

Abstract
The 5-HT3B subunit was first cloned in 1999, and co-expression with the 5-HT3A subunit results in heteromeric 5-HT3AB receptors that are functionally distinct from homomeric 5-HT3A receptors. The affinities of competitive ligands at the two receptor subtypes are usually similar, but those of non-competitive antagonists that bind in the pore often differ. A competitive ligand and allosteric modulator that distinguishes 5-HT3A from 5-HT3AB receptors has recently been described, and the number of non-competitive antagonists identified with this ability has increased in recent years. In this review, we discuss the differences between 5-HT3A and 5-HT3AB receptors and describe the possible sites of action of compounds that can distinguish between them.
Keywords: 5-HT3, ligand gated, ion channel, 5-HT3A, 5-HT3B, 5-HT3AB, antagonist, Cys-loop, allosteric, ligand
5-HT3 receptor subtypes
There are seven families of 5-HT receptor (5-HT1 to 5-HT7), several containing multiple receptors, which are classified primarily on amino acid similarities and structural properties. The majority of these are GCPRs, with the 5-HT3 receptor (5-HT3R) being the only ligand-gated ion channel. The 5-HT3R also belongs to the Cys-loop receptor family, a group of membrane proteins that include nicotinic ACh (nACh), GABA and glycine receptors, which are responsible for fast synaptic neurotransmission in the CNS and peripheral nervous system (PNS). The first 5-HT3R subunit to be cloned was 5-HT3A, but when the protein was expressed in recombinant systems the functional response did not match those seen in some native tissues (Maricq et al., 1991). Many of the differences were resolved when the 5-HT3B subunit was later cloned (Davies et al., 1999; Dubin et al., 1999). While this subunit could not form functional receptors when expressed alone, in combination with 5-HT3A subunits it assembled as functional heteromers with altered properties. Since then, heteromeric receptors containing 5-HT3C, 5-HT3D or 5-HT3E subunits have been studied, but these receptors have 5-HT concentration–response curves and biophysical properties that are similar to homomeric 5-HT3A receptors, and only minimal pharmacology has been described to date (Niesler et al., 2007; Holbrook et al., 2009). Consequently, only the 5-HT3A and 5-HT3AB receptors will be reviewed here (see Niesler, 2011 and Walstab et al., 2010 for reviews on subunits 5-HT3C to 5-HT3E).
Currently, the main therapeutic application of 5-HT3R antagonists is in the management of chemotherapy-induced, radiation-induced, and post-operative nausea and vomiting, where a range of antagonists exhibit high levels of anti-emetic activity with a low incidence of serious adverse effects. There has been limited use of an antagonist for treating Irritable Bowel Syndrome, and partial agonists are also being explored for the same disorder (Gershon and Tack, 2007; Manning et al., 2011). There is also evidence that the 5-HT3R is involved in depression, drug and alcohol abuse, pruritis, cognitive and psychotic disorders, and pain (reviewed in Thompson and Lummis, 2007; Walstab et al., 2010). 5-HT3R ligands therefore have considerable therapeutic potential, and subtype-specific ligands could possibly target different conditions to provide a means for improved clinical benefit. Polymorphisms in HT3A and HT3B genes have been associated with several of these disorders, and to the clinical response to drug treatments, and therefore genetics may also provide opportunities for diagnostics and improved patient care (reviewed in Walstab et al., 2010). The 5-HT3R antagonists that are currently used in the clinic freely cross the blood brain barrier, making them suitable for treating neurological disorders, while non-permeant compounds could be used to target 5-HT3R subtypes in the PNS. To this end, Cappelli and co-workers have modified the physicochemical properties of 5-HT3R ligands to prevent them from crossing the blood brain barrier (Butini et al., 2009; Morelli et al., 2009; Cappelli et al., 2010; Modica et al., 2010). Ligands with activities at more than one receptor have also been described by the same group, and have the potential for treating disorders with complex aetiologies (Morelli et al., 2009; Cappelli et al., 2011). New ligands are still being discovered, and fragment library screens have recently been used to successfully identify novel allosteric modulators and competitive antagonists, with at least one showing 5-HT3R subtype selectivity. These studies demonstrate that the search for new 5-HT3R ligands is still valuable over 30 years after the discovery of the first antagonists, and that there may be several ways of targeting the more recently identified 5-HT3R subtypes (Thompson et al., 2010b; Trattnig et al., 2012; Verheij et al., 2012).
Differences between 5-HT3A and 5-HT3AB receptors
5-HT3AB receptors have been extensively investigated in heterologous systems, and when compared with homomeric 5-HT3R they have differing 5-HT concentration–response curves (increased EC50 values and shallower Hill slopes), increased single channel conductance (5-HT3A = sub-pS; 5-HT3AB = 16–30 pS), an increased rate of desensitization, reduced relative Ca2+ permeability and different current–voltage relationships (5-HT3A is inwardly rectifying, 5-HT3AB is linear) (Davies et al., 1999; Kelley et al., 2003; Livesey et al., 2008; Peters et al., 2010). However, it is worth noting that the extent of some of these differences can vary depending upon the species studied; for example, human 5-HT3A and 5-HT3AB receptors have a larger difference in their EC50 values than their rat or mouse counterparts (e.g. Hanna et al., 2000; Hayrapetyan et al., 2005; Stevens et al., 2005; Thompson and Lummis, 2008; Lochner and Lummis, 2010).
Early work suggested that the 5-HT3B subunit was only present in the PNS. Now there is evidence for its distribution in the human CNS, but expression in rodents is still disputed (reviewed in Jensen et al., 2008; Barnes et al., 2009; Walstab et al., 2010). In these studies, subtype-specific ligands could have provided a convenient method of probing the character of receptors, but it was only recently that ligands were identified that could distinguish 5-HT3A from 5-HT3AB receptors (Table 1). The majority of these are non-competitive antagonists (NCAs) that bind in the receptor pore, and their differences in potency reflect the differing pore-lining residues of the 5-HT3A and 5-HT3B subunits.
Table 1.
IC50 values derived from electrophysiological measurements at 5-HT3A and 5-HT3AB receptors
| Compound a | 5-HT3A IC50 (μM) | 5-HT3AB IC50 (μM) | Mode of action | Reference |
|---|---|---|---|---|
| 5-Hydroxyindole b | – | – | Competitive and non-competitive | Deiml et al., 2004; Hu and Lovinger, 2008 |
| α-Thujone b | 60 | – | Non-competitive | Deiml et al., 2004 |
| Bilobalide | 468 | 3100 | Non-competitive | Thompson et al., 2011a |
| Chloroquine | 24.3 | 23.6 | Competitive | Thompson and Lummis, 2008 |
| Diltiazem | 21.4 | 302 | Competitive and non-competitive | Thompson et al., 2011a |
| d-Tubocurarine | 3.4 | 14.2 | Competitive | Davies et al., 1999 |
| Etomidate | 180 | 140 | Non-competitive | Rusch et al., 2007 |
| Ginkgolide B | 727 | 3900 | Non-competitive | Thompson et al., 2011a |
| Irinotecan | 5.37 | 14.0 | Competitive | Nakamura et al., 2011 |
| Methadone | 14.1 | 41.1 | Competitive and non-competitive | Deeb et al., 2009 |
| Mefloquine | 0.66 | 2.70 | Competitive and non-competitive | Thompson and Lummis, 2008 |
| Morphine | 0.33 | 1.15 | Competitive and non-competitive | Baptista-Hon et al., 2012 |
| Pentobarbital | 520 | 270 | Non-competitive | Rusch et al., 2007 |
| Picrotoxin | 41.2c | 1135c | Non-competitive | Das and Dillon, 2005 |
| Picrotoxinin | 10.7 | 63.1 | Non-competitive | Thompson et al., 2011a |
| Propofol | 370 | 300 | Non-competitive | Rusch et al., 2007 |
| Quinine | 1.06 | 15.8 | Competitive and non-competitive | Thompson and Lummis, 2008 |
| VUF10166 | ND | 0.04 | Competitive and allosteric | Thompson et al., 2012b |
| Topotecan | 114.1 | 8.5d | Competitive | Nakamura et al., 2013 |
The affinities of the listed compounds may be different if measured by radioligand methods.
5-Hydroxyindole and α-thujone have complex actions on 5-HT3R function and concentration dependence has not been enumerated for some subtypes.
Values from mouse receptors; all others are from human receptors. ND = inhibition not determined as the dissociation of this ligand is too slow to make equilibrium measurements. It should be noted that several of these compounds also have effects at other receptor types; for example, diltiazem is better known as a voltage-gated calcium channel blocker for use in hypertension; irinotecan is an anti-cancer agent; and chloroquine, mefloquine and quinine are anti-malarial drugs.
EC50 value as topotecan potentiates 5-HT3AB receptor responses.
In contrast to the growing list of NCAs that can distinguish between the two 5-HT3R subtypes, the affinities of competitive antagonists are usually similar at 5-HT3A and 5-HT3AB receptors (Brady et al., 2001). The apparent affinities of agonists are slightly reduced at 5-HT3AB receptors, possibly as a result of their efficacy, but the differences are too small to be of practical use (Colquhoun, 1998). However, the first reports of competitive antagonist that discriminate between the two receptor subtypes were recently described, and we shall discuss the properties of these below. Between species, the affinities of competitive ligands can vary because of their differing binding site residues. One notable example is 3-(2-hydoxyl,4-methoxybenzylidine)-anabasine, which is a partial agonist at the human 5-HT3R and an antagonist at mouse receptors (Miyake et al., 1995; Hope et al., 1999; Zhang et al., 2006; 2007).
Ligand-binding sites in 5-HT3R
The 5-HT3R consists of five subunits that surround a central ion-conducting pore. Each of the subunits contains three functional domains. The extracellular domain (ECD) is responsible for agonist binding, the transmembrane domain (TMD) forms a channel that allows ions to cross the cell membrane, and the intracellular domain influences channel conductance, receptor trafficking and intracellular modulation (Thompson et al., 2010a). The majority of ligands described to date bind at two main regions of the 5-HT3R: the orthosteric binding site (where the native agonist binds) and the channel (Figure 1).
Figure 1.

Binding sites in 5-HT3R. (A) The 5-HT3R consists of five subunits that surround a central ion-conducting pore that is shown here from the side (left) and from the extracellular side (right) of the cell membrane. The orthosteric binding site (red) is located in the extracellular domain at the interface of two subunits (green and blue). The transmembrane domain consists of four α-helices (M1–M4) from each subunit, and the pore is formed by the convergence of five M2 α-helices (yellow); M1–M4 of the two facing subunits have been removed to view the pore more clearly. Competitive antagonists bind to the orthosteric site and the majority of non-competitive antagonists to the channel. Hydrophobic ligands may bind in inter-subunit cavities at the top of transmembrane domain α-helices. The orthosteric binding site is seen in more detail in Figure 2. (B) An alignment of amino acids that form the M2 α-helices (left) in a range of receptors, and their locations in the pore (right). Channel-lining residues mentioned in the text are highlighted as white on grey. The box shows the extent of the M2 α-helix as described by Hilf and Dutzler (2008). Accession numbers for the alignment are: human 5-HT3A P46098, mouse 5-HT3A Q6J1J7, human 5-HT3B O95264, mouse 5-HT3B Q9JHJ5, human GABA σ P24046, human GABA α1 P14867, GABA β2 P47870, GABA γ2 P18507, Glycine α1 P23415, Glycine β P48167, GluCl G5EBR3.
The orthosteric binding site is located in the ECD at the interface of two adjacent subunits, and is formed by six distinct peptide loops (Figures 1A & 2). These are loops A–C from one subunit (termed the principal or ‘ + ’ face) face and loops D–E from the other (complementary or ‘–’). The molecular determinants of agonists and competitive antagonists that bind here are reviewed in Thompson et al. (2010a).
Figure 2.
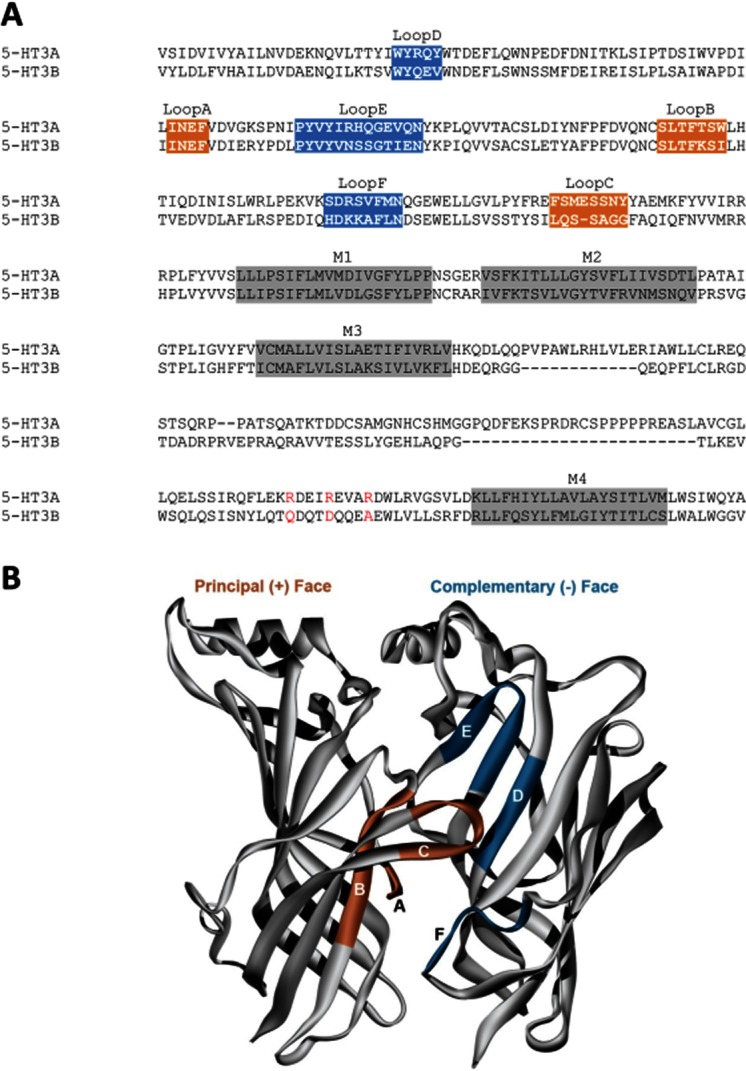
Human 5-HT3A and 5-HT3B subunits. (A) A protein sequence alignment highlighting the binding loops of the ECD and the α-helices of the transmembrane domain (M1–M4). The orange and blue colours show the residues that are shown in (B); a description of specific ligand–receptor interactions in the orthosteric binding site of the 5-HT3R can be found in a review by Thompson et al. (2010a). The channel can also be seen in Figure 1. Accession numbers for the human sequences are 5-HT3A P46098, 5-HT3B O95264. (B) A homology model of the 5-HT3A receptor extracellular domain, showing binding loops A–C of the principal (orange) face and D–F of the complementary (blue) face. Only two of the five subunits are shown for clarity.
Compounds that interact at locations other than the orthosteric binding site often do so in the TMD. The TMD of each subunit consists of four α-helices (M1–M4), with M2 from each subunit coming together to form the central ion-conducting pore (Figure 1B). To facilitate comparisons between the M2 regions of different subunits, residues are referred to by an index number, with 0′ being a conserved positively charged residue at the cytoplasmic end of the α-helices. Many of the NCAs that bind in the pore inhibit the 5-HT3R by blocking the channel, but allosteric modulators can also influence channel gating by binding to sites such as the inter-subunit region at the extracellular ends of the TMD α-helices (Trattnig et al., 2012).
The location of orthosteric binding sites in 5-HT3AB receptors
The pentameric structure of the 5-HT3R was first observed using electron microscopy in 1995 (Boess et al., 1995). Homomeric receptors consist of five identical 5-HT3A subunits, but the stoichiometry of heteromeric 5-HT3AB receptors has been more difficult to determine. In 2005, it seemed that this problem had finally been solved when atomic force microscopy (AFM) was used to measure angles between antibodies on dual-labelled receptors consisting of co-expressed epitope-tagged human 5-HT3A-myc and 5-HT3B-V5 subunits (Barrera et al., 2005). For 5-HT3AB receptors the results were unequivocal, showing that when samples were labelled with anti-Myc, the angle between two antibodies was 144°, with angles of both 72° and 144° being observed with anti-V5 antibodies. These results showed that in heteromeric receptors, more than one copy of both 5-HT3A and 5-HT3B subunits were present in the receptor, that 5-HT3A-myc subunits were always separated by another subunit, and that 5-HT3B-V5 subunits were either adjacent or separated by another subunit. The only possible arrangement of the five subunits was B-B-A-B-A, reminiscent of the α and non-α subunit arrangements in the heteromeric nACh (e.g. δ-β1-α1-ε-α1, β2-β2-α4-β2-α4) and GABAA (e.g. γ2-β2-α1–2β-α1) receptors.
However, it is difficult to understand how such a large diversity of competitive antagonists could have similar affinities at 5-HT3A and 5-HT3AB receptor-binding sites when the B-B-A-B-B arrangement does not contain the A+A− interface found in 5-HT3A receptors. Additionally, both homomeric and heteromeric receptors are activated by the same agonists despite the different binding sites this stoichiometry imposes. One proposal suggested that the A+B− interfaces of the heteromeric receptor are equivalent to the A+A− binding sites found in the homomer (Moura Barbosa et al., 2010), but a more likely reason is that a common A+A− binding site exists in both receptor types. To investigate this, Lochner and Lummis (2010) exchanged residues in mouse 5-HT3A and 5-HT3B subunits to determine the effects these substitutions had on 5-HT3R radioligand binding and function. They found that substitutions in the 5-HT3A subunit altered antagonist binding and 5-HT activation, but equivalent substitutions in the 5-HT3B subunits had no effects, indicating that only 5-HT3A subunits form the orthosteric binding site. Thompson et al. (2011c) supported this conclusion using disulphide trapping between cysteines on either side of the binding pocket of the human 5-HT3R. Several residue combinations were tested and pairings between loops C–E and loops C–F were identified. These 5-HT3A subunit double mutants were expressed as both homomeric receptors and in combination with 5-HT3B subunits, and in both receptor types a response to 5-HT was not seen until the disulphide bonds were reduced by DTT. Removal of DTT allowed a gradual reduction in peak current amplitude of subsequent 5-HT responses as the disulphide bonds reformed. These experiments demonstrated that an A+A− interface is essential for agonist activation in both receptor types, an interface that is absent from the B-B-A-B-A stoichiometry that was reported using AFM. However, there are several other possible reasons for the differences between these studies, including external factors such as temperature, ratios of subunit DNA transfected, expression systems, added epitope tags and differences in endogenous levels of chaperones. It is also possible that while the studies of Lochner and Lummis (2010) and Thompson et al. (2011c) examined only functional cell-surface receptors, AFM may have detected both intracellular and cell-surface receptors that could differ in their stoichiometries.
Other experiments have also supported the presence of an A+A− interface in both receptor types (Thompson et al., 2011c). Single cysteine substitutions to binding-site residues on the principal or complementary faces of 5-HT3A subunits showed that the majority affected the 5-HT EC50 and the binding affinity of the 5-HT3-specific competitive antagonist [3H]granisetron, regardless of whether they were expressed as homomeric or heteromeric receptors. Further changes were seen when (2-aminoethyl)methanethiosulfonate hydrobromide (MTSEA) was applied to these mutant receptors, a reagent that covalently modifies cysteine residues, adding bulk and thereby limiting access to the ligand-binding site; co-applying MTSEA with 5-HT3R ligands protected the residues, confirming their location in the binding site. In contrast, the 5-HT3B subunit substitutions did not alter 5-HT function or granisetron binding, suggesting that neither the principal nor the complementary faces of this subunit bind ligand or activate the receptor. These results support a stoichiometry that contains an A+A− interface in heteromeric receptors. However, to provide further support for this stoichiometry, evidence from other techniques is needed, such as that from high-resolution structural data Förster resonance energy transfer, total internal reflection fluorescence microscopy and reporter mutations that have been used on other Cys-loop receptors (Chang et al., 1996; Boorman et al., 2000; Durisic et al., 2012; Srinivasan et al., 2012).
Competitive antagonists with differing properties at 5-HT3A and 5-HT3AB receptors
The majority of competitive ligands have similar affinities at 5-HT3A and 5-HT3AB receptors (Table 1, Figure 3; Brady et al., 2001; Low et al., 2001). The ligand VUF10166, however, is unusual because it distinguishes between the two receptor types via their binding sites. VUF10166 displaces [3H]granisetron with sub-nanomolar affinity at 5-HT3A receptors, and shows surmountable effects on [3H]granisetron saturation binding curves, indicating competitive antagonism (Thompson et al., 2012b). Dissociation of [3H]granisetron in the presence of excess VUF10166 is best fit by a single exponential decay, suggesting that there is a single population of binding sites. When the 5-HT3B subunit is co-expressed these properties are changed. The affinity of VUF10166 is lower at 5-HT3AB receptors, and [3H]granisetron saturation-binding curves are insurmountable, indicating a non-competitive behaviour. [3H]granisetron dissociation in the presence of excess VUF10166 is also altered; 5-HT3AB receptors have two rates, one that is similar to 5-HT3A receptors and another that is more rapid. The faster dissociation rate is eliminated when substitutions are made to the complementary face of the 5-HT3B subunit (B–), but is unaffected by substitutions to the principal face (B+), indicating an interaction of VUF10166 at an A+B− interface.
Figure 3.

Examples of electrophysiological ( ) and radioligand binding (
) and radioligand binding ( ) measurements at human 5-HT3A and 5-HT3AB receptors. (A) Concentration–response curves differ at human 5-HT3A and 5-HT3AB receptors. Higher concentrations of 5-HT are needed to elicit a current response at 5-HT3AB receptors and the slope of the curves differs. Parameters derived from these curves are: 5-HT3A, pEC50 = 5.76 ± 0.03, EC50 = 1.74 μM, nH = 2.3, n = 6 and 5-HT3AB, pEC50 = 4.53 ± 0.04, EC50 = 29.5 μM, nH = 1.0, n = 6. (B) Saturation binding with the radioligand [3H]granisetron shows that like many other competitive ligands it has the same affinity at 5-HT3A and 5-HT3AB receptors. Kd values for these representative curves were 0.21 and 0.19 nM for 5-HT3A and 5-HT3AB receptors respectively. (C) Like many other non-competitive antagonists, the sensitivity of 5-HT3R currents to PTX differs at the two receptor types. Parameters derived from these curves are: 5-HT3A, pIC50 = 5.02 ± 0.09, IC50 = 9.55 μM, nH = 0.7, n = 9 and 5-HT3AB, pIC50 = 4.26 ± 0.06, IC50 = 55.0 μM, nH = 0.7, n = 5. (D) VUF10166 is unusual as this competitive antagonist has differing affinities at 5-HT3A and 5-HT3AB receptors. Kd values for these representative curves were 0.08 and 12.6 nM for 5-HT3A and 5-HT3AB receptors respectively. (E) Similar to many other NCAs, the sensitivity of 5-HT3R currents to DTZ also differs at 5-HT3A and 5-HT3AB receptors. Mutagenesis has shown that DTZ has a pore-binding site in the 5-HT3A receptor that is responsible for its increased potency relative to 5-HT3AB receptors. Parameters derived from these curves are: 5-HT3A, pIC50 = 4.68 ± 0.07, IC50 = 20.9 μM, nH = 0.8, n = 7 and 5-HT3AB, pIC50 = 3.53 ± 0.01, IC50 = 295 μM, nH = 0.8, n = 5. (F) In contrast to the electrophysiological measurements shown in panel (E), radioligand competition binding studies show that the binding affinity of DTZ is the same at 5-HT3A and 5-HT3AB receptors. This is consistent with the majority of other competitive antagonists that also have similar binding affinities at the two receptor types. Ki values for these representative curves were 180 μM for 5-HT3A receptors and 169 μM for 5-HT3AB receptors.
) measurements at human 5-HT3A and 5-HT3AB receptors. (A) Concentration–response curves differ at human 5-HT3A and 5-HT3AB receptors. Higher concentrations of 5-HT are needed to elicit a current response at 5-HT3AB receptors and the slope of the curves differs. Parameters derived from these curves are: 5-HT3A, pEC50 = 5.76 ± 0.03, EC50 = 1.74 μM, nH = 2.3, n = 6 and 5-HT3AB, pEC50 = 4.53 ± 0.04, EC50 = 29.5 μM, nH = 1.0, n = 6. (B) Saturation binding with the radioligand [3H]granisetron shows that like many other competitive ligands it has the same affinity at 5-HT3A and 5-HT3AB receptors. Kd values for these representative curves were 0.21 and 0.19 nM for 5-HT3A and 5-HT3AB receptors respectively. (C) Like many other non-competitive antagonists, the sensitivity of 5-HT3R currents to PTX differs at the two receptor types. Parameters derived from these curves are: 5-HT3A, pIC50 = 5.02 ± 0.09, IC50 = 9.55 μM, nH = 0.7, n = 9 and 5-HT3AB, pIC50 = 4.26 ± 0.06, IC50 = 55.0 μM, nH = 0.7, n = 5. (D) VUF10166 is unusual as this competitive antagonist has differing affinities at 5-HT3A and 5-HT3AB receptors. Kd values for these representative curves were 0.08 and 12.6 nM for 5-HT3A and 5-HT3AB receptors respectively. (E) Similar to many other NCAs, the sensitivity of 5-HT3R currents to DTZ also differs at 5-HT3A and 5-HT3AB receptors. Mutagenesis has shown that DTZ has a pore-binding site in the 5-HT3A receptor that is responsible for its increased potency relative to 5-HT3AB receptors. Parameters derived from these curves are: 5-HT3A, pIC50 = 4.68 ± 0.07, IC50 = 20.9 μM, nH = 0.8, n = 7 and 5-HT3AB, pIC50 = 3.53 ± 0.01, IC50 = 295 μM, nH = 0.8, n = 5. (F) In contrast to the electrophysiological measurements shown in panel (E), radioligand competition binding studies show that the binding affinity of DTZ is the same at 5-HT3A and 5-HT3AB receptors. This is consistent with the majority of other competitive antagonists that also have similar binding affinities at the two receptor types. Ki values for these representative curves were 180 μM for 5-HT3A receptors and 169 μM for 5-HT3AB receptors.
Functional studies also reveal differences. VUF10166 potently inhibits 5-HT-induced responses at 5-HT3A and 5-HT3AB receptors expressed in oocytes, but recovery from inhibition is much faster at 5-HT3AB receptors, consistent with the more rapid dissociation seen in radioligand-binding studies (Table 1). At homomeric receptors, VUF10166 also elicits a partial agonist response (Rmax = 0.24) at micromolar concentrations, followed by a long-lived inhibition of subsequent responses, possibly due to receptors slowly accumulating in a ligand-bound desensitized state, as has been observed for other 5-HT3R agonists (van Hooft and Vijverberg, 1996). Similar to the binding described above, substitutions to the complementary face of the 5-HT3B subunit (B–) produce receptors with recovery rates more similar to those from 5-HT3A receptors containing only A+A− binding sites, supporting the hypothesis that the interaction of VUF10166 at an A+B− interface is responsible for the observed differences between the homomeric and heteromeric receptors. Therefore, at 5-HT3A and 5-HT3AB receptors, VUF10166 binds in the orthosteric binding site formed at A+A− interfaces, but at 5-HT3AB receptors it also binds to an A+B− binding site from where it may allosterically increase the dissociation of ligands bound to the A+A− binding site (Thompson et al., 2012b). Differing effects of topotecan at 5-HT3A and 5-HT3AB receptors were also reported during the preparation of this review. At high micromolar concentrations 5-HT3A receptors currents are competitively inhibited by topotecan while 5-HT3AB receptor currents are potentiated, a property that is influenced by a 5-HT3B subunit mutation (Y129C) that lies outside of the binding site (Nakamura et al., 2013).
Other ligands that may bind to sites other than the orthosteric binding site include d-tubocurarine and azasetron. These ligands inhibit 5-HT3A receptor currents with differing potencies than those from 5-HT3AB receptors, but radioligand binding shows they have the same affinities at both 5-HT3A and 5-HT3AB receptors (Davies et al., 1999; Dubin et al., 1999; Brady et al., 2001). As the binding and functional studies give different results, it is possible that these ligands also bind elsewhere or they are slow to reach equilibrium, meaning that current responses desensitize before the full antagonist effects are seen, a property that could particularly influence inhibition at the more rapidly desensitizing 5-HT3AB receptor.
Non-competitive antagonists
A range of NCAs can discriminate between 5-HT3A and 5-HT3AB receptors (Table 1, Figure 3). Picrotoxin (PTX) is a well-known GABAAR channel blocker that blocks many other Cys-loop receptors, and was one of the first to be studied in detail at the 5-HT3R (Das et al., 2003a). Its potency at 5-HT3AB receptors is lower than at 5-HT3A receptors, and substitution of 5-HT3A subunit channel-lining residues has shown that PTX binds close to the 6′ position of M2 (Das and Dillon, 2003b; Thompson et al., 2011a). PTX binding is also influenced by substitutions at the 9′ and 12′ residues, which may affect the passage of this NCA as it descends through the narrowest region (9′–13′) of the pore to its binding site at the 6′ position (Thompson et al., 2011a). In GABAA, glycine and glutamate-gated chloride channels (GluCl), PTX acts at or close to the −2′, 2′ and 6′ residues, demonstrating that it can reach below the channel gate (9′) to exert its actions in all PTX-sensitive Cys-loop receptors (Ffrench-Constant et al., 1993; Gurley et al., 1995; Hawthorne et al., 2006; Hibbs and Gouaux, 2011).
Other channel-blocking compounds might similarly be expected to distinguish 5-HT3A and 5-HT3AB receptors. This is indeed the case for bilobalide (BB) and ginkgolide B (GB) that have binding sites that overlap with the structurally related PTX and are ∼6-fold less potent at 5-HT3AB than at 5-HT3A receptors (Thompson et al., 2011b). It is possible that differing residues in the channel of the 5-HT3B subunit are responsible for the lower potency at heteromers because substitution of the 6′ and 12′ positions of the 5-HT3A abolishes BB and GB inhibition. Additionally, an alanine substitution at the 2′ position in 5-HT3A receptors causes GB to be trapped in the channel following pore closure, and is only relieved following several subsequent agonist applications. Similar ligand trap has been reported for BB, GA and PTX at glycine receptors, highlighting the similarities of action that NCAs often share at other members of the Cys-loop family (Hawthorne and Lynch, 2005; Bali and Akabas, 2007).
Diltiazem (DTZ) is another inhibitor that has a lower potency (∼14-fold) at 5-HT3AB receptors than 5-HT3A receptors in functional studies. It has mixed (competitive and non-competitive) antagonism, and probing the competitive component using [3H]granisetron binding reveals similar DTZ affinity at both receptor types, consistent with most other competitive antagonists. The non-competitive component is only present in the 5-HT3A receptor and is due to DTZ acting in the pore at higher concentrations. This component is voltage dependent and substitution of the 7′ and 12′ positions abolishes the non-competitive antagonism found in homomeric receptors (Gunthorpe and Lummis, 2011a).
Morphine also shows mixed antagonism consisting of a low-affinity (μM) competitive component and higher affinity non-competitive component (Baptista-Hon et al., 2012). At both 5-HT3A and 5-HT3AB receptors, inhibition of 5-HT currents is surmountable when morphine is co-applied, and radioligand competition reveals similar binding affinities (Brady et al., 2001; Baptista-Hon et al., 2012). The non-competitive component becomes apparent when morphine is pre-applied, and its potency is reduced ∼4-fold in the presence of the 5-HT3B subunit. However, the binding site for the non-competitive component is unlikely to be in the channel because inhibition is not voltage dependent and substitution of the whole of the 5-HT3A subunit M2 region with the aligning 5-HT3B sequence does not affect morphine potency. Therefore, the location of the binding site for the non-competitive component is still unknown. Methadone is an analogue of morphine that is also fourfold less potent at 5-HT3AB receptors than at 5-HT3A receptors in functional studies. Its inhibition at 5-HT3A receptors is surmountable, but at 5-HT3AB receptors it is insurmountable and voltage dependent, suggesting that unlike morphine, methadone may act in the pore of heteromeric receptors (Deeb et al., 2009).
The antimalarial compounds quinine and mefloquine also show mixed antagonism (Thompson and Lummis, 2008). Quinine is a competitive antagonist at 5-HT3A receptors, but its potency is 15-fold less at 5-HT3AB receptors where it also has non-competitive actions (Table 1). In comparison, mefloquine has mixed competitive and non-competitive effects at both 5-HT3A and 5-HT3AB receptors, but is fourfold less potent at the heteromer. For both quinine and mefloquine the non-competitive activity is dependent upon pre-application, and for mefloquine a small voltage dependence in the 5-HT3A receptor (5-HT3AB was not tested) suggests channel binding (Thompson et al., 2007). The closely related antimalarial chloroquine is solely competitive at both receptor types, and does not display differences in potency, consistent with competitive antagonists having similar affinities at both receptor types.
The fact that NCAs can distinguish between 5-HT3A and 5-HT3AB receptors provides a practical means of discriminating between the 5-HT3R subtypes in functional studies, such as electrophysiological experiments in transfected cells (e.g. Thompson et al., 2011c), and their relatively low potency means that agonist responses often recover quickly following inhibition. These compounds, however, are unlikely to have therapeutic applications as many also affect other members of the Cys-loop family, or entirely different receptor classes and cellular pathways.
Modulators of 5-HT3R function
Allosteric modulators bind to regions that are distinct to the orthosteric binding site and can alter agonist sensitivity, agonist efficacy and channel kinetics. There are a number of allosteric modulators that affect the 5-HT3R including n-alcohols, anaesthetics, antidepressants, cannabinoids, opioids, steroids and natural compounds, many of which also modulate other Cys-loop receptors (see reviews by Davies, 2011, Machu, 2011 and Walstab et al., 2010). Specific effects on different 5-HT3R subtypes have not been widely explored, although alcohols and inhaled anaesthetics have been shown to have reduced sensitivity at 5-HT3AB receptors, and the potencies of the intravenous anaesthetics etomidate, propofol and pentobarbital, and the modulatory compound PU02, are similar at the two receptor types (Table 1; Hayrapetyan et al., 2005; Solt et al., 2005; Stevens et al., 2005; Rusch et al., 2007). All of these compounds are likely to bind in an inter-subunit binding cavity at the top of the TMD which imposes an upper size limit upon them (Stevens et al., 2005; Nury et al., 2011). Although we still do not have high-resolution structural information on 5-HT3R, we can hypothesize that incorporation of the 5-HT3B subunit is likely to alter the size, shape and number of these binding cavities. This is supported by the finding that mutating this region has significant effects on the properties of some 5-HT3R allosteric modulators (Trattnig et al., 2012).
Other compounds reported to modulate the 5-HT3R may have distinct modes of action. For example, the convulsant α-thujone shows a subunit-dependent inhibition of 5-HT3R responses. Interpretation of its affects is complicated as α-thujone may alter 5-HT3A and 5-HT3AB receptor desensitization rates, which are already quite different at the two receptor types (Höld et al., 2000; Deiml et al., 2004). However, it does not compete with the 5-HT3R-selective antagonist [3H]GR65630 at 5-HT3A receptors, showing that it does not bind to the orthosteric binding site, and as tail currents (brief currents that appear after removal of high concentrations of 5-HT in functional studies) are not inhibited by the continued presence of α-thujone it is thought to be not directly blocking the channel (Hapfelmeier et al., 2001). It has been suggested that it increases the likelihood of auto-inhibition by 5-HT channel blockade rather than block by α-thujone itself, but further work is needed to clarify this. The binding site of α-thujone is unknown, but similar changes in desensitization are seen in the presence of 5-hydroxyindole, where mutation of the 15' position in 5-HT3 A receptors abolishes the potentiating effects of 5-OHi (Kooyman et al., 1994; Hu and Lovinger, 2008). Mutation of the 15′ position in 5-HT3A receptors abolishes the potentiating effect of 5-OHi, suggesting an action of this compound at the 15′ position (Hu and Lovinger, 2008). At higher concentrations, 5-OHi competes with [3H]GR65630, which is unsurprising given that 5-OHi shares much of its molecular structure with 5-HT, and could also provide an explanation why 5-OHi can elicit agonist responses in some mutant receptors (Hu and Lovinger, 2008). However, caution may be needed when interpreting the effects of the 15′ mutation as they are conspicuously similar to those of alcohols and channel mutations that enhance ligand efficacy rather than directly affect a specific binding site (Lovinger and White, 1991; Downie et al., 1995; Palma et al., 1996). Therefore, the similar properties of α-thujone and 5-OHi at lower concentrations could reflect a common binding site at the channel 15′ position, but further work is needed to rule out the possibility of broader effects on channel gating.
5-HT3R homology models
Without a crystal structure of the 5-HT3R, researchers have used homology models based on crystal structures of homologous proteins. At 5-HT3A receptors, these have been used to support mutagenesis, to identify residues important for ligand binding, and for studies that used mutant cycle analysis and molecular rulers to define the geometry of the binding region (Yan and White, 2002; 2005; Nyce et al., 2010; Thompson et al., 2010a). The similar effects that 5-HT3A subunit mutations have in homomeric and heteromeric receptors have shown that these homology models of A+A− binding sites are relevant to both receptor types (Thompson et al., 2011c). For homology models containing 5-HT3B subunits, Lochner and Lummis (2010) predicted possible ligand interactions at A+B–, B+A− and B+B− interfaces, which were subsequently tested using mutagenesis and shown to be unlikely to exist. The same study found that mutation of identified residues in the 5-HT3A subunit altered 5-HT activation and [3H]granisetron, supporting the proposal that heteromeric receptors are activated via an A+A− interface. Moura Barbosa et al. (2010) and De Rienzo et al. (2012) provided comprehensive computational validations of their homology models, and known binding site interactions for 5-HT and granisetron were present when these ligands were docked into the A+A− binding site of their models. However, experimental validation is still required for their binding sites containing the 5-HT3B subunit as many of the residues with predicted ligand interactions in B+A− binding sites are known to not effect 5-HT activation or granisetron binding (Lochner and Lummis, 2010; Thompson et al., 2011c). The finding that VUF10166 binds to the A+B− interface presents another opportunity for evaluating interactions at the A+B− interface (Thompson et al., 2012b).
At other Cys-loop receptors, homology models have also been used to probe ligand interactions in the pore (e.g. Zhorov and Bregestovski, 2000; Jensen et al., 2010). At 5-HT3R, there has been limited use of similar homology models, and these have been restricted to 5-HT3A receptors alone (Thompson et al., 2012a). With the identification of increasing numbers of channel-binding ligands and allosteric compounds, we anticipate these heteromeric models will see further utility in the future and could help us understand the differences between the properties of ligands at 5-HT3A and 5-HT3AB receptors.
Therapeutic implications
The first 5-HT3R antagonists were described in the 1950s, but it was several decades before that the first antagonists were licensed for clinical use. These competitive antagonists are now widely used for the treatment of nausea and vomiting arising from chemotherapy, radiotherapy and general anaesthesia. There is also potential for other therapeutic applications as genetic and physiological evidence indicates that the 5-HT3R may be associated with several other disorders (Walstab et al., 2010). These drugs target the A+A− orthosteric binding site shared by 5-HT3A and 5-HT3AB receptors, but as we report in this review, there are now competitive and non-competitive ligands that can distinguish between 5-HT3A and 5-HT3AB receptors. As yet, none of these are clinically used to target 5-HT3R, and NCAs are unlikely to be of therapeutic value because they lack specificity. However, competitive antagonists and allosteric modulators are more selective, and the recent descriptions of competitive ligands with receptor subtype specificity show it is possible to target the different 5-HT3Rs. Allosteric ligands provide further possibilities, and with a growing number of subunit variants (including those containing the more recently identified 5-HT3C-E subunits) there is a now wider range of potential sites at which these modulators could bind. The remaining challenge is to determine the physiological roles of the different 5-HT3R subtypes, develop specific ligands and then determine their therapeutic value. High throughput assay has recently been used to identify novel, potent and selective 5-HT3R ligands (Thompson et al., 2010b; Trattnig et al., 2012; Verheij et al., 2012), showing that there is still chemical space in which 5-HT3R ligands reside, and given the widespread distribution of these receptors, there are still considerable therapeutic opportunities for 5-HT3R ligands that are waiting to be realized.
Acknowledgments
We would like to thank Prof Martin Lochner for his kind support during the preparation of this manuscript. A. J. T. is supported by Wellcome Trust grant 81295 (to S. C. R. L.) and S. C. R. L. holds a Wellcome Trust Senior Research Fellowship in Basic Biomedical Science.
Glossary
- 5-HT
5-hydroxytryptamine
- AFM
atomic force microscopy
- BB
bilobalide
- DTZ
diltiazem
- ECD
extracellular domain
- GB
ginkgolide B
- M2
second transmembrane α-helix
- nACh
nicotinic ACh
- NCA
non-competitive antagonist
- PTX
picrotoxin
- TMD
transmembrane domain
Conflict of interest
None.
References
- Bali M, Akabas MH. The location of a closed channel gate in the GABAA receptor channel. J Gen Physiol. 2007;129:145–159. doi: 10.1085/jgp.200609639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista-Hon DT, Deeb TZ, Othman NA, Sharp D, Hales TG. The 5-HT3B subunit affects high-potency inhibition of 5-HT3 receptors by morphine. Br J Pharmacol. 2012;165:693–704. doi: 10.1111/j.1476-5381.2011.01582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes NM, Hales TG, Lummis SC, Peters JA. The 5-HT3 receptor – the relationship between structure and function. Neuropharmacology. 2009;56:273–284. doi: 10.1016/j.neuropharm.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera NP, Herbert P, Henderson RM, Martin IL, Edwardson JM. Atomic force microscopy reveals the stoichiometry and subunit arrangement of 5-HT3 receptors. Proc Natl Acad Sci U S A. 2005;102:12595–12600. doi: 10.1073/pnas.0503253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boess FG, Beroukhim R, Martin IL. Ultrastructure of the 5-hydroxytryptamine3 receptor. J Neurochem. 1995;64:1401–1405. doi: 10.1046/j.1471-4159.1995.64031401.x. [DOI] [PubMed] [Google Scholar]
- Boorman JP, Groot-Kormelink PJ, Sivilotti LG. Stoichiometry of human recombinant neuronal nicotinic receptors containing the β3 subunit expressed in Xenopus oocytes. J Physiol. 2000;529:565–577. doi: 10.1111/j.1469-7793.2000.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Stanford IM, Ali I, Lin L, Williams JM, Dubin AE, et al. Pharmacological comparison of human homomeric 5-HT3A receptors versus heteromeric 5-HT3A/3B receptors. Neuropharmacology. 2001;41:282–284. doi: 10.1016/s0028-3908(01)00074-0. [DOI] [PubMed] [Google Scholar]
- Butini S, Budriesi R, Hamon M, Morelli E, Gemma S, Brindisi M, et al. Novel, potent, and selective quinoxaline-based 5-HT3 receptor ligands. 1. Further structure-activity relationships and pharmacological characterization. J Med Chem. 2009;52:6946–6950. doi: 10.1021/jm901126m. [DOI] [PubMed] [Google Scholar]
- Cappelli A, Butini S, Brizzi A, Gemma S, Valenti S, Giuliani G, et al. The interactions of the 5-HT3 receptor with quipazine-like arylpiperazine ligands. The journey track at the end of the first decade of the third millennium. Curr Top Med Chem. 2010;10:504–526. doi: 10.2174/156802610791111560. [DOI] [PubMed] [Google Scholar]
- Cappelli A, Manini M, Paolino M, Gallelli A, Anzini M, Mennuni L, et al. Bivalent ligands for the serotonin 5-HT3 receptor. ACS Med Chem Lett. 2011;2:571–576. doi: 10.1021/ml2000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Wang R, Barot S, Weiss DS. Stoichiometry of a recombinant GABAA receptor. J Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure- activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Dillon GH. The 5-HT3B subunit confers reduced sensitivity to picrotoxin when co-expressed with the 5-HT3A receptor. Brain Res Mol Brain Res. 2003b;119:207–212. doi: 10.1016/j.molbrainres.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Das P, Dillon GH. Molecular determinants of picrotoxin inhibition of 5-hydroxytryptamine type 3 receptors. J Pharmacol Exp Ther. 2005;314:320–328. doi: 10.1124/jpet.104.080325. [DOI] [PubMed] [Google Scholar]
- Das P, Bell-Horner CL, Machu TK, Dillon GH. The GABAA receptor antagonist picrotoxin inhibits 5-hydroxytryptamine type 3A receptors. Neuropharmacology. 2003a;44:431–438. doi: 10.1016/s0028-3908(03)00032-7. [DOI] [PubMed] [Google Scholar]
- Davies PA. Allosteric modulation of the 5-HT3 receptor. Curr Opin Pharmacol. 2011;11:75–80. doi: 10.1016/j.coph.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PA, Pistis M, Hanna MC, Peters JA, Lambert JJ, Hales TG, et al. The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature. 1999;397:359–363. doi: 10.1038/16941. [DOI] [PubMed] [Google Scholar]
- De Rienzo F, Del Cadia M, Menziani MC. A first step towards the understanding of the 5-HT3 receptor subunit heterogeneity from a computational point of view. Phys Chem Chem Phys. 2012;14:12625–12636. doi: 10.1039/c2cp41028a. [DOI] [PubMed] [Google Scholar]
- Deeb TZ, Sharp D, Hales TG. Direct subunit-dependent multimodal 5-hydroxytryptamine3 receptor antagonism by methadone. Mol Pharmacol. 2009;75:908–917. doi: 10.1124/mol.108.053322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiml T, Haseneder R, Zieglgansberger W, Rammes G, Eisensamer B, Rupprecht R, et al. α-thujone reduces 5-HT3 receptor activity by an effect on the agonist-reduced desensitization. Neuropharmacology. 2004;46:192–201. doi: 10.1016/j.neuropharm.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Downie DL, Hope AG, Belelli D, Lambert JJ, Peters JA, Bentley KR, et al. The interaction of trichloroethanol with murine recombinant 5-HT3 receptors. Br J Pharmacol. 1995;114:1641–1651. doi: 10.1111/j.1476-5381.1995.tb14952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin AE, Huvar R, D'Andrea MR, Pyati J, Zhu JY, Joy KC, et al. The pharmacological and functional characteristics of the serotonin 5-HT3A receptor are specifically modified by a 5-HT3B receptor subunit. J Biol Chem. 1999;274:30799–30810. doi: 10.1074/jbc.274.43.30799. [DOI] [PubMed] [Google Scholar]
- Durisic N, Godin AG, Wever CM, Heyes CD, Lakadamyali M, Dent JA. Stoichiometry of the human glycine receptor revealed by direct subunit counting. J Neurosci. 2012;32:12915–12920. doi: 10.1523/JNEUROSCI.2050-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ffrench-Constant RH, Rocheleau TA, Steichen JC, Chalmers AE. A point mutation in a drosophila GABA receptor confers insecticide resistance. Nature. 1993;363:449–451. doi: 10.1038/363449a0. [DOI] [PubMed] [Google Scholar]
- Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397–414. doi: 10.1053/j.gastro.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Lummis SCR. Diltiazem causes open channel block of recombinant 5-HT3 receptors. J Physiol. 1999;519:713–722. doi: 10.1111/j.1469-7793.1999.0713n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley D, Amin J, Ross PC, Weiss DS, White G. Point mutations in the M2 region of the α, β, or γ subunit of the GABAA channel that abolish block by picrotoxin. Receptors Channels. 1995;3:13–20. [PubMed] [Google Scholar]
- Hanna MC, Davies PA, Hales TG, Kirkness EF. Evidence for expression of heteromeric serotonin 5-HT3 receptors in rodents. J Neurochem. 2000;75:240–247. doi: 10.1046/j.1471-4159.2000.0750240.x. [DOI] [PubMed] [Google Scholar]
- Hapfelmeier G, Haseneder R, Kochs E, Beyerle M, Zieglgansberger W. Coadministered nitrous oxide enhances the effect of isoflurane on GABAergic transmission by an increase in open-channel block. J Pharmacol Exp Ther. 2001;298:201–208. [PubMed] [Google Scholar]
- Hawthorne R, Lynch JW. A picrotoxin-specific conformational change in the glycine receptor M2-M3 loop. J Biol Chem. 2005;280:35836–35843. doi: 10.1074/jbc.M506645200. [DOI] [PubMed] [Google Scholar]
- Hawthorne R, Cromer BA, Ng HL, Parker MW, Lynch JW. Molecular determinants of ginkgolide binding in the glycine receptor pore. J Neurochem. 2006;98:395–407. doi: 10.1111/j.1471-4159.2006.03875.x. [DOI] [PubMed] [Google Scholar]
- Hayrapetyan V, Jenschke M, Dillon GH, Machu TK. Co-expression of the 5-HT3B subunit with the 5-HT3A receptor reduces alcohol sensitivity. Brain Res Mol Brain Res. 2005;142:146–150. doi: 10.1016/j.molbrainres.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- Holbrook JD, Gill CH, Zebda N, Spencer JP, Leyland R, Rance KH, et al. Characterisation of 5-HT3C, 5-HT3D and 5-HT3E receptor subunits: evolution, distribution and function. J Neurochem. 2009;108:384–396. doi: 10.1111/j.1471-4159.2008.05775.x. [DOI] [PubMed] [Google Scholar]
- Höld KM, Sirisoma NS, Ikeda T, Narahashi T, Casida JE. α-Thujone (the active component of absinthe): γ-aminobutyric acid type A receptor modulation and metabolic detoxification. Proc Natl Acad Sci U S A. 2000;97:3826–3831. doi: 10.1073/pnas.070042397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hooft JA, Vijverberg HP. Selection of distinct conformational states of the 5-HT3 receptor by full and partial agonists. Br J Pharmacol. 1996;117:839–846. doi: 10.1111/j.1476-5381.1996.tb15269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope AG, Belelli D, Mair ID, Lambert JJ, Peters JA. Molecular determinants of (+)-tubocurarine binding at recombinant 5-hydroxytryptamine3A receptor subunits. Mol Pharmacol. 1999;55:1037–1043. doi: 10.1124/mol.55.6.1037. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Lovinger DM. The L293 residue in transmembrane domain 2 of the 5-HT3A receptor is a molecular determinant of allosteric modulation by 5-hydroxyindole. Neuropharmacology. 2008;54:1153–1165. doi: 10.1016/j.neuropharm.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Davies PA, Brauner-Osborne H, Krzywkowski K. 3B but which 3B? And that's just one of the questions: the heterogeneity of human 5-HT(3) receptors. Trends Pharmacol Sci. 2008;29:437–444. doi: 10.1016/j.tips.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen AA, Bergmann ML, Sander T, Balle T. Ginkgolide X is a potent antagonist of anionic Cys-loop receptors with a unique selectivity profile at glycine receptors. J Biol Chem. 2010;285:10141–10153. doi: 10.1074/jbc.M109.079319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley SP, Dunlop JI, Kirkness EF, Lambert JJ, Peters JA. A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature. 2003;424:321–324. doi: 10.1038/nature01788. [DOI] [PubMed] [Google Scholar]
- Kooyman AR, van Hooft JA, Vanderheijden PM, Vijverberg HP. Competitive and non-competitive effects of 5-hydroxyindole on 5-HT3 receptors in N1E-115 neuroblastoma cells. Br J Pharmacol. 1994;112:541–546. doi: 10.1111/j.1476-5381.1994.tb13107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey MR, Cooper MA, Deeb TZ, Carland JE, Kozuska J, Hales TG, et al. Structural determinants of Ca2+ permeability and conduction in the human 5-hydroxytryptamine type 3A receptor. J Biol Chem. 2008;283:19301–19313. doi: 10.1074/jbc.M802406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochner M, Lummis SC. Agonists and antagonists bind to an A-A interface in the heteromeric 5-HT3AB receptor. Biophys J. 2010;98:1494–1502. doi: 10.1016/j.bpj.2009.12.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, White G. Ethanol potentiation of 5-hydroxytryptamine3 receptor-mediated ion current in neuroblastoma cells and isolated adult mammalian neurons. Mol Pharmacol. 1991;40:263–270. [PubMed] [Google Scholar]
- Low PB, Lambert JJ, Peters JA. A comparative study of the pharmacological properties of homo-oligomeric and hetero-oligomeric human recombinant 5-hydroxytryptamine type-3 (5-HT3) receptors. Br J Pharmacol. 2001;133:144P. [Google Scholar]
- Machu TK. Therapeutics of 5-HT3 receptor antagonists: current uses and future directions. Pharmacol Ther. 2011;130:338–347. doi: 10.1016/j.pharmthera.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning D, Wierschke JD, Barnes NM, Moore N. 5-HT3 receptor partial agonist modulation, a novel approach to the treatment of diarrhea predominant irritable bowel syndrome (IBS-D) Gastroenterology. 2011;140:S616–S616. [Google Scholar]
- Maricq AV, Peterson AS, Brake AJ, Myers RM, Julius D. Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science. 1991;254:432–437. doi: 10.1126/science.1718042. [DOI] [PubMed] [Google Scholar]
- Miyake A, Mochizuki S, Takemoto Y, Akuzawa S. Molecular cloning of human 5-hydroxytryptamine3 receptor: heterogeneity in distribution and function among species. Mol Pharmacol. 1995;48:407–416. [PubMed] [Google Scholar]
- Modica MN, Pittala V, Romeo G, Salerno L, Siracusa MA. Serotonin 5-HT3 and 5-HT4 ligands: an update of medicinal chemistry research in the last few years. Curr Med Chem. 2010;17:334–362. doi: 10.2174/092986710790192730. [DOI] [PubMed] [Google Scholar]
- Morelli E, Gemma S, Budriesi R, Campiani G, Novellino E, Fattorusso C, et al. Specific targeting of peripheral serotonin 5-HT3 receptors. Synthesis, biological investigation, and structure-activity relationships. J Med Chem. 2009;52:3548–3562. doi: 10.1021/jm900018b. [DOI] [PubMed] [Google Scholar]
- Moura Barbosa AJ, De Rienzo F, Ramos MJ, Menziani MC. Computational analysis of ligand recognition sites of homo- and heteropentameric 5-HT3 receptors. Eur J Med Chem. 2010;45:4746–4760. doi: 10.1016/j.ejmech.2010.07.039. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Ishida Y, Yamada T, Kondo M, Shimada S. Subunit-dependent inhibition and potentiation of 5-HT3 receptor by the anticancer drug, topotecan. J Neurochem. 2013;125:7–15. doi: 10.1111/jnc.12146. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Ishida Y, Yamada T, Shimada S. Anticancer drug irinotecan inhibits homomeric 5-HT3A and heteromeric 5-HT3AB receptor responses. Biochem Biophys Res Commun. 2011;415:416–420. doi: 10.1016/j.bbrc.2011.10.084. [DOI] [PubMed] [Google Scholar]
- Niesler B. 5-HT3 receptors: potential of individual isoforms for personalised therapy. Curr Opin Pharmacol. 2011;11:81–86. doi: 10.1016/j.coph.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Niesler B, Walstab J, Combrink S, Moller D, Kapeller J, Rietdorf J, et al. Characterization of the novel human serotonin receptor subunits 5-HT3C,5-HT3D, and 5-HT3E. Mol Pharmacol. 2007;72:8–17. doi: 10.1124/mol.106.032144. [DOI] [PubMed] [Google Scholar]
- Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, et al. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- Nyce HL, Stober ST, Abrams CF, White MM. Mapping spatial relationships between residues in the ligand-binding domain of the 5-HT3 receptor using a molecular ruler. Biophys J. 2010;98:1847–1855. doi: 10.1016/j.bpj.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E, Mileo AM, Eusebi F, Miledi R. Threonine-for-leucine mutation within domain M2 of the neuronal α7 nicotinic receptor converts 5-hydroxytryptamine from antagonist to agonist. Proc Natl Acad Sci U S A. 1996;93:11231–11235. doi: 10.1073/pnas.93.20.11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JA, Cooper MA, Carland JE, Livesey MR, Hales TG, Lambert JJ. Novel structural determinants of single channel conductance and ion selectivity in 5-hydroxytryptamine type 3 and nicotinic acetylcholine receptors. J Physiol. 2010;588:587–596. doi: 10.1113/jphysiol.2009.183137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch D, Braun HA, Wulf H, Schuster A, Raines DE. Inhibition of human 5-HT(3A) and 5-HT(3AB) receptors by etomidate, propofol and pentobarbital. Eur J Pharmacol. 2007;573:60–64. doi: 10.1016/j.ejphar.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt K, Stevens RJ, Davies PA, Raines DE. General anesthetic-induced channel gating enhancement of 5-hydroxytryptamine type 3 receptors depends on receptor subunit composition. J Pharmacol Exp Ther. 2005;315:771–776. doi: 10.1124/jpet.105.090621. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Richards CI, Dilworth C, Moss FJ, Dougherty DA, Lester HA. Forster resonance energy transfer (FRET) correlates of altered subunit stoichiometry in Cys-loop receptors, exemplified by nicotinic α4β2. Int J Mol Sci. 2012;13:10022–10040. doi: 10.3390/ijms130810022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens R, Rusch D, Solt K, Raines DE, Davies PA. Modulation of human 5-hydroxytryptamine type 3AB receptors by volatile anesthetics and n-alcohols. J Pharmacol Exp Ther. 2005;314:338–345. doi: 10.1124/jpet.105.085076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lummis SC. Antimalarial drugs inhibit human 5-HT3 and GABAA but not GABAC receptors. Br J Pharmacol. 2008;153:1686–1696. doi: 10.1038/bjp.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lummis SCR. The 5-HT3 receptor as a therapeutic target. Expert Opin Ther Targ. 2007;11:527–540. doi: 10.1517/14728222.11.4.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lochner M, Lummis SC. The antimalarial drugs quinine, chloroquine and mefloquine are antagonists at 5-HT3 receptors. Br J Pharmacol. 2007;151:666–677. doi: 10.1038/sj.bjp.0707238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lester HA, Lummis SC. The structural basis of function in Cys-loop receptors. Q Rev Biophys. 2010a;43:449–499. doi: 10.1017/S0033583510000168. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Verheij MH, Leurs R, De Esch IJ, Lummis SC. An efficient and information-rich biochemical method design for fragment library screening on ion channels. Biotechniques. 2010b;49:822–829. doi: 10.2144/000113538. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Duke RK, Lummis SC. Binding Sites for bilobalide, diltiazem, ginkgolide, and picrotoxinin at the 5-HT3 receptor. Mol Pharmacol. 2011a;80:183–190. doi: 10.1124/mol.111.071415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Jarvis GE, Duke RK, Johnston GA, Lummis SC. Ginkgolide B and bilobalide block the pore of the 5-HT3 receptor at a location that overlaps the picrotoxin binding site. Neuropharmacology. 2011b;60:488–495. doi: 10.1016/j.neuropharm.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Price KL, Lummis SC. Cysteine modification reveals which subunits form the ligand binding site in human heteromeric 5-HT3AB receptors. J Physiol. 2011c;589:4243–4257. doi: 10.1113/jphysiol.2011.208439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, McGonigle I, Duke R, Johnston GA, Lummis SC. A single amino acid determines the toxicity of Ginkgo biloba extracts. FASEB J. 2012a;26:1884–1891. doi: 10.1096/fj.11-192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Verheij MH, de Esch IJ, Lummis SC. VUF10166, a novel compound with differing activities at 5-HT3A and 5-HT3AB receptors. J Pharmacol Exp Ther. 2012b;341:350–359. doi: 10.1124/jpet.111.190769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trattnig SM, Harpsoe K, Thygesen SB, Rahr LM, Ahring PK, Balle T, et al. Discovery of a novel allosteric modulator of 5-HT3 receptors: inhibition and potentiation of Cys-loop receptor signalling through a conserved transmembrane intersubunits site. J Biol Chem. 2012;287:25241–25254. doi: 10.1074/jbc.M112.360370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij MH, Thompson AJ, van Muijlwijk-Koezen JE, Lummis SC, Leurs R, de Esch IJ. Design, synthesis, and structure-activity relationships of highly potent 5-HT3 receptor ligands. J Med Chem. 2012;55:8603–8614. doi: 10.1021/jm300801u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walstab J, Rappold G, Niesler B. 5-HT3 receptors: role in disease and target of drugs. Pharmacol Ther. 2010;128:146–169. doi: 10.1016/j.pharmthera.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Yan D, White MM. Interaction of d-tubocurarine analogs with mutant 5-HT3 receptors. Neuropharmacology. 2002;43:367–373. doi: 10.1016/s0028-3908(02)00125-9. [DOI] [PubMed] [Google Scholar]
- Yan D, White MM. Spatial orientation of the antagonist granisetron in the ligand-binding site of the 5-HT3 receptor. Mol Pharmacol. 2005;68:365–371. doi: 10.1124/mol.105.011957. [DOI] [PubMed] [Google Scholar]
- Zhang R, White NA, Soti FS, Kem WR, Machu TK. N-terminal domains in mouse and human 5-hydroxytryptamine3A receptors confer partial agonist and antagonist properties to benzylidene analogs of anabaseine. J Pharmacol Exp Ther. 2006;317:1276–1284. doi: 10.1124/jpet.106.101485. [DOI] [PubMed] [Google Scholar]
- Zhang R, Wen X, Militante J, Hester B, Rhubottom HE, Sun H, et al. The role of loop F residues in determining differential d-tubocurarine potencies in mouse and human 5-hydroxytryptamine 3A receptors. Biochemistry. 2007;46:1194–1204. doi: 10.1021/bi0616100. [DOI] [PubMed] [Google Scholar]
- Zhorov BS, Bregestovski PD. Chloride channels of glycine and GABA receptors with blockers: Monte Carlo minimization and structure-activity relationships. Biophys J. 2000;78:1786–1803. doi: 10.1016/S0006-3495(00)76729-4. [DOI] [PMC free article] [PubMed] [Google Scholar]