

Abstract
Hypophosphatasia is a rare inherited disorder caused by deficient tissue-nonspecific alkaline phosphatase activity. It is classified into 6 subtypes, and the perinatal lethal form of hypophosphatasia is the most severe. Patients with this form suffer from various symptoms, including respiratory failure, premature craniosynostosis, rachitic changes in the metaphyses, convulsions and hypercalcemia. This report presents 6 cases of the perinatal lethal form of hypophosphatasia. All of the patients showed shortening of the long bones in utero in ultrasonographic examinations. Two of the six patients died at birth because they could not establish spontaneous breathing. Three of the remaining four patients also died before 1 yr of age. The major cause of death was respiratory failure due to hypoplastic lung. All of the patients, except for the two who died at birth, experienced convulsions in their clinical courses. Vitamin B6 therapy effectively reduced the frequency and severity of convulsions. However, it could not always make the patients convulsion free. Three patients underwent a genetic analysis. The 1559delT mutation, which abolishes Alkaline Phosphatase (ALP) activity, was a hot spot. A homozygous 1559delT mutation was observed in two patients. However, they differed in severity of symptoms. Although a good genotype-phenotype correlation has been reported in hypophosphatasia, the genotype alone does not always predict the life span of the patients. These cases therefore suggested the importance of genetic counseling.
Keywords: Hypophosphatasia, perinatal lethal form, alkaline phosphatase, ALP, ALPL
Introduction
Hypophosphatasia is caused by deficient tissue-nonspecific alkaline phosphatase (TNSALP) activity, thus resulting in hypomineralization of bone. The prevalence of severe forms is estimated at 1/100,000 (1). It has been classified into five subtypes based on the age of onset and the severity as follows: perinatal, infantile, childhood, adult and odontohypophosphatasia. Recently, the perinatal type was divided into lethal and benign types (2). Patients affected with the perinatal lethal form of hypophosphatasia tend to die around the time of birth due to impaired development of the lung and severe hypomineralization of their bones. On the other hand, patients with the perinatal benign form have a spontaneous improvement of skeletal defects despite the prenatal symptoms (3, 4).
Patients with the perinatal form of hypophosphatasia suffer from various symptoms, including respiratory failure associated with a narrow chest, premature craniosynostosis, rachitic changes in the metaphyses, uncontrollable convulsions and hypercalcemia. X-ray examination reveals shortening of the long bones, osteochondral spurs protruding from the forearms or legs and rachitic deformities of the chest (5). Laboratory examination shows markedly reduced serum alkaline phosphatase (ALP) activity as a characteristic finding. Furthermore, an increased urine phosphoethanolamine (PEA) levels are also a supportive finding.
Hypophosphatasia is caused by mutations in the ALPL gene, which encodes the TNSALP. More than 100 mutations have been reported in ALPL (1). Although some patients with the mild phenotype present autosomal dominant inheritance, the severe type of hypophosphatasia, including the perinatal lethal form, presents autosomal recessive inheritance (2).
This report presents 6 cases with the perinatal lethal form of hypophosphatasia.
Patient Reports
The characteristics of the 6 patients with the perinatal lethal form of hypophosphatasia are summarized in Table 1. None of the patients had an obvious family history suggesting hypophosphatasia, except for mildly low levels of serum ALP activity. After obtaining written informed consent during genetic counseling, a genomic analysis was performed in 3 patients. Genetic analysis was not performed in patient 1, 2 and 5 because we could not obtain approval for genetic analysis from their parents.
Table 1. Summary of the patients.
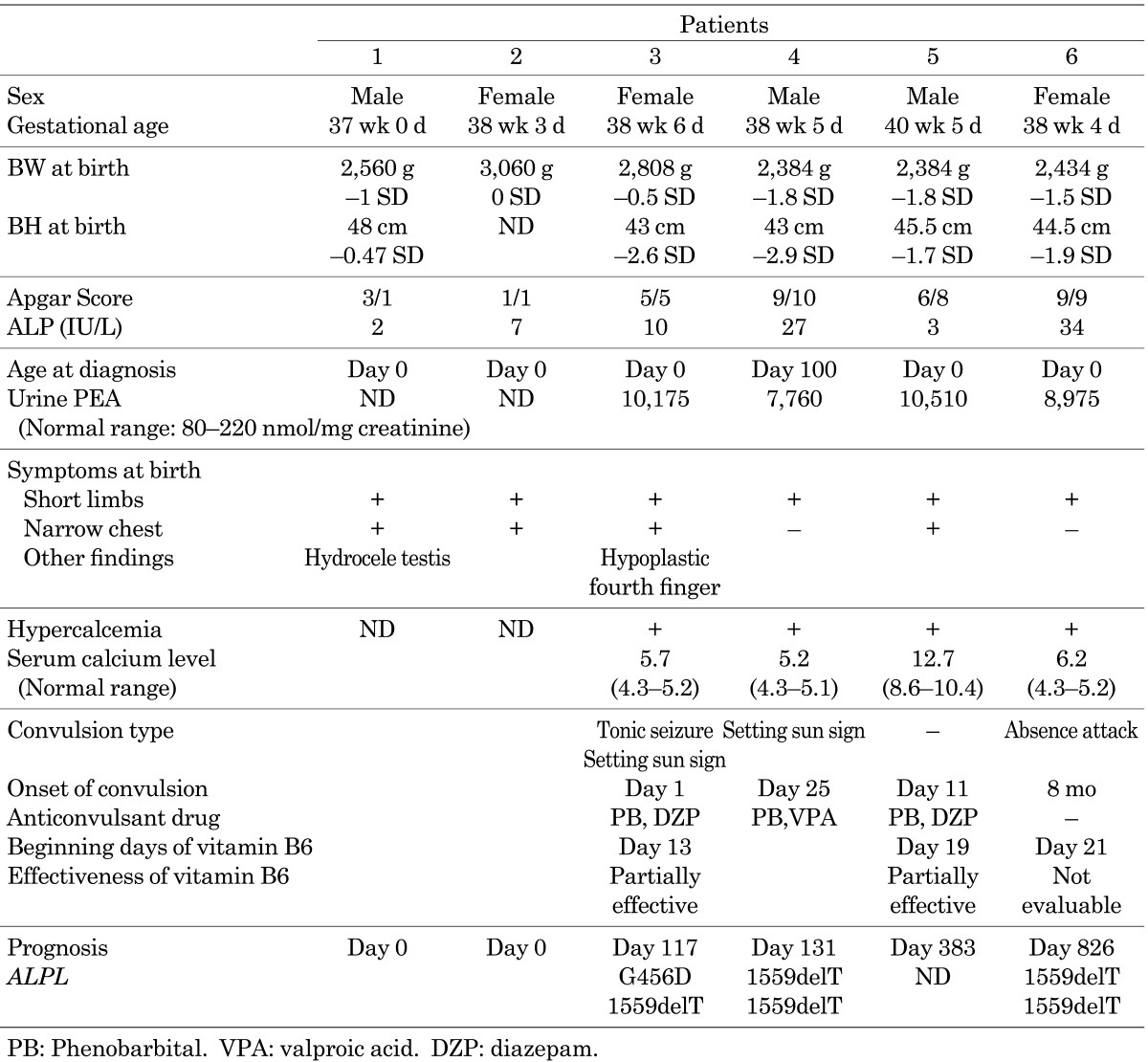
Shortening of the long bones and hydramnion was detected in Patient 1 in utero at 28 wk of gestation in an ultrasonographic examination. Aspiration of the amniotic fluid was performed at 36 wk of gestation. He was delivered by natural childbirth method at 37 wk of gestation. He presented bowing of the limbs, a narrow chest and hydrocele testis at birth. He could not establish spontaneous breathing and died within several hours. His serum ALP was 2 IU/l (normal range: 419–1,110). Severe hypomineralization of his bones was detected by X-ray and CT examination at birth.
Patient 2 manifested limb shortening, an enlarged biparietal diameter and hydramnion in utero at 29 wk of gestation in an ultrasonographic examination. She was born at 38 wk of gestation by Cesarean section. She presented bowing of the limbs and a narrow chest at birth. Her serum ALP was 7 IU/l. Her mother’s ALP was 175 IU/l. She could not establish spontaneous breathing and died within several hours.
Patient 3 had shortening of the long bones, which was detected in utero at 19 wk of gestation in an ultrasonographic examination. She was born at 38 wk of gestation by natural childbirth. She presented with bowed limbs, a narrow chest, an enlarged anterior fontanelle and a short right fourth finger at birth. Skeletal radiographs showed a narrow chest, undermineralized bones and fraying metaphyses. Her serum ALP was 10 IU/l on day 0. Her urine PEA was 10,174 nmol/mg creatinine (normal range: 80–220 nmol/mg creatinine). Artificial respiration was needed from birth to treat respiratory failure. She presented with sunset phenomenon and generalized seizures on day 1 and was treated with phenobarbital (PB). Vitamin B6 therapy (20 mg/kg/d) was started at 13 d of age. The vitamin B6 therapy was effective in reducing the frequency and severity of convulsion. She underwent a tracheotomy at 62 d of age. Despite the intensive treatment, she developed a pulmonary hemorrhage on day 102 and died from respiratory failure at 117 d of age. Sequence analyses revealed that the patient had compound heterozygous mutations, a single base pair substitution, G to A, at nucleotide 1418, 1418G>A (G456D) and a single base pair deletion at nucleotide 1559, 1559delT, in ALPL. And, her parents were heterozygous carrier.
Patient 4 was found to have short limbs in utero in an ultrasonographic examination. He was born at 38 wk of gestation. He presented with shortening of the limbs and was suspected to have osteogenesis imperfecta (OI). His first convulsion was observed on day 25. Artificial respiration was started at that time. Although multiple antiepileptic drugs, including valproic acid (VPA), PB, lidocaine and clonazepam were administered, they could not sufficiently suppress the convulsions. A low level of serum ALP (27 IU/l) was noted on day 100. Unfortunately, this was the first sampling of the serum ALP level. This data led to diagnosis of hypophosphatasia. His urine PEA level (7,760 nmol/mg creatinine) was elevated, and he and his parents were subjected to genetic testing. A sequence analysis revealed that the patient had homozygous 1559delT mutations in ALPL, and his parents were heterozygous carrier. In spite of the treatment, he died from respiratory failure at 131 d.
Patient 5 had short-limbs, which were detected in utero at 28 wk of gestation in an ultrasonographic examination. He was born at 40 wk of gestation by Cesarean section. He could not establish spontaneous breathing and was resuscitated by intubation. He presented a narrow chest and bowing of the limbs at birth. His serum ALP was 3 IU/l day 0, and his urine PEA level (10,510 nmol/mg creatinine) was elevated. The serum ALP level of the mother was 53 IU/l. His first convulsion was observed on day 11. Although multiple antiepileptic drugs, including VPA, PB and diazepam, were administered, none could sufficiently suppress his convulsions. Thereafter, vitamin B6 therapy was started on day 19. The vitamin B6 therapy was able to effectively reduce the frequency and severity of convulsion. He underwent a tracheotomy at 62 d of age. He died from respiratory failure at 383 d of age.
Patient 6 had short limbs and intra-uterine growth retardation, which were detected in utero at 32 wk gestation in an ultrasonographic examination. She was born at 38 wk of gestation by Cesarean section. Bowing of the long bones and rachitic changes in the metaphysis were detected by X-ray examination on day 1. She presented with short-limb syndrome and an enlarged anterior frontalle at birth. She had low levels of serum ALP (34 IU/l) and an elevated level of urine PEA (8,975 nmol/mg creatinine). Vitamin B6 therapy was started on day 21 to prevent convulsions. On day 87, she fractured a right rib. Subsequently, her respiratory distress progressed, and artificial ventilation was started on day 121. She received a tracheotomy on day 131. Her first convulsion was observed on day 144. The dose of vitamin B6 was increased from 10 mg/kg/d to 20 mg/kg/d. Thereafter, her convulsions were controlled. At 4 mo of age, she developed an enlarged liver and exhibited a deterioration of hepatic function. A prolonged prothrombin time and low level in the hepaplastin test were observed at 6 mo of age. Very low levels of clotting factor activity in V, VII, IX, XI and XII were observed. Vitamin K did not improve the low clotting activity. The laboratory findings for the levels of cholinesterase, fibrinogen, serum albumin and total bilirubin did not reflect severe cirrhosis of the liver, except for a relatively low platelet count (around 100,000 /µl). Therefore, the cause of the low clotting activity was unclear. In spite of these laboratory data, her general conditions gradually became stable at around 1 yr of age. She was able to eat food and hold toys with her hands at 2 yr of age. Digitate impressions were observed by cranial X-ray examinations at 2 yr old. A head CT scan revealed that she had a narrow cerebral ventricle and a brain fissure. Bulging of the anterior fontanelle was also observed. These findings suggested that the patient had developed craniostenosis. On day 822, she began to vomit and had a poor appetite without fever. She subsequently experienced intractable convulsions. A CT image taken after this convulsion episode did not show any remarkable changes from the previous study. In addition, an electroencephalogram study showed hypsarrhythmia. Therefore, it was not clear that the convulsions originated from either an elevation of the intracranial pressure or complications due to the primary disease. She died on day 826 due to respiratory failure associated with convulsions and a lung hemorrhage. A necropsy was not performed because consent could not be obtained from her parents. Sequence analyses revealed that the patient had homozygous deletions of 1559delT in ALPL. And, her parents were heterozygous carrier.
Discussion
This manuscript summarized 6 cases presenting with the perinatal lethal form of hypophosphatasia. All of the patients demonstrated findings such as shortening of the long bones, and 2 patients had hydramnion in utero according to ultrasonographic examinations. Zanki et al. noted that osseous spurs, a patchy ossification pattern, undermineralization of the thoracic spine and cupped metaphyses are specific ultrasonographic features of this condition (6). Patient 5 was suspected to have OI at birth. Unfortunately, the serum ALP level was not measured in this patient until day 100. Therefore, the diagnosis of hypophosphatasia was late. OI, rickets, achondrogenesis and hypochondrogenesis are thought to be among the major differential diagnoses of hypophosphatasia (2, 6). The lower levels of serum ALP are a helpful indicator to identify hypophosphatasia. Therefore, measurement of serum ALP activity is very important when ultrasonography reveals shortening of the long bones in utero.
The characteristic symptoms of the perinatal lethal form are respiratory failure, hypercalcemia and uncontrollable convulsion. Two of the six patients died at birth because they could not establish spontaneous breathing. The other 4 patients also needed artificial ventilation during their clinical courses. In spite of intensive care, including artificial ventilation, 3 of the remaining 4 patients died before one year of age. The major cause of death was respiratory failure. Hypercalcemia was observed in all of the patients except for the patients that died at birth. The use of low-calcium milk was effective in improving hypercalcemia. No other treatments of hypercalcemia, e.g., calcitonin and diuretic drugs, were needed to control hypercalcemia in the current study.
Three of the four patients, patients 3, 4 and 5, demonstrated refractory convulsions in their clinical courses. Such symptoms are thought to be a severe and frequent complication in patients with the perinatal lethal form of hypophosphatasia. Vitamin B6 therapy was selected for patients 3, 5 and 6. In order to control the convulsions, patients 3 and 5 were administered vitamin B6 in addition to other anticonvulsants. The vitamin B6 therapy effectively reduced the frequency and severity of their convulsions, but it could not always achieve complete resolution of the convulsions. Vitamin B6 was also administered to patient 6 to prevent convulsions. This patient experienced her first convulsions at day 144. The convulsions were suppressed by an increased dose of vitamin B6. Her first convulsions occurred later than those of the other 3 patients. However, considering the comparatively mild clinical course of this patient, it was not possible to verify whether the vitamin B6 therapy effectively prevented convulsions. Litmanovitz et al. reported a case of the perinatal lethal form that was effectively treated with vitamin B6 (7). The convulsions of the patient were immediately suppressed, and the vitamin B6 therapy resulted in her electroencephalogram to normal. The mechanisms of vitamin B6 action have not yet been fully elucidated; however, TNSALP is known to play a role in vitamin B6 metabolism (7, 8). Vitamin B6 therapy should therefore be considered one of the alternative treatments against uncontrollable convulsions associated with hypophosphatasia.
Craniostenosis is thought to be the one of major complications, and it was observed in one patient. Kozlowski et al. conducted a radiographic analysis of 24 cases of hypophosphatasia and suggested that decreased growth of the skull, a tense fontanelle and a palpable sutural ridge should be regarded as signs of craniostenosis, whatever the radiographic findings (9). They suggested a craniectomy before craniostenosis. Although the sudden exacerbation of the general condition of patient 6 was not suspected to be related to the uncontrollable elevated intracranial pressure based on a CT study, she may have benefitted from an early craniectomy.
We performed a genomic analysis for three patients, and all three of them had biallelic mutations in ALPL. Two of the patients had homozygous mutations of 1559delT, and one patient had compound heterozygous mutations of G456D/1559delT. These results suggest that the 1559delT mutation is a hot spot among Japanese patients with the perinatal lethal form of hypophosphatasia. Michigami et al. reported that the F310L and 1559delT mutations are hot spots in hypophosphatasia in Japan (1). In that study, three patients were identified with homozygous 1559delT mutations, and all three died shortly after birth. Their ALP enzymatic activity in vitro proved that the 1559delT mutation completely eliminated the activity of this enzyme (1). Previous reports have indicated a good correlation between the severity and in vitro enzymatic activity of the mutant protein (2). However, in the current study, homozygous 1559delT mutations were also identified in patient 6, who presented with a relatively mild phenotype of the perinatal lethal form of hypophosphatasia. The clinical course of patient 6 indicated that the prognosis could not always be predicted based on the results of a genetic analysis alone. In addition, similar phenomena have also been reported in previous studies. Ozono et al. reported sibling cases carrying the same ALPL compound heterozygous mutations, but who demonstrated different clinical courses from each other (10). Brun-Heath et al. reported the 1133A>T mutation in ALPL to be associated with an increase in the level of ALPL mRNA (11). Based on these data, an mRNA analysis, e.g., quantification of mRNA, should therefore be considered when the nature of a mutation cannot be fully explained the phenotype. Furthermore, Fauvert et al. reported that a particular haplotype, which derived from a sequence variation of the ALPL gene, could play a role as an aggravating factor (12). These results suggest that an additional genetic modifier and/or an environmental factor may therefore influence the severity in patients. Therefore, genetic counseling for hypophosphatasia should be carefully carried out.
This manuscript detailed 6 cases of the perinatal lethal form of hypophosphatasia. All of the cases exhibited shortening of the long bones and hydramnion in utero in ultrasonographic examinations. Measurement of serum ALP was a helpful indicator for diagnosis of hypophosphatasia. Although there is no curative treatment for hypophosphatasia, some treatments are useful for treating its symptoms. Recently, other challenging effective treatments, e.g., enzyme replacement therapy, transplantation therapy using bone fragments and cultured osteoblasts, have been reported (2, 13). Further study is needed to improve the prognosis and quality of life of patients with hypophosphatasia.
Acknowledgments
This work was supported in part by a grant from the Ministry of Health, Labour and Welfare of Japan (to K.O.).
References
- 1.Michigami T, Uchihashi T, Suzuki A, Tachikawa K, Nakajima S, Ozono K. Common mutations F310L and T1559del in the tissue-nonspecific alkaline phosphatase gene are related to distinct phenotypes in Japanese patients with hypophosphatasia. Eur J Pediatr 2005;164: 277–82 [DOI] [PubMed] [Google Scholar]
- 2.Mornet E. Hypophosphatasia. Orphanet J Rare Dis 2007;2: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore CA, Curry CJ, Henthorn PS, Smith JA, Smith JC, O’Lague P, et al. Mild autosomal dominant hypophosphatasia: in utero presentation in two families. Am J Med Genet 1999;86: 410–5 [DOI] [PubMed] [Google Scholar]
- 4.Pauli RM, Modaff P, Sipes SL, Whyte MP. Mild hypophosphatasia mimicking severe osteogenesis imperfecta in utero: bent but not broken. Am J Med Genet 1999;86: 434–8 [DOI] [PubMed] [Google Scholar]
- 5.Shohat M, Rimoin DL, Gruber HE, Lachman RS. Perinatal lethal hypophosphatasia: clinical, radiologic and morphologic findings. Pediatr Radiol 1991;21: 421–7 [DOI] [PubMed] [Google Scholar]
- 6.Zankl A, Mornet E, Wong S. Specific ultrasonographic features of perinatal lethal hypophosphatasia. Am J Med Genet A 2008;146A: 1200–4 [DOI] [PubMed] [Google Scholar]
- 7.Litmanovitz I, Reish O, Dolfin T, Arnon S, Regev R, Grinshpan G, et al. Glu274Lys/Gly309Arg mutation of the tissue-nonspecific alkaline phosphatase gene in neonatal hypophosphatasia associated with convulsions. J Inherit Metab Dis 2002;25: 35–40 [DOI] [PubMed] [Google Scholar]
- 8.Whyte MP, Mahuren JD, Fedde KN, Cole FS, McCabe ER, Coburn SP. Perinatal hypophosphatasia: tissue levels of vitamin B6 are unremarkable despite markedly increased circulating concentrations of pyridoxal-5’-phosphate. Evidence for an ectoenzyme role for tissue-nonspecific alkaline phosphatase. J Clin Invest 1988;81: 1234–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kozlowski K, Sutcliffe J, Barylak A, Harrington G, Kemperdick H, Nolte K, et al. Hypophosphatasia. Review of 24 cases. Pediatr Radiol 1976;5: 103–17 [DOI] [PubMed] [Google Scholar]
- 10.Ozono K, Yamagata M, Michigami T, Nakajima S, Sakai N, Cai G, et al. Identification of novel missense mutations (Phe310Leu and Gly439Arg) in a neonatal case of hypophosphatasia. J Clin Endocrinol Metab 1996;81: 4458–61 [DOI] [PubMed] [Google Scholar]
- 11.Brun-Heath I, Chabrol E, Fox M, Drexler K, Petit C, Taillandier A, et al. A case of lethal hypophosphatasia providing new insights into the perinatal benign form of hypophosphatasia and expression of the ALPL gene. Clin Genet 2008;73: 245–50 [DOI] [PubMed] [Google Scholar]
- 12.Fauvert D, Brun-Heath I, Lia-Baldini AS, Bellazi L, Taillandier A, Serre JL, et al. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterogosity for severe and moderate alleles. BMC Medical Genetics 2009;10: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whyte MP, Kurtzberg J, McAlister WH, Mumm S, Podgornik MN, Coburn SP, et al. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res 2003;18: 624–36 [DOI] [PubMed] [Google Scholar]