

Abstract
Background and Purpose
The development of potent and selective inhibitors of the biosynthesis of the endocannabinoid 2-arachidonoylglycerol (2-AG) via DAG lipases (DAGL) α and β is just starting to be considered as a novel and promising source of pharmaceuticals for the treatment of disorders that might benefit from a reduction in endocannabinoid tone, such as hyperphagia in obese subjects.
Experimental Approach
Three new fluorophosphonate compounds O-7458, O-7459 and O-7460 were synthesized and characterized in various enzymatic assays. The effects of O-7460 on high-fat diet intake were tested in mice.
Key Results
Of the new compounds, O-7460 exhibited the highest potency (IC50 = 690 nM) against the human recombinant DAGLα, and selectivity (IC50 > 10 μM) towards COS-7 cell and human monoacylglycerol lipase (MAGL), and rat brain fatty acid amide hydrolase. Competitive activity-based protein profiling confirmed that O-7460 inhibits mouse brain MAGL only at concentrations ≥10 μM, and showed that this compound has only one major ‘off-target’, that is, the serine hydrolase KIAA1363. O-7460 did not exhibit measurable affinity for human recombinant CB1 or CB2 cannabinoid receptors (Ki > 10 μM). In mouse neuroblastoma N18TG2 cells stimulated with ionomycin, O-7460 (10 μM) reduced 2-AG levels. When administered to mice, O-7460 dose-dependently (0–12 mg·kg−1, i.p.) inhibited the intake of a high-fat diet over a 14 h observation period, and, subsequently, slightly but significantly reduced body weight.
Conclusions and Implications
O-7460 might be considered a useful pharmacological tool to investigate further the role played by 2-AG both in vitro and in vivo under physiological as well as pathological conditions.
Linked Articles
This article is part of a themed section on Cannabinoids. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2013.169.issue-4 & http://dx.doi.org/10.1111/bph.2012.167.issue-8
Keywords: inhibitor, diacylglycerol, cannabinoid, 2-arachidonoylglycerol, serine lipase, anti-obesity effects
Introduction
The endocannabinoid 2-arachidonoylglycerol (2-AG) (Mechoulam et al., 1995; Sugiura et al., 1995) exerts its physiological action by binding and functionally activating the two cannabinoid receptor subtypes identified to date, CB1 and CB2 (Pertwee, 2008). The tissue levels of 2-AG, as well as those of the other proposed endocannabinoid, anandamide, undergo remodelling during either acute or chronic pathological conditions. The general strategy of these changes is to afford protection, usually by aiming at restoring physiological homeostasis (Di Marzo, 2008). However, endocannabinoid plasticity can lose specificity and contribute to the symptoms or progression of certain diseases. In this case, the pharmacological reduction of 2-AG tissue levels, by blocking its biosynthesis, might be therapeutically valuable. Obesity, liver fibrosis, and Alzheimer's and Parkinson's disease are only some of the pathological conditions that might benefit from a reduction in endocannabinoid tone (Di Marzo, 2008). In fact, the use of inhibitors of 2-AG formation might have pharmacological consequences and therapeutic applications similar to those of ‘neutral’ CB1 receptor antagonists, which have been suggested to produce fewer side effects than CB1 receptor inverse agonists (Janero, 2012), such as rimonabant (Ward and Raffa, 2011). The latter compound was in clinical development until 2008 and even reached the market as an anti-obesity drug before being discontinued due to potentially serious psychiatric effects. Indeed, obesity is characterized by pathologically elevated endocannabinoid levels in both the CNS and peripheral tissues involved in energy homeostasis, and rimonabant, as well as other CB1 inverse agonists, proved very effective at reducing body weight (BW) and several parameters of the metabolic syndrome, although these compounds were not devoid of undesired adverse side effects (Di Marzo and Després, 2009).
The endocannabinoid 2-AG is biosynthesized from diacylglycerol (DAG) precursors by the action of specific sn-1 selective DAG lipases (DAGL). Two sn-1 DAGL isozymes (DAGLα and DAGLβ) have been cloned and enzymatically characterized, and at least one of them, DAGLα, is localized to the postsynaptic dendritic spines establishing synapses with CB1-expressing axons, supporting the proposed role for 2-AG as a retrograde signal mediating depolarization-induced suppression of neurotransmission and heterosynaptic plasticity (Bisogno et al., 2003; Ludányi et al. 2011). Since 2-AG biosynthetic enzymes have been identified only recently, still relatively little information on possible selective inhibitors for these proteins is available. We have previously described the development of two fluorophosphonate inhibitors of DAGLα, O-3640 and O-3841, with excellent selectivity for DAGLα over the other proteins of the endocannabinoid system tested (Bisogno et al., 2006). These compounds have been successfully used in vitro but, unfortunately, they are not suitable for systemic use in vivo due to their lack of stability and poor permeability through the plasma membrane, which have been only partially solved by the design, a few years later, of O-5596, a more stable and slightly more potent and cell membrane permeable DAGL inhibitor (Figure 1) (Bisogno et al., 2009a). Interestingly, O-5596 was shown to inhibit a typical endocannabinoid-mediated effect in vivo, since its administration to mice reduced selectively the intake of palatable food (Bisogno et al., 2009a). Here we report three new fluorophosphonate compounds, that is O-7458, O-7459 and O-7460 (Figure 1), and their in vitro and in vivo pharmacological characterization as potential selective inhibitors of DAGLα.
Figure 1.
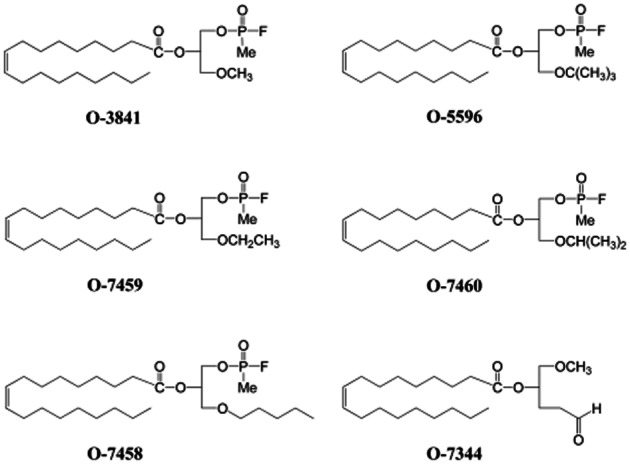
Chemical structures of new and already known fluorophosphonate inhibitors of 2-AG biosynthesis by DAGLα.
