vector pComb3XTT as the template. Second, equimolar quantities of VH and CH1 PCR products were used in the overlap PCR to create the heavy chain Fd (Fd), while equimolar quantities of VL and CL PCR products were used to create the light chain. Third, equimolar quantities of Fd and light chain PCR products were used in the overlap PCR to create the Fab DNA. Then, LF8-Fab DNA was cloned into pComb3XSS following single digestion of restriction endonuclease Sfi I. The recombinant vector pComb3X/LF8-Fab was sequenced using an ABI 3700-capillary electrophoresis DNA sequencer. Sequences were further analyzed using the VBASE2 database (http://www.vbase2.org/).
2.3. Expression and Purification of Human/Murine Chimeric LF8-Fab
The recombinant vector pComb3X/LF8-Fab was transformed into competent E. coli HB2151. The LF8-Fab expression was induced by 1 mM isopropyl-β-D-thiogalactoside (IPTG) overnight at 30°C. Individual clones were identified by Western blotting using goat antihuman IgG (Fab-specific) and IRDye 800CW donkey antigoat IgG in an Odyssey infrared image system (LI-COR Biosciences, Lincoln, NE). Soluble LF8-Fab in the cell lysate was purified by immobilized metal affinity chromatography (IMAC) using a 5 mL His-trap HP column, followed by ion exchange chromatography (IEC) using a 5 mL Q sepharose column as described previously [24]. The purity of the LF8-Fab was examined by SDS-PAGE (10%) with coomassie blue staining.
2.4. Affinity Calculation of the LF8-Fab
The affinity of the LF8-Fab was calculated by noncompetitive ELISA [25]. Ninety-six-well EIA plates were coated overnight at 4°C with LF at two different concentrations, 4 μg/mL and 2 μg/mL. After the plate was blocked, serial concentrations of the LF8-Fab were added (4 replicated wells for each concentration) as the primary antibody. The alkaline phosphatase- (AP-) conjugated antihuman Fab severed as the secondary antibody. The absorbance was read at 405 nm after color development. The concentration of the LF8-Fab and the absorbance at 405 nm were plotted to two hyperbolic curves by GraphPad Prism software version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Affinity constant (K aff ) was calculated using SPSS statistical software version 13.0 (SPSS Inc., Chicago, IL, USA) by equation K aff = 1/(2[Ab′]t − [Ab]t), where [Ab′]t was the free mAb concentration at the OD50 of 2 μg/mL coated antigen, while [Ab]t was the free mAb concentration at the OD50 of 4 μg/mL coated antigen.
2.5. In Vitro LeTx Neutralization Assay
The LeTx in vitro neutralization assay was performed as described previously [26]. Murine macrophage J774A.1 cells, cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10% FBS, were seeded in 96-well plates (2 × 104 cells in 100 μL medium per well), 12 h prior to the assay. The LF8-Fab was serially diluted and mixed with LeTx (0.5 μg/mL of PA plus 0.1 μg/mL of LF). The mixture was applied to cells (3 replicated wells for each dilution). Untreated cells and cells with only LeTx acted as controls. Plate was incubated for 3 h at 37°C. Cell viability was determined using a CellTiter 96 AQueous assay. The concentration of the LF8-Fab and the percentage of viability were plotted to a hyperbolic curve by GraphPad Prism software. SPSS statistical software was used to calculate the 50% effective concentration (EC50).
2.6. In Vitro LeTx Challenge Study
To determine the protection effect of the LF8-Fab under prophylactic and postexposure conditions, cells were treated with either LeTx prior to the LF8-Fab or the converse at various time points (–120, –60, –30, –15, –5, 0, +5, +15, +30, +60, +75, +90, +105, and +120 min). Minus (–) means mAb treatment prior to LeTx, while plus (+) means LeTx prior to mAb treatment. The percentage of cell viability was calculated in the same way as above.
3. Results
3.1. Construction of Expression Vector
Total RNA was extracted from the LF8 hybridoma cells, and cDNA was synthesized. As expected, murine VH, VL, and human CH1 products were about 350 bp, while human CL product was about 400 bp. GAPDH severing as the internal control of RT-PCR was also amplified. Then, chimeric heavy chain Fd (about 750 bp), light chain (about 800 bp), and Fab (about 1500 bp) products were consequently achieved (Figure 1(a)). The recombinant vector pComb3X/LF8-Fab was constructed successfully according to sequencing analysis and could be recut by Sfi I (Figure 1(b)), indicating the integrity of the vector. The complementary determining region (CDR) and framework region (FR) of VH and VL were determined by VBASE2 database (Figure 2). The VH sequence was one member of the Igh-V15 VH15 family, while the VL sequence belonged to IGKV4/5 subgroup.
Figure 1.
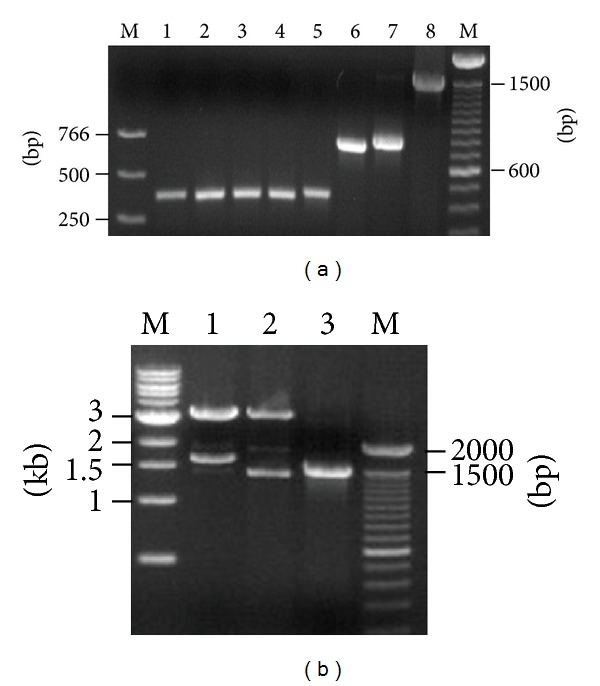
Construction of expression vector. (a) PCR products from three-round amplification. M, DNA marker; lane 1, GAPDH as internal control; lane 2, VH; lane 3, VL; lane 4, CH1; lane 5, CL; lane 6, heavy chain Fd; lane 7, light chain; lane 8, Fab. (b) Recombinant vector pComb3X/LF8-Fab recut by Sfi I. M, DNA marker; lane 1, pComb3XSS cut by Sfi I; lane 2, pComb3X/LF8-Fab recut by Sfi I; lane 3, PCR product of Fab.
Figure 2.

Nucleotide and deduced amino acid sequences of VH and VL. The CDRs are underlined based on the analysis of VBASE2 database. (a) Nucleotide sequence of VH and deduced amino acid sequence of VH. (b) Nucleotide sequence of VL and deduced amino acid sequence of VL.
3.2. Expression and Purification of Human/Murine Chimeric LF8-Fab
Recombinant vector pComb3X/LF8-Fab was transfected into competent E. coli HB2151. The LF8-Fab expression was induced by 1 mM IPTG overnight at 30°C and then identified by Western blotting in the Odyssey infrared image system. Both heavy chain Fd and light chain were expressed as the expected sizes (Figure 3(a)). The theoretical pI was 7.92, calculated by the Compute pI/Mw tool at ExPASy (http://web.expasy.org/compute_pi/). The optimized IEC eluent was start buffer plus 150 mM NaCl. SDS-PAGE (10%) followed by Coomassie Blue staining showed that heavy chain Fd and light chain were equally expressed, and the purity was above 95% (Figure 3(b)). The output level was about 1 mg purified protein from 1 L E. coli culture by BCA protein assay.
Figure 3.
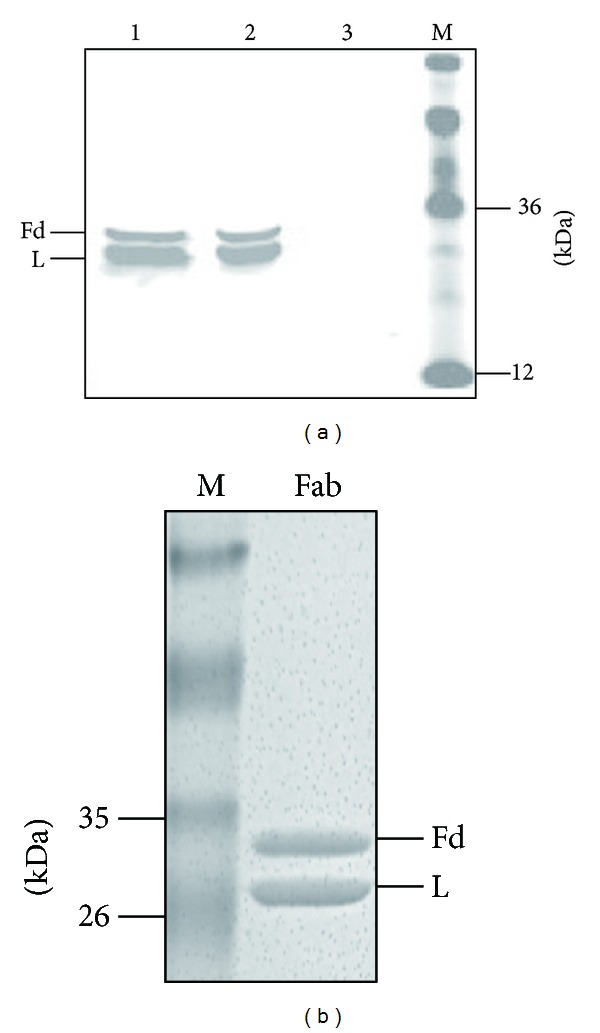
Expression and purification of the LF8-Fab. (a) A goat antihuman Fab and IRDye 800CW donkey antigoat IgG were used to detect Fab expression in Western blotting. Both heavy chain Fd (Fd) and light chain (L) were expressed. Lane 1, supernatant of sonicated lysate; lane 2, pellet of sonicated lysate; lane 3, lysate of untranfected E. coli HB2151 as negative control; lane 4, protein marker. (b) Coomassie blue staining showed that heavy chain Fd (Fd) and light chain (L) of the LF8-Fab were equally expressed after the purification of IMAC and IEC.
3.3. Affinity Calculation of the LF8-Fab
The concentration of the LF8-Fab and the absorbance at 405 nm were plotted to two hyperbolic curves using GraphPad Prism software (Figure 4). The LF8-Fab could identify LF (either at 4 μg/mL or 2 μg/mL) in a dose-dependent manner. Using SPSS statistical software, [Ab′]t was 20.5 nM and [Ab]t was 12.1 nM. According to the equation K aff = 1/(2[Ab′]t − [Ab]t), the K aff of the LF8-Fab was 3.46 × 107 L/mol.
Figure 4.
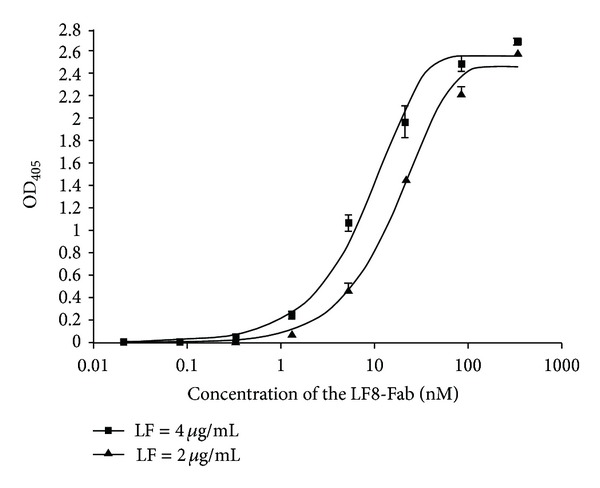
Affinity calculation of the LF8-Fab. Affinity was measured by noncompetitive ELISA. The concentration of the LF8-Fab and the absorbance at 405 nm were plotted to two hyperbolic curves using GraphPad Prism software. The LF8-Fab could identify LF in a dose-dependent manner with an affinity constant of 3.46 × 107 L/mol according to the calculation of SPSS statistical software.
3.4. In Vitro LeTx Neutralization Assay
The in vitro neutralization assay showed that the LF8-Fab could protect cells against LeTx in a dose-dependent manner and could offer 100% protection at a concentration of 5 μM (Figure 5(a)). In contrast, the irrelevant Fab could not protect cells in the presence of LeTx. The EC50 of the LF8-Fab was 85 μg/mL, according to the calculation of SPSS statistical software.
Figure 5.
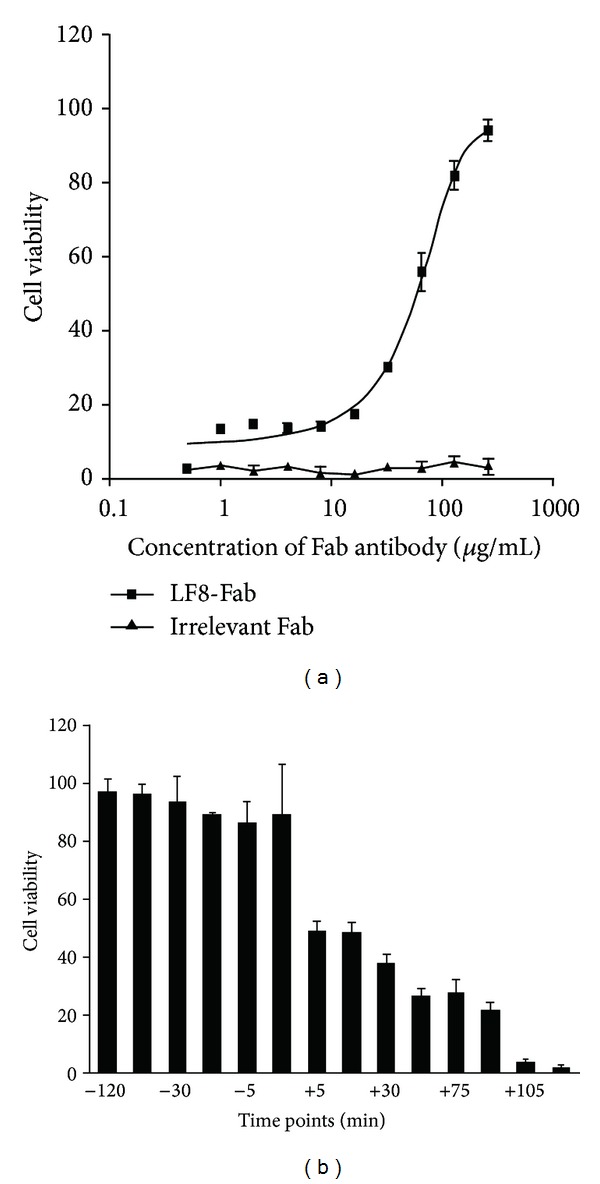
In vitro protection of J774A.1 cells against LeTx challenge. (a) In vitro neutralization assay showed that LF8-Fab could protect cells against LeTx in a dose-dependent manner, with an EC50 of 85 μg/mL according to calculation of SPSS statistical software. The percentage of viability was demonstrated by GraphPad Prism software. (b) In vitro LeTx challenge study to test neutralization effect of LF8-Fab before or after LeTx exposure of J774A.1 cells at different time points. Minus (−) means LF8-Fab prior to LeTx, while plus (+) means LeTx prior to LF8-Fab. LF8-Fab could completely neutralize LeTx and protect cells as long as the treatment was prior to LeTx addition. As for the converse situation, protection effect declined in a time-dependent manner.
3.5. In Vitro LeTx Challenge Study
In order to determine the protection effect under prophylactic and postexposure conditions, the LF8-Fab was chosen at a concentration of 100% protection (5 μM). LeTx and the LF8-Fab were added to cells at different time points. The results of cell viability indicate that the LF8-Fab could completely neutralize LeTx and protect cells if the mAb treatment was prior to LeTx addition. As for the converse situation when LeTx challenge was prior to the mAb treatment, protection effect declined in a time-dependent manner and had 37.8% protection at +30 min and 26.8% protection at +60 min (Figure 5(b)).
4. Discussion
In the present study, we have produced a human/mouse chimeric anti-LF Fab (LF8-Fab) in Escherichia coli which could specifically identify LF with an affinity of 3.46 × 107 L/mol and could neutralize LeTx in vitro with an EC50 of 85 μg/mL. Even after LeTx challenge prior to the mAb treatment, the LF8-Fab demonstrated protection of J774A.1 cells in vitro.
Some of anti-LF mAbs (5/9) listed in Table 1 are murine ones. The others include human, chimpanzee, and chimpanzee/human chimeric mAbs. Only one is Fab, while the rest mAbs are all IgGs. Chimeric mAb, like LF8-Fab, keeps a balance between murine mAb which has high affinity and human mAb which has low immunogenicity. Undoubtedly, human mAbs are the most desirable source for clinical application in human body, while molecular modifications of murine mAbs, including the chimeric and humanized mAbs, can reduce the immunogenicity and can retain the similar affinity and stability of murine mAbs as well. Compared to humanized mAb, generation of chimeric one is relatively time and labor saving. Chimeric mAb with two isoforms, Fab and IgG, has equivalent therapeutic effect and less side effect as humanized mAb, especially in the treatment of infection diseases. Fab mAb, consisting of light chain and heavy chain Fd, lacks Fc region which is not necessary for antibody binding. Moreover, Fc region may bind to Fc receptors (FcRs) on the surface of certain cells, including leukocytes, epithelial cells, endothelial cells, and hepatocytes [32]. Thus, Fc region may attenuate the specificity of IgG mAb and increase the dosage of antibody. Fab mAb has the advantage of eliminating nonspecific binding between Fc region and FcRs. Moreover, Fab mAb may penetrate tissues more efficiently, as it has smaller size than IgG mAb (only 1/3 the size of IgG mAb) [33].
Table 1.
List of neutralizing mAbs against anthrax LF.
| Authors | Source | Isoform | Reference |
|---|---|---|---|
| Little et al. (1990) | Murine | IgG1 | [27] |
| Zhao et al. (2003) | Murine | IgG | [20] |
| Lim et al. (2005) | Murine | IgG1 | [8] |
| Albrecht et al. (2007) | Human | IgG1 | [28] |
| Staats et al. (2007) | Murine | IgG1 | [29] |
| Chen et al. (2009) | Chimpanzee | Fab | [15] |
| Chen et al. (2009) | Chimpanzee/human | IgG1 | [15] |
| Kulshreshtha and Bhatnagar (2011) | Murine | IgG2b | [30] |
| vor dem Esche et al. (2011) | Human | IgG1 | [31] |
LF: lethal factor.
VBASE2 is used to analyze sequences of VH and VL of LF8-Fab. This is an integrative database of germ-line variable genes from the immunoglobulin loci of human and mouse, while all variable gene sequences are extracted from the EMBL-Bank [34]. It is often used to analyze variable regions of antibody sequences with intact FR4 information. According to this database, CDRs and FRs of VH and VL were determined. The VH sequence was a member of the IGH15 family, while the VL sequence belonged to IGKV4/5 subgroup. Sequences were also examined in the V-QUEST tool at IMGT database (http://www.imgt.org/), and similar results were achieved. The only differentia was that the VH sequence belonged to the IGH14 family by IMGT. This might be caused by the criterion of different databases. The pComb3X vectors (GenBank accession number AF268281) are phagemids for phage display and antibody expression [35]. In this study, pComb3XTT vector was employed as PCR template to amplify human CH1 and CL. The other vector, pComb3XSS, was used for Fab expression. The light chain and heavy chain Fd were expressed, respectively, and assembled in the periplasm of bacterium, and soluble Fab mAb was obtained. A number of techniques have used to purify Fab mAb, including antigen affinity, IMAC, IEC, protein L affinity, gel filtration, and so on [36]. As vector pComb3XSS has the His6 tag, so IMAC was carried out first. However, the relatively low purity of LF8-Fab of IMAC (about 60%) was mainly caused by the endogenous histidine residues in E. coli HB2151. So, it was necessary to make a secondary purification of IEC by Q sepharose column. According to the preliminary experiment of small scale, the optimized IEC eluent was start buffer plus 150 mM NaCl. This was consistent with the calculation by the Compute pI/Mw tool at ExPASy. With the combination of IMAC and IEC, the purity rose to 95% and could be used in further experiments.
Several methods have been established to measure affinity of mAbs, such as surface plasmon resonance (SPR) [37] and quartz crystal microbalance (QCM) [38]. Here we utilized a simple while reliable method based on noncompetitive ELISA, which utilizes the dose-response curve to calculate an affinity constant. This method is based upon the Law of Mass Action, using total antibody concentration added per well rather than the bound-to-free antigen ratio. It compares the OD50 of two sigmoid curves of antibody serial dilutions on a plate coated with the same antigen at two different concentrations. It is generally believed that the Fab (and other monovalents) fragment displays a relatively low affinity than the divalent constructs, such as the F (ab′)2 and the full-length immunoglobulin [39]. Compared to the affinity of the LF8 (data not shown), LF8-Fab revealed a moderate affinity against LF.
In in vitro assay, the molar ratio of PA and LF consisting of LeTx is the key to LeTx challenge. According to other studies [40] and our preliminary test, 0.5 μg/mL of PA to 0.1 μg/mL of LF is an appropriate ratio of LeTx. Under these concentrations, cell viability of J774A.1 cells dropped to zero in less than 2 h. Hence, employing LeTx 120 min prior to mAb treatment was the last time point under postexposure condition. Neutralization activities in vitro demonstrated that the LF8-Fab could protect J774A.1 cells well against LeTx challenge. When mixed with LeTx, LF8-Fab could protect cells against LeTx in a dose-dependent manner and offer 100% protection at a concentration of 5 μM. And then this concentration (5 μM) of the LF8-Fab was used to determine the protection effect under prophylactic and postexposure conditions. LeTx and the LF8-Fab were added to cells at different time points. As expected, the LF8-Fab could completely neutralize LeTx and protect J774A.1 cells if the mAb treatment was prior to LeTx challenge. On the reverse situation when the mAb treatment was posterior to exposure to LeTx, the LF8-Fab could partly retrieve the cells in a time-dependent manner. There was about 37.8% protection at +30 min and 26.8% protection at +60 min after exposure.
Further study will focus on several aspects of this mAb. First, the ability of the LF8-Fab to protect mice against LeTx challenge will be evaluated under both prophylactic and postexposure conditions. Second, the affinity needs to be improved by affinity maturation. Third, this mAb will be characterized in detail (i.e., specificity, toxicity studies, autoantigen testing, etc.). Last, epitope mapping and structure-function analysis of the murine LF8 mAb have been performed. Generally, it is believed that conversion from murine mAb to chimeric one may not change the interaction of antigen and antibody. However, the LF8-Fab lacks Fc region, compared to the LF8. So, mAb epitope analysis of the LF8-Fab is still worth investigating. The epitope mapping of Fab-LF interaction will bring out a better understanding of the neutralization mechanism of the LF8-Fab.
In summary, we report herein the development of a human/murine chimeric Fab mAb, the LF8-Fab, to reduce murine immunogenicity. The LF8-Fab can identify LF specifically with moderate affinity and can neutralize LeTx and protect the macrophage cells in vitro. Thus, we believe that LF8-Fab might be further characterized (i.e., specificity, toxicity studies, autoantigen testing, etc.) and potentially be used alone or in combination with other neutralizing mAbs for medical therapy of anthrax infection.
Conflict of Interests
All authors have declared that there is no financial conflict of interests in regard to this work.
Acknowledgments
The authors wish to thank Dr. David Petillo for DNA sequencing and Mr. David Nadziejka for editorial reading of the paper. This work was supported by Van Andel Research Institute.
References
- 1.Tournier J, Ulrich RG, Quesnel-Hellmann A, Mohamadzadeh M, Stiles BG. Anthrax, toxins and vaccines: a 125-year journey targeting Bacillus anthracis . Expert Review of Anti-Infective Therapy. 2009;7(2):219–236. doi: 10.1586/14787210.7.2.219. [DOI] [PubMed] [Google Scholar]
- 2.Jernigan JA, Stephens DS, Ashford DA, et al. Anthrax bioterrorism investigation team. Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerging Infectious Diseases. 2001;7(6):933–944. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweeney DA, Hicks CW, Cui X, Li Y, Eichacker PQ. Anthrax infection. American Journal of Respiratory and Critical Care Medicine. 2011;184(12):1333–1341. doi: 10.1164/rccm.201102-0209CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bromberg-White J, Lee C, Duesbery N. Consequences and utility of the zinc-dependent metalloprotease activity of anthrax lethal toxin. Toxins. 2010;2(5):1038–1053. doi: 10.3390/toxins2051038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scobie HM, Young JAT. Interactions between anthrax toxin receptors and protective antigen. Current Opinion in Microbiology. 2005;8(1):106–112. doi: 10.1016/j.mib.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Moayeri M, Leppla SH. The roles of anthrax toxin in pathogenesis. Current Opinion in Microbiology. 2004;7(1):19–24. doi: 10.1016/j.mib.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Kronhardt A, Rolando M, Beitzinger C, et al. Cross-reactivity of anthrax and C2 toxin: protective antigen promotes the uptake of botulinum C2I toxin into human endothelial cells. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0023133.e23133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim NK, Kim J, Oh MS, et al. An anthrax lethal factor-neutralizing monoclonal antibody protects rats before and after challenge with anthrax toxin. Infection and Immunity. 2005;73(10):6547–6551. doi: 10.1128/IAI.73.10.6547-6551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z, Moayeri M, Zhao H, Crown D, Leppla SH, Purcell RH. Potent neutralization of anthrax edema toxin by a humanized monoclonal antibody that competes with calmodulin for edema factor binding. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(32):13487–13492. doi: 10.1073/pnas.0906581106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klinman DM, Tross D. A single-dose combination therapy that both prevents and treats anthrax infection. Vaccine. 2009;27(12):1811–1815. doi: 10.1016/j.vaccine.2009.01.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bienek DR, Loomis LJ, Biagini RE. The anthrax vaccine: no new tricks for an old dog. Human Vaccines. 2009;5(3):184–189. doi: 10.4161/hv.5.3.7308. [DOI] [PubMed] [Google Scholar]
- 12.Cao S, Guo A, Liu Z, et al. Investigation of new dominant-negative inhibitors of anthrax protective antigen mutants for use in therapy and vaccination. Infection and Immunity. 2009;77(10):4679–4687. doi: 10.1128/IAI.00264-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scobie HM, Thomas D, Marlett JM, et al. A soluble receptor decoy protects rats against anthrax lethal toxin challenge. Journal of Infectious Diseases. 2005;192(6):1047–1051. doi: 10.1086/432731. [DOI] [PubMed] [Google Scholar]
- 14.Riddle V, Leese P, Blanset D, Adamcio M, Meldorf M, Lowy I. Phase I study evaluating the safety and pharmacokinetics of MDX-1303, a fully human monoclonal antibody against Bacillus anthracis protective antigen, in healthy volunteers. Clinical and Vaccine Immunology. 2011;18(12):2136–2142. doi: 10.1128/CVI.05059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z, Moayeri M, Crown D, et al. Novel chimpanzee/human monoclonal antibodies that neutralize anthrax lethal factor, and evidence for possible synergy with anti-protective antigen antibody. Infection and Immunity. 2009;77(9):3902–3908. doi: 10.1128/IAI.00200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pohl MA, Rivera J, Nakouzi A, Chow SK, Casadevall A. Combinations of monoclonal antibodies to anthrax toxin manifest new properties in neutralization assays. Infection & Immunity. 2013;81(6):1880–1888. doi: 10.1128/IAI.01328-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azinovic I, DeNardo GL, Lamborn KR, et al. Survival benefit associated with human anti-mouse antibody (HAMA) in patients with B-cell malignancies. Cancer Immunology, Immunotherapy. 2006;55(12):1451–1458. doi: 10.1007/s00262-006-0148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clementi N, Mancini N, Solforosi L, Castelli M, Clementi M, Burioni R. Phage display-based strategies for cloning and optimization of monoclonal antibodies directed against human pathogens. International Journal of Molecular Sciences. 2012;13(7):8273–8292. doi: 10.3390/ijms13078273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Migone T, Subramanian GM, Zhong J, et al. Raxibacumab for the treatment of inhalational anthrax. The New England Journal of Medicine. 2009;361(2):135–144. doi: 10.1056/NEJMoa0810603. [DOI] [PubMed] [Google Scholar]
- 20.Zhao P, Liang X, Kalbfleisch J, Koo H, Cao B. Neutralizing monoclonal antibody against anthrax lethal factor inhibits intoxication in a mouse model. Human Antibodies. 2003;12(4):129–135. [PubMed] [Google Scholar]
- 21.Bromberg-White JL, Duesbery NS. Biological and biochemical characterization of anthrax lethal factor, a proteolytic inhibitor of MEK signaling pathways. Methods in Enzymology. 2008;438:355–365. doi: 10.1016/S0076-6879(07)38025-7. [DOI] [PubMed] [Google Scholar]
- 22.Li C, Zhang F, Lin H, et al. Generation and characterization of the human neutralizing antibody fragment Fab091 against rabies virus. Acta Pharmacologica Sinica. 2011;32(3):329–337. doi: 10.1038/aps.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu J, Kren BT, Park CW, Bilgim R, Wong PY-P, Steer CJ. Erythroid-specific expression of β-globin by the sleeping beauty transposon for sickle cell disease. Biochemistry. 2007;46(23):6844–6858. doi: 10.1021/bi6024484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Zhu J, Zhao P, et al. In vitro efficacy of immuno-chemotherapy with anti-EGFR human Fab-Taxol conjugate on A431 epidermoid carcinoma cells. Cancer Biology and Therapy. 2007;6(6):980–986. doi: 10.4161/cbt.6.6.4197. [DOI] [PubMed] [Google Scholar]
- 25.Beatty JD, Beatty BG, Vlahos WG. Measurement of monoclonal antibody affinity by non-competitive enzyme immunoassay. Journal of Immunological Methods. 1987;100(1-2):173–179. doi: 10.1016/0022-1759(87)90187-6. [DOI] [PubMed] [Google Scholar]
- 26.Abboud N, Casadevall A. Immunogenicity of Bacillus anthracis protective antigen domains and efficacy of elicited antibody responses depend on host genetic background. Clinical and Vaccine Immunology. 2008;15(7):1115–1123. doi: 10.1128/CVI.00015-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Little SF, Leppla SH, Friedlander AM. Production and characterization of monoclonal antibodies against the lethal factor component of Bacillus anthracis lethal toxin. Infection and Immunity. 1990;58(6):1606–1613. doi: 10.21236/ada216203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albrecht MT, Li H, Williamson ED, et al. Human monoclonal antibodies against anthrax lethal factor and protective antigen act independently to protect against Bacillus anthracis infection and enhance endogenous immunity to anthrax. Infection and Immunity. 2007;75(11):5425–5433. doi: 10.1128/IAI.00261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staats HF, Alam SM, Scearce RM, et al. In vitro and in vivo characterization of anthrax anti-protective antigen and anti-lethal factor monoclonal antibodies after passive transfer in a mouse lethal toxin challenge model to define correlates of immunity. Infection and Immunity. 2007;75(11):5443–5452. doi: 10.1128/IAI.00529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulshreshtha P, Bhatnagar R. Inhibition of anthrax toxins with a bispecific monoclonal antibody that cross reacts with edema factor as well as lethal factor of Bacillus anthracis . Molecular Immunology. 2011;48(15-16):1958–1965. doi: 10.1016/j.molimm.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 31.vor dem Esche U, Huber M, Zgaga-Griesz A, et al. Passive vaccination with a human monoclonal antibody: generation of antibodies and studies for efficacy in Bacillus anthracis infections. Immunobiology. 2011;216(7):847–853. doi: 10.1016/j.imbio.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Kacskovics I, Mayer B, Kis Z, et al. Cloning and characterization of the dromedary (Camelus dromedarius) neonatal Fc receptor (drFcRn) Developmental and Comparative Immunology. 2006;30(12):1203–1215. doi: 10.1016/j.dci.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 33.Kameyama S, Okada R, Kikuchi T, et al. Distribution of immunoglobulin Fab fragment conjugated with HIV-1 REV peptide following intravenous administration in rats. Molecular Pharmaceutics. 2006;3(2):174–180. doi: 10.1021/mp050064m. [DOI] [PubMed] [Google Scholar]
- 34.Lim TS, Mollova S, Rubelt F, et al. V-gene amplification revisited—an optimised procedure for amplification of rearranged human antibody genes of different isotypes. New Biotechnology. 2010;27(2):108–117. doi: 10.1016/j.nbt.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Chassagne S, Laffly E, Drouet E, Hérodin F, Lefranc M, Thullier P. A high-affinity macaque antibody Fab with human-like framework regions obtained from a small phage display immune library. Molecular Immunology. 2004;41(5):539–546. doi: 10.1016/j.molimm.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y, Gutshall L, Jiang H, et al. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: papain digestion of mAb and transient expression in mammalian cells. Protein Expression and Purification. 2009;67(2):182–189. doi: 10.1016/j.pep.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Acchione M, Lipschultz CA, DeSantis ME, et al. Light chain somatic mutations change thermodynamics of binding and water coordination in the HyHEL-10 family of antibodies. Molecular Immunology. 2009;47(2-3):457–464. doi: 10.1016/j.molimm.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang D, Zhang D, Tang D, Ai H. Amplification of the antigen-antibody interaction from quartz crystal microbalance immunosensors via back-filling immobilization of nanogold on biorecognition surface. Journal of Immunological Methods. 2006;316(1-2):144–152. doi: 10.1016/j.jim.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 39.Huang J, Liang J, Tang Q, et al. An active murine-human chimeric Fab antibody derived from Escherichia coli, potential therapy against over-expressing VEGFR2 solid tumors. Applied Microbiology and Biotechnology. 2011;91(5):1341–1351. doi: 10.1007/s00253-011-3335-y. [DOI] [PubMed] [Google Scholar]
- 40.Wild MA, Xin H, Maruyama T, et al. Human antibodies from immunized donors are protective against anthrax toxin in vivo . Nature Biotechnology. 2003;21(11):1305–1306. doi: 10.1038/nbt891. [DOI] [PubMed] [Google Scholar]