Methods
Nomenclature
Drug and receptor nomenclature have been used according to BJP's ‘Guide to receptors and channels’ (Alexander et al., 2011).
Synthesis of O-7460
Compound O-7460 was synthesized from commercially available 1-chloro-3-isopropoxypropan-2-ol (1) in four steps in an overall yield of 8% (see Supporting Information Figure S1). Coupling of compound 1 with oleic acid in the presence of EDCI/DMAP gave compound 2 (84%), which on treatment with NaI/dimethyl methylphosphonate in a sealed tube at 135°C for 2 days yielded pure compound 3 (30%) after chromatography. Demethylation of 3 with TMSBr followed by chromatography yielded compound 4 (80%), which on treatment with methyl DAST followed by chromatography yielded pure target compound O-7460 (40%). Similar procedures were applied for the synthesis of O-7458 and O-7459, and will be described, together with the synthesis of O-7344, in a separate report in a specialist journal.
DAGLα assay
DAGL activity was assessed as previously reported (Bisogno et al., 2003) by using membrane preparations (70 μg of protein) obtained from COS-7 cells overexpressing the human recombinant DAGLα, and 1-[14C]oleoyl-2-arachidonoylglycerol [1.0 mCi·mmol–1, 25 μM, synthesized as reported previously (Bisogno et al., 2003)] as substrate in the presence of vehicle or increasing concentrations of test compounds in Tris–HCl buffer, pH = 7 for 20 min. After the incubation, lipids were extracted with two volumes of CHCl3/CH3OH (2:1, v · v-1). The organic extracts, lyophilized under vacuum, were purified by using TLC on silica on polypropylene plates eluted in CHCl3/CH3OH/NH4OH (85:15:0.1, v · v-1) as the eluting solvent. Bands corresponding to [14C]-oleic acid were cut and their radioactivity was measured with a β-counter. In order to investigate the irreversibility of the inhibitory action of O-7460, the most active inhibitor tested in this study, membrane preparations obtained from COS-7 cells overexpressing the human recombinant DAGLα were incubated in the presence of vehicle or 0.5 or 5 μM concentrations of the compound in Tris–HCl buffer, pH = 7 for 20 min. After incubation, DAGLα membrane preparations were washed three times with either Tris–HCl buffer, 50 μM pH = 7 (buffer A) or Tris–HCl buffer, 50 μM pH = 7 containing 0.03% BSA (buffer B), using centrifugations at 10 000× g. The DAGLα membrane preparations were than resuspended in Tris–HCl buffer, 50 μM pH = 7, and DAGL activity was re-assessed as reported above in the presence of 1-[14C]-oleoyl-2-arachidonoylglycerol.
Monoacylglycerol lipase (MAGL) assay
The 10 000 g cytosolic fraction (100 μg of protein) and human recombinant MAGL (0.05 μg, Cayman Chemicals, Ann Arbor, MI, USA) were incubated in Tris–HCl 50 μM, at pH 7.0 at 37°C for 20 min, with 2-arachidonoyl-[3H]-glycerol (40 Ci·mmol−1, St. Louis, MO, USA) diluted with non-radiolabelled 2-AG (Cayman Chemicals) to 20 μM, as previously reported (Bisogno et al., 2009b), in the presence of vehicle or increasing concentrations of test compounds. After the incubation, the amount of [3H]-glycerol produced was measured by scintillation counting of the aqueous phase after extraction of the incubation mixture with 2 volumes of CHCl3/MeOH 1:1 (v · v-1).
CB1 and CB2 binding assays
Membranes from HEK-293 cells transfected with either the human recombinant CB1 or CB2 cDNA (Perkin-Elmer, Monza, Italy) were incubated with increasing concentrations of the test compounds and [3H]-CP-55940 (KD = 0.12 nM for CB1 and KD = 0.18 nM for CB2) as the high affinity ligand, as described by the manufacturer (Perkin-Elmer). Displacement curves were generated by incubating drugs with a fixed concentration of [3H]-CP-55940 (0.4 nM for CB1 and 0.53 nM for CB2 binding assay). Non-specific binding was determined using 1 μM WIN55312-2 (Tocris, Bristol, UK).
Fatty acid amide hydrolase (FAAH) assay
The effect of compounds on the enzymatic hydrolysis of [14C]-anandamide (2.4 μM, 5.0 mCi·mmol−1) was studied as previously reported (Cascio et al., 2004) by using membranes prepared from rat brain, incubated with increasing concentrations of test compounds in 50 μM Tris–HCl, pH = 9, for 30 min at 37°C. [14C]-Ethanolamine produced from [14C]-anandamide hydrolysis was measured by scintillation counting of the aqueous phase after the extraction of the incubation mixture with two volumes of CHCl3/CH3OH (1:1, v · v-1).
Affinity-based protein profiling assay
The screening of possible ‘off-target’ serine hydrolases for O-7458, O-7459 and O-7460 was performed with mouse brain proteomes (Long et al., 2009). Proteomes (mouse membrane fractions) (50 μL, 1.0 mg·mL−1 total protein concentration) were pre-incubated with varying concentrations of inhibitors at 37°C. After 30 min, fluorophosphonate rhodamine (1.0 μL, 50 μM in DMSO) was added, and the mixture was incubated for another 30 min at room temperature. Reactions were quenched with SDS loading buffer (35 μL) and run on SDS-PAGE, and visualized in gel with a Hitachi FMBio lle flatbed fluorescence scanner (MiraiBio, San Francisco, CA, USA). The selective MAGL inhibitor, JZL184, was used as a positive control for MAGL inhibition.
Effect of O-7460 on endocannabinoid levels in intact N18TG2 cells
Confluent N18TG2 cells were stimulated for 20 min at 37°C with ionomycin (3 μM) with or without O-7460 (10 μM). After stimulation, cells plus medium were extracted with CHCl3/CH3OH 2:1 (v · v-1). Each extract was purified by open bed chromatography and 2-AG was quantified by LC-MS as previously reported (Marsicano et al., 2002).
Animals
Seven-week-old male C57BL/6N inbred mice (n = 16, weighing 20 ± 0.5 g on arrival) were purchased from Charles River (Calco, CO, Italy). Mice were housed in individual cages (26.7 × 20.7 × 14 cm), which were kept in a room at 22–24°C with a 50–60% relative humidity, 12 h light/12 h dark cycle, and maintained with standard diet (diet no. 4RF21; Mucedola s.r.l., Milan, Italy) and water available ad libitum. Animal husbandry and experiments were carried out in accordance with the Council Directive of the European Community (86/EEC) of the Italian D.L. 116 (27 January 1992) and approved by veterinarian supervision. Before starting the evaluation of the effects of O-7460 administration on high-fat diet (HFD) intake, mice were habituated for 10 days to the transition from the standard 4RF21 diet to a HFD dietary regimen containing 45% fat (6% of soybean oil and 39% of lard, kcal) and providing about 4.73 kcal·g−1 of energy level (diet no. D12451; Research Diets, New Brunswick, NJ, USA). Daily HFD intake and BW were monitored during the habituation phase (data not shown). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Effects of O-7460 on HFD intake and BW in mice
O-7460 was dissolved in ethanol : saline (1:1) solution and administered i.p. at the following doses, 0 mg·kg−1 (n = 6), 6 mg·kg−1 (n = 5), and 12 mg·kg−1 (n = 5), in a volume of 1 mL 100 g−1 BW. Animals were not deprived of food and the different doses of O-7460 were administered just before the 12 h dark cycle (19:00 h) and HFD intake assessed at the following time intervals: 30 min, 60 min and 14 h after O-7460 treatment. HFD consumption was measured as the difference between the initial quantity of food allotted (g) in each cage and the weight of the remaining food 30– 60 min and 14 h after O-7460 administration, respectively. BW was also collected at baseline before drug administration (0 min) and 14 h after O-7460 administration. Each measurement was made by a Mettler ML802E precision balance (Toledo, OH, USA) providing an accuracy of 0.01g.
Effects of O-7460 on motor activity and body temperature in mice
Seven mice maintained on the same dual diet regimen (either 4RF21 or HFD) were administered O-7460 i.p. (1 mL 100 g-1 BW) at 0 mg·kg−1 (n = 3) and 12 mg·kg−1 (n = 4), and then individually placed into activity chambers equipped with infrared photo sensors (InfraMot system® by TSE Systems, Bad Homburg, Germany) suitable for measuring, over time, the animal's horizontal and vertical spatial movement. Motor activity was recorded for a total time of 60 min. Body temperature was measured 30 min after drug administration with a digital thermometer (model BAT7001H) connected to a thermostat rectal probe (model RET-3) both produced by Physitemp Instruments, Inc. (Clifton, NJ, USA).
Effects of HFD exposure with or without O-7460 administration on 2-AG levels in hypothalamus, liver and white adipose tissue (WAT)
Seven mice were habituated (10 days) to the transition from the standard 4RF21 diet to the HFD (D12451) while another six mice were maintained on regular diet. After an additional 10 days of dual diet regimen, seven mice were administered either O-7460 (12 mg·kg−1; n = 4) or vehicle (0 mg·kg−1; n = 3) whereas the other six mice (4RF21, n = 3 and HFD, n = 3) were left drug-free. At day 10, 60 min after O-7460 administration, both groups were killed and tissue samples of WAT, liver and hypothalamus were collected. WAT (epididymal), liver and brain were quickly removed and flash-frozen in liquid nitrogen. For hypothalami extraction, each brain was fixed vertically on the freeze plate of a freezing microtome. Micro-punches of both hemispheres were obtained from brain slices (coronal sections) no thicker than 300 μm. A probe stainless steel needle of 1.3 mm (i.d.) was used for tissue punching. The coordinates were taken according to the atlas of Franklin and Paxinos (1998) as follows: three slices from −1.28 to −2.12 mm (coronal sections from Bregma, to include lateral, ventromedial hypothalamus and arcuate nucleus). Tissues were stored at −80°C up to the day of analysis. Tissues were homogenized in 5 vol of chloroform/methanol/Tris–HCl 50 μM (2:1:1) containing 100 pmol of d5-2-AG and extracted three times with chloroform. Each extract was purified by open bed chromatography and 2-AG was quantified by LC-MS as previously reported (Marsicano et al., 2002).
Data analysis
IC50 values, means ± SEM of n = 3 experiments, were obtained using GraphPad Prism4® (GraphPad Software, Inc., La Jolla, CA, USA). Ki values were calculated by applying the Cheng–Prusoff equation to the IC50 values (obtained using GraphPad Prism4). Results from the in vitro studies and 2-AG content were analysed by anova followed by the Bonferroni post hoc test. Results from in vivo studies were analysed by anova followed by Tukey's honestly significant difference (HSD) as post hoc test. Differences between means were deemed to be significant when P < 0.05.
Results
Effects of compounds on human DAGLα
The possible inhibitory effect of compounds on DAGL was tested in COS-7 cells overexpressing human DAGLα. Compound O-7344 exhibited no significant inhibitory activity up to 10 μM. Under the same conditions, O-7458 exhibited a moderate inhibitory action on DAGLα (IC50 = 3.26 ± 0.09 μM), whereas O-7459 and O-7460 exhibited higher potency as DAGLα inhibitors with IC50 values of 1.17 ± 0.16 μM and 0.69 ± 0.087 μM, respectively (Figure 2A–C and Table 1).
Figure 2.
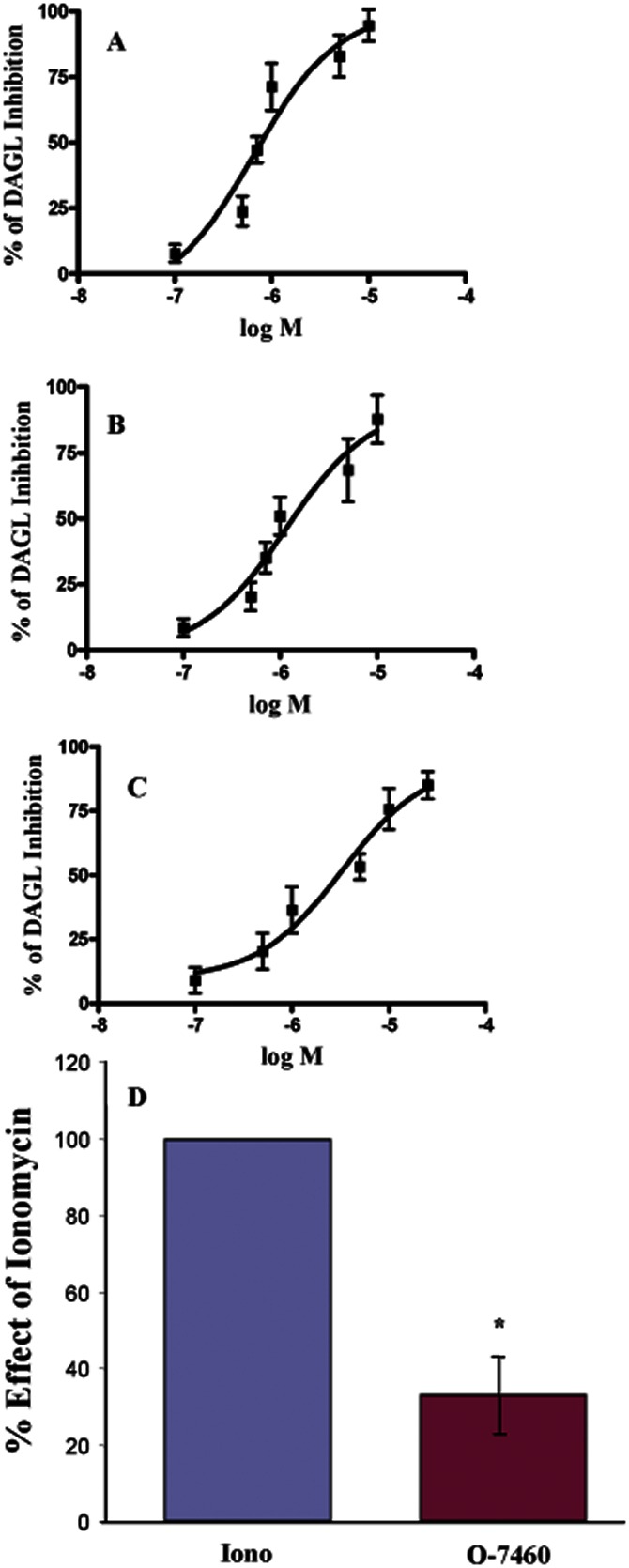
Concentration–response curves for inhibition of DAGLα activity by O-7460 (A), O-7459 (B) and O-7458 (C). The effect of increasing concentrations of drugs is expressed as percentage of inhibition, and IC50 values (reported in Table 1) were obtained with GraphPad Prism4. Data are means of n = 3 determinations. (D) Ionomycin-induced formation of 2-AG in intact N18TG2 cells. The effect of O-7460 (10 μM) is reported as percentage of the maximum effect observed with ionomycin 3 μM plus vehicle. Data are means ± SEM of n = 3 determinations (means were compared by anova followed by the Bonferroni test. *P < 0.05).
Table 1.
Screening of the new compounds on enzymes of the endocannabinoid system
| DAGLα human recombinant (IC50 μM) | MAGL COS cell cytosol (IC50 μM) | Human recombinant MAGL | FAAH rat brain membranes (IC50 μM) | CB1 human recombinant (Ki μM) | CB2 human recombinant (Ki μM) | |
|---|---|---|---|---|---|---|
| O-7344 | >10 | >10 | n.t. | >50 | >10 | 1.21 ± 0.20 |
| O-7458 | 3.26 ± 0.09 | >10 | n.t. | >50 | >10 | >10 |
| O-7459 | 1.17 ± 0.16 | >10 | n.t. | >50 | >10 | >10 |
| O-7460 | 0.69 ± 0.09 | >10 | >10 | >50 | >10 | >10 |
| JZL184 | n.t. | 0.12 ± 0.08 | n.t. | n.t. | n.t. | n.t. |
| URB597 | n.t. | n.t. | n.t. | 0.05 ± 0.02 | n.t. | n.t. |
| WIN55212-2 | n.t | n.t | n.t | n.t | 0.00267 ± 0.0004 | 0.001 ± 0.00029 |
Data are means ± SEM of n = 3 experiments (see Figure 2). Ki values were calculated by applying the Cheng–Prusoff equation to the IC50 values (obtained using GraphPad Prism4). n.t., not tested.
Moreover, O-7460 inhibited DAGLα in a seemingly irreversible way since its inhibitory effect persisted after repeated washing of the DAGLα membrane preparations with buffer A or buffer B. In fact, the % of DAGLα inhibition by O-7460 at the concentrations of 0.5 and 5 μM changed from 24 ± 5 to 24 ± 8 and 13 ± 6 and from 80 ± 8 to 70 ± 7 and 66 ± 5 after no washing or three washings with buffer A or buffer B respectively. Data are means ± SEM, n = 3.
Effect of compounds on rat brain FAAH
The effect of the compounds on FAAH was studied by using membrane preparations from rat brain. None of the compounds exerted any significant inhibition of anandamide hydrolysis up to a concentration of 50 μM (Table 1).
Effects of compounds on MAGL
The effect of the compounds on MAGL was studied using cytosolic fractions of COS-7 cells, or, for O-7460 only, on human recombinant MAGL, and by using 2-arachidonoyl-[3H]-glycerol as substrate. None of the compounds exhibited any significant inhibitory activity in any of the enzymatic preparations up to 10 μM (Table 1).
Effect of compounds in an affinity-based protein profiling assay
As shown in Figure 3, and as denoted by the progressive reduction in intensity of the labelling of some proteins with increasing concentrations of the compounds, when tested in this assay, O-7458, O-7459 and O-7460 were found to ‘hit’ particularly two types of serine hydrolases: MAGL (bands at 33–35 kDa), but only at the highest concentration tested (10 μM), and KIAA 1363 (bands at ∼45 kDa), even at the 0.1 μM concentration with an estimated potency of 0.5 < IC50 < 1 μM.
Figure 3.
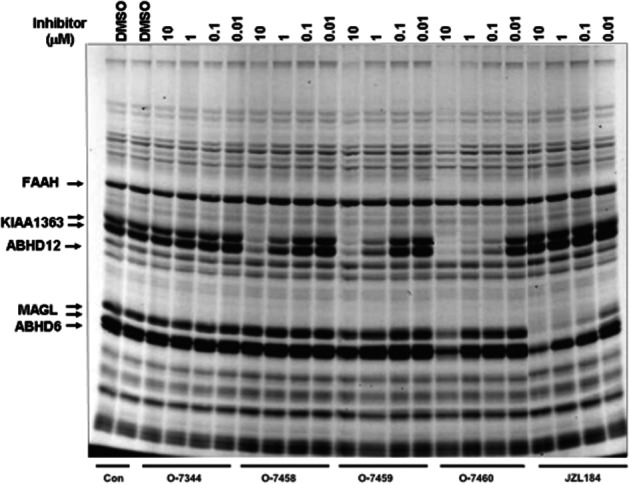
Affinity-based proteomic profiling assay of O-7460, O-7459 and O-7458 using the mouse brain proteome. MAGL, KIAA1363, FAAH, ABHD6 and ABHD12 bands are shown by arrows. The effect of the selective MAGL inhibitor JZL184 is also shown. Con: control incubation with no inhibitor. These results are representative of three separate experiments.
Binding affinity for human recombinant CB1 and CB2
Radioligand competition binding studies were performed by using membranes from HEK-293 cells transfected with human recombinant CB1 or CB2 receptors. These data are summarized in Table 1. None of the compounds exhibited any significant affinity for cannabinoid receptors up to 10 μM except for compound O-7344, which showed some affinity for CB2 receptors with a Ki of 1.21 ± 0.20 μM.
Experiments in intact cells
The effect of O-7460 (10 μM, corresponding approximately to an IC80 concentration of the compound) on 2-AG formation stimulated by ionomycin was studied in intact N18TG2 cells where the enzymatic formation of 2-AG has been previously reported (Bisogno et al., 2003). As reported in Figure 2D, O-7460 decreased the ionomycin-induced formation of 2-AG in intact cells (down to 33 ± 10% of the maximum effect observed with ionomycin 3 μM).
Effects of O-7460 on HFD intake and BW in mice
Repeated-measures anova performed on total HFD intake (g) 30 min, 60 min and 14 h after O-7460 administration revealed a significant main effect of treatment [F(2,13) = 81.76; P < 0.0001] and time [F(2,26) = 833.83; P < 0.0001], as well as a significant treatment × time interaction [F(4,26) = 43.70; P < 0.0001]. The administration of O-7460 (Figure 4) induced a time and dose-dependent decrease in HFD intake as compared with vehicle-treated mice: 30 and 60 min, O-7460 6 mg·kg−1, Tukey's HSD n.s.; 14 h, 6 mg·kg−1, Tukey's HSD, P < 0.05; 30 min, 60 min and 14 h, O-7460 12 mg·kg−1, Tukey's HSD, P < 0.05.
Figure 4.
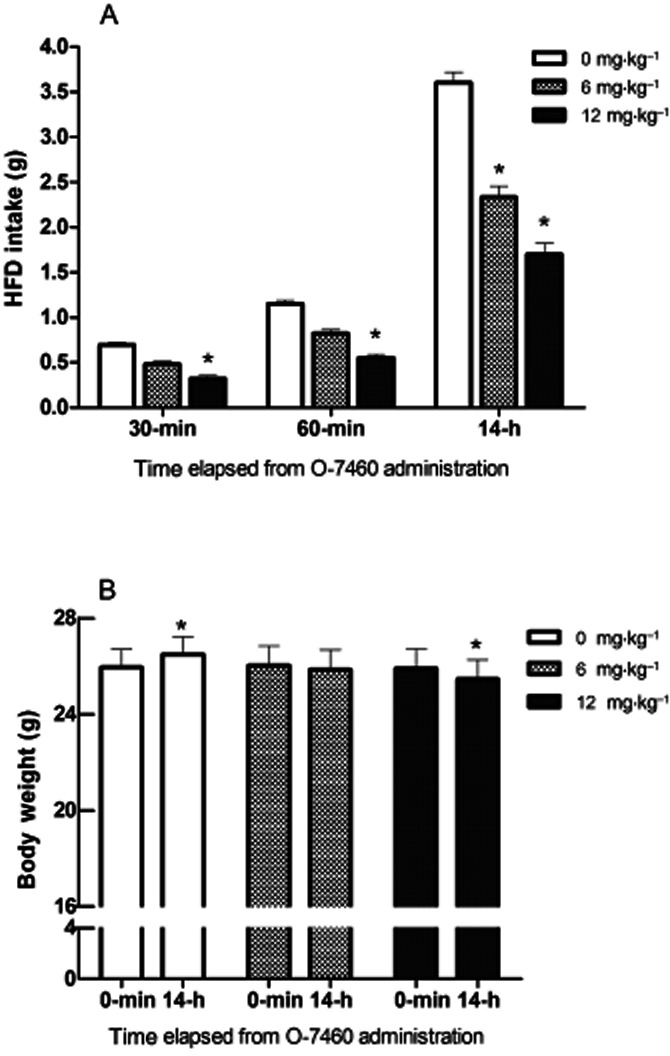
Effects of O-7460 on HFD intake (A) and body weight (B) in mice. HFD cumulative food intake (HFD-I) and body weight (BW) evolution were analysed by separate two-way anovas with drug (three doses) and three or two time points as HFD-I (30 min, 60 min and 14 h) and BW (0 min and 14 h) factors respectively. A P-value below 0.05 was considered statistically significant as compared with vehicle group (HFD-I) or baseline BW. Post hoc analyses were carried out by Tukey's HSD test comparisons. All analyses were executed by STATISTICA version 8.0 (data analysis software system, StatSoft, Inc., Tulsa, OK, USA, 2007).
Repeated-measures anova carried out on BW (g) 0 min (baseline) and 14 h after O-7460 administration did not reveal any significant treatment [F(2,13) = 0.12; n.s.] or time [F(1,13) = 1.52; n.s.] effect, although a significant treatment × time interaction was found [F(2,13) = 80.37; P < 0.0001]. Post hoc analysis (Figure 4) revealed the progressive enhancement of BW only for vehicle-treated animals (0 min and 14 h; O-7460, 0 mg·kg−1, Tukey's HSD, P < 0.05), whereas the intermediate dose (O-7460, 6 mg·kg−1) did not affect the increase in BW. However (Figure 4), in contrast to the time-dependent increase in BW observed in vehicle-treated animals, the administration of the highest dose (O-7460, 12 mg·kg−1) significantly counteracted the BW increase.
Effects of O-7460 on motor activity and body temperature in mice
In order to exclude the possibility that the effect of O-7460 on food intake could be due to overt changes in the normal behaviour of the mice, we investigated both motor activity and body temperature after O-7460 administration (12 mg·kg−1, i.p.) in HFD mice. Motor activity was recorded for a total time of 60 min and O-7460 administration exhibited no effect on total locomotor activities (1717 ± 720.22 and 1618 ± 719.22 counts in vehicle- and O-7460-treated mice, respectively). Furthermore, O-7460 did not modify body temperature in HFD mice (36.76 ± 0.2°C in vehicle and 36.85 ± 0.18°C in treated mice). Data are means ± SEM of n = 3 or 4 mice.
Effects of HFD exposure or O-7460 administration on 2-AG levels in the hypothalamus, liver and WAT
We next wanted to assess whether the effect of O-7460 observed on food intake and body weight in HFD mice correlated with a reduction in 2-AG levels at the both central and peripheral levels. In the hypothalamus, 10 days of HFD caused an up-regulation of 2-AG levels (from 3.50 ± 0.59 to 5.03 ± 0.71 pmol·mg−1 tissue in standard diet and HFD, respectively), which was significantly counteracted after 60 min from O-7460 (12 mg·kg−1) i.p. injection (5.06 ± 0.004 to 3.42 ± 0.65 pmol·mg−1 tissue in vehicle- and O-7460-treated HFD mice respectively). Furthermore, when peripheral tissues were analysed for 2-AG content, no differences were observed in the WAT, whereas the 2-AG concentration in liver was modified, as in the hypothalamus. In fact, 2-AG levels in the liver were up-regulated after 10 days of HFD (from 3.39 ± 0.25 to 6.41 ± 1.25 pmol·mg−1 tissue in standard diet and HFD respectively), and were significantly reduced after 60 min from O-7460 (12 mg·kg−1) i.p. injection (6.22 ± 1.42 to 3.46 ± 0.03 pmol·mg−1 tissue in vehicle- and O-7460-treated mice respectively). Data are means ± SEM of n = 3 or 4 mice, with P < 0.05 for all the differences described above, as assessed by one-way anova followed by the Bonferroni test.
Discussion
We have reported here the synthesis and pharmacological activity of O-7460, a novel potent and relatively selective inhibitor of the biosynthesis of the endocannabinoid 2-AG, and have confirmed, through the use of this compound in mice fed with HFD, that inhibition of DAGLα might represent a new strategy to counteract hyperphagia and obesity. We have previously described a series of fluorophosphonate inhibitors of 2-AG biosynthesis by DAGLα (Bisogno et al., 2006), and showed more recently that the substitution, in the most potent of these compounds, that is, O-3841, of its methoxyl moiety with a tert-butoxyl group yielded O-5596, a relatively more stable and cell membrane permeant DAGLα inhibitor, with some inhibitory activity in vivo against palatable versus normal chow intake in mice (Bisogno et al., 2009a). However, compounds obtained by replacing all H atoms of the methoxyl moiety of O-3841 had not been investigated yet. Hence, in this study we synthesized three new compounds, O-7459, O-7460, O-7458, in which one or two H atoms of O-3841 were replaced with one or two methyl groups or an n-butane group respectively. Furthermore, we also synthesized O-7344, that is, an O-3841 analogue without the phosphonate moiety, which we reasoned would allow us to establish the importance of this group for inhibitory activity. Indeed, among these new compounds, O-7344 exhibited no DAGLα inhibitory activity up to 10 μM, confirming that the fluoro-methyl-phosphinoyloxy group present in the chemical structure of both O-3841 and its tert-butoxymethyl derivative, O-5596 (Bisogno et al., 2006; 2009a), is strictly required for these compounds to covalently inhibit this enzyme. Accordingly, O-7459 and O-7460 inhibited DAGLα and were selective for CB1 and CB2 receptors as well as for FAAH and MAGL, but were less potent than the previously reported O-5596. Indeed, the rank order of potency against DAGLα of this previous compound and the new compounds reported here was O-7459 < O-7460 < O-5596, and hence increased with the increasing number of methyl groups and, therefore, with the steric hindrance of the substituent. Thus, replacement of all the H atoms in the methoxyl moiety of O-3841 with methyl groups is required to increase the potency of this phosphonate derivative of oleic acid as a DAGLα inhibitor. On the other hand, the substitution of its methoxyl moiety with a pentyloxy group, as in O-7458, yielded a less-potent inhibitor of the enzyme, again suggesting that a reduction in the steric hindrance, due in this case to the presence of a linear C5-aliphatic chain instead of a tert-butyl group, has a negative effect on its potency against DAGLα. Importantly, and as expected from this class of compounds, O-7460 inhibited DAGLα in a seemingly irreversible way, because its effect could not be removed by repeated washing of the DAGLα membrane preparations.
We showed here that the most potent compound developed in this study, O-7460, is ∼20-fold selective towards both human and primate (COS-7) MAGL, rat FAAH, and human CB1 and CB2 receptors [please note that some linear aliphatic methyl-fluoro-phosphonates were also previously found to potently and irreversibly bind to CB1 receptors (Fernando and Pertwee, 1997; Martin et al., 2000)]. However, possibly the best way to rule out ‘off targets’ for newly developed inhibitors of serine hydrolases is through the use of affinity-based proteomic profiling (Long et al., 2009), which we performed here using a mouse brain proteome. We show that O-7460, up to a concentration of 10 μM, performed rather well in this screen as we could recognize only one other serine hydrolase, KIAA1363, which this compound inhibited at concentrations comparable to those needed to inhibit DAGLα, and the function of which in conditions other than cancer and cholesteryl ester hydrolysis is still poorly understood (Chang et al., 2011). The assay also confirmed that O-7460 inhibited MAGL only at concentrations ≥10 μM, and showed that O-7459, which is slightly less potent as an inhibitor of DAGLα than O-7460, is also less potent as a KIAA1363 inhibitor, but perhaps slightly more potent at inhibiting MAGL, whereas O-7458 was completely inactive at inhibiting this latter enzyme.
We also showed here that O-7460, like other DAGLα inhibitors, inhibits 2-AG de novo biosynthesis in intact cells. In mouse neuroblastoma N18TG2 cells stimulated with ionomycin, a nearly maximally effective (IC80) concentration of the compound was found to cause a reduction in 2-AG levels comparable to that induced, under identical experimental conditions, by a IC80 concentration of the more potent OMDM188 (Ortar et al., 2008), and stronger than that induced by a IC80 concentration of O-5596 (Bisogno et al., 2009a), perhaps suggesting that plasma membrane permeability of a phosphonate DAGLα inhibitor like O-7460 might be lower than that of a non-phosphonate inhibitor such as OMDM188, but higher than that of the other phosphonate, O-5596. On the other hand, OMDM188 was developed starting from tetrahydrolipstatin, a lipase inhibitor with low reported permeability through the gastrointestinal tract, which limits its bioavailability after oral administration (Ballinger, 2000). Hence, the necessity to develop DAGL inhibitors belonging to other chemical classes but still suitable for in vivo use.
When administered i.p. to mice, O-7460 dose-dependently inhibited the intake of HFD, as one would expect from a ‘suppressor’ of central endocannabinoid tone, and it did so at doses comparable to those previously shown to be necessary for CB1 receptor inverse agonists to suppress food intake when administered i.p. In fact, rimonabant and AM251 (Simiand et al., 1998; Mathes et al., 2008), two widely used CB1 inverse agonists, as well as O-5596 (Bisogno et al., 2009a), an inhibitor of 2-AG biosynthesis, have been previously reported to reduce the intake of palatable food in mice. Perhaps more relevant to our present results, a HFD-induced increase in hypothalamic 2-AG levels, and the subsequent overactivation of CB1, were reported to be responsible for mouse preference for HFD, because the intake of this diet was suppressed by a neutral CB1 antagonist, O-2050 (Higuchi et al., 2010; 2011). Here, we not only confirmed that such an increase in hypothalamic 2-AG concentrations also occurred in our HFD mice, but showed that the high-fat dietary regimen also elevates the levels of this endocannabinoid in the liver. Importantly, concomitant with its effect on food intake, O-7460 also reduced 2-AG levels in both the hypothalamus and liver. Thus, it also likely that in the mice used in our study, which were habituated to HFD consumption for several days, up-regulated hypothalamic 2-AG levels contribute to stronger HFD consumption and preference, and that O-7460 reduces the consumption of the HFD both 30 and 60 min after administration, as well as over a period of 14 h, by producing an acute inhibition of DAGLα, an enzyme abundantly expressed in the hypothalamus (Suárez et al., 2011).
We also showed here, for the first time, that acute DAGLα inhibition causes a reduction in BW. In fact, mice treated with the highest dose of O-7460, unlike mice treated with vehicle, not only did not gain weight over the observation period, but even experienced a small but statistically significant reduction in BW. It is likely that this latter effect is due to the reduction of HFD consumption described above, although other possible mechanisms cannot be ruled out. In fact, very recently, 2-AG was suggested to participate in the regulation of the activity of forebrain neural circuits implicated in the control of energy dissipation, based on the observation that transgenic mice overexpressing MAGL in forebrain neurons are resistant to BW gain if fed with a HFD (Jung et al., 2012). Interestingly, although the effects of chronically reduced brain 2-AG levels in a transgenic mouse following congenital up-regulation of 2-AG degradation in forebrain neurons, and the transient reduction of whole body 2-AG levels following acute systemic inhibition of 2-AG biosynthesis, may not necessarily yield similar results, MAGL-overexpressing mice did not eat less HFD than their wild-type littermates (Jung et al., 2012). This may indicate that the effects of O-7460 on HFD consumption and BW, described here, were also exerted at the peripheral level, as suggested also by our present finding of reduced hepatic 2-AG levels in HFD mice following administration of the compound.
In conclusion, we have reported here the synthesis and pharmacological characterization in vitro and in vivo of O-7460, a novel selective inhibitor of DAGLα, belonging to the class of fluorophosphonates. Our data suggest that O-7460 might be considered a useful pharmacological tool to further investigate the role played by 2-AG in physiological and pathological conditions, and particularly in the framework of endocannabinoid function in the control of food intake and energy homeostasis. Since DAGLα inhibitors have been shown to only counteract the stimulated biosynthesis of 2-AG, with no effect on the basal levels of this endocannabinoid (Bisogno et al., 2003; 2006; present in vitro data; see Alger and Kim, 2011, for review), these compounds, like ‘neutral’ CB1 agonists, and unlike CB1 ‘inverse agonists’, might reduce endocannabinoid tone only when and where this is pathologically altered. They might, therefore, be used to treat conditions determined by pathologically elevated 2-AG levels, with fewer side effects than compounds like rimonabant and taranabant (Di Marzo and Després, 2009).
Acknowledgments
We thank Dr. Fabiana Piscitelli, Institute of Biomolecular Chemistry, C.N.R. Pozzuoli (Napoli), Italy, for valuable help and advice.
We are grateful to the NIH for partly funding this study (Grant No. DA009789).
Glossary
- 2-AG
2-arachidonoylglycerol
- CB1
cannabinoid receptor subtype 1
- CB2
cannabinoid receptor subtype 2
- COS-7
monkey kidney fibroblast cell line
- CP-55940
(1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl) phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol
- DAG
diacylglycerol
- DAGL
diacylglycerol lipase
- FAAH
fatty acid amide hydrolase
- HFD
high-fat diet
- MAGL
monoacylglycerol lipase
- N18TG2
mouse neuroblastoma cell line
- O-3640
octadec-9-enoic acid-1-(fluoro-methyl-phosphoryloxymethyl)-propylester
- O-3841
octadec-9-enoic acid 1-methoxymethyl-2-(fluoro-methyl-phosphinoyloxy)-ethyl ester
- O-5596
octadec-9-enoic acid 1-tert-butoxymethyl-2(fluoro-methyl-phosphinoyloxy)-ethyl ester
- O-7344
(S)-1-methoxy-5-oxopentan-2-yl oleate
- O-7458
1-((fluoro(methyl)phosphoryl)oxy)-3-(penthyloxy)propan-2-yl oleate
- O-7459
1-ethoxy-3-((fluoro(methyl)phosphoryl)oxy)propan-2-yl oleate
- O-7460
1-((fluoro(methyl)phosphoryl)oxy)-3-isopropoxypropan-2-yl oleate
- OMDM188
N-formyl-L-isoleucine-(1S)-1-[[(2S,3S)-3-hexyl-4-oxo-2-oxetanyl]-methyl]dodecyl ester
- WIN55312-2
(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenyl methanone mesylate
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Synthesis, purification and spectroscopic characterization of O-7460.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE, Kim J. Supply and demand for endocannabinoids. Trends Neurosci. 2011;34:304–315. doi: 10.1016/j.tins.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger A. Orlistat in the treatment of obesity. Expert Opin Pharmacother. 2000;1:841–847. doi: 10.1517/14656566.1.4.841. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, et al. Cloning of the first sn-1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Cascio MG, Saha B, Mahadevan A, Urbani P, Minassi A, et al. Development of the first potent and specific inhibitors of endocannabinoid biosynthesis. Biochim Biophys Acta. 2006;176:205–212. doi: 10.1016/j.bbalip.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Burston JJ, Rai R, Allarà M, Saha B, Mahadevan A, et al. Synthesis and pharmacological activity of a potent inhibitor of the biosynthesis of the endocannabinoid 2-arachidonoylglycerol. ChemMedChem. 2009a;4:946–950. doi: 10.1002/cmdc.200800442. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Ortar G, Petrosino S, Morera E, Palazzo E, Nalli M, et al. Development of a potent inhibitor of 2-arachidonoylglycerol hydrolysis with antinociceptive activity in vivo. Biochim Biophys Acta. 2009b;1791:53–60. doi: 10.1016/j.bbalip.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Cascio MG, Minassi A, Ligresti A, Appendino G, Burstein S, Di Marzo V. A structure-activity relationship study on N-arachidonoyl-amino acids as possible endogenous inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Commun. 2004;314:192–196. doi: 10.1016/j.bbrc.2003.12.075. [DOI] [PubMed] [Google Scholar]
- Chang JW, Nomura DK, Cravatt BF. A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem Biol. 2011;18:476–484. doi: 10.1016/j.chembiol.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V. Targeting the endocannabinoid system: to enhance or reduce? Nat Rev Drug Discov. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Després JP. CB1 antagonists for obesity – what lessons have we learned from rimonabant? Nat Rev Endocrinol. 2009;5:633–638. doi: 10.1038/nrendo.2009.197. [DOI] [PubMed] [Google Scholar]
- Fernando SR, Pertwee RG. Evidence that methyl arachidonyl fluorophosphonate is an irreversible cannabinoid receptor antagonist. Br J Pharmacol. 1997;121:1716–1720. doi: 10.1038/sj.bjp.0701303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin K, Paxinos G. the mouse brain in stereotaxic co-ordinates. San Diego: Academic Press; 1998. [Google Scholar]
- Higuchi S, Irie K, Mishima S, Araki M, Ohji M, Shirakawa A, et al. The cannabinoid 1-receptor silent antagonist O-2050 attenuates preference for high-fat diet and activated astrocytes in mice. J Pharmacol Sci. 2010;112:369–372. doi: 10.1254/jphs.09326sc. [DOI] [PubMed] [Google Scholar]
- Higuchi S, Ohji M, Araki M, Furuta R, Katsuki M, Yamaguchi R, et al. Increment of hypothalamic 2-arachidonoylglycerol induces the preference for a high-fat diet via activation of cannabinoid 1 receptors. Behav Brain Res. 2011;216:477–480. doi: 10.1016/j.bbr.2010.08.042. [DOI] [PubMed] [Google Scholar]
- Janero DR. Cannabinoid-1 receptor (CB1R) blockers as medicines: beyond obesity and cardiometabolic disorders to substance abuse/drug addiction with CB1R neutral antagonists. Expert Opin Emerg Drugs. 2012;17:17–29. doi: 10.1517/14728214.2012.660916. [DOI] [PubMed] [Google Scholar]
- Jung KM, Clapper JR, Fu J, D'Agostino G, Guijarro A, Thongkham D, et al. 2-arachidonoylglycerol signaling in forebrain regulates systemic energy metabolism. Cell Metab. 2012;15:299–310. doi: 10.1016/j.cmet.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludányi A, Hu SS, Yamazaki M, Tanimura A, Piomelli D, Watanabe M, et al. Complementary synaptic distribution of enzymes responsible for synthesis and inactivation of the endocannabinoid 2-arachidonoylglycerol in the human hippocampus. Neuroscience. 2011;174:50–63. doi: 10.1016/j.neuroscience.2010.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Martin BR, Beletskaya I, Patrick G, Jefferson R, Winckler R, Deutsch DG, et al. Cannabinoid properties of methylfluorophosphonate analogs. J Pharmacol Exp Ther. 2000;294:1209–1218. [PubMed] [Google Scholar]
- Mathes CM, Ferrara M, Rowland NE. Cannabinoid-1 receptor antagonists reduce caloric intake by decreasing palatable diet selection in a novel dessert protocol in female rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R67–R75. doi: 10.1152/ajpregu.00150.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Compton, identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Ortar G, Bisogno T, Ligresti A, Morera E, Nalli M, Di Marzo V. Tetrahydrolipstatin analogues as modulators of endocannabinoid 2-arachidonoylglycerol metabolism. J Med Chem. 2008;51:6970–6979. doi: 10.1021/jm800978m. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addict Biol. 2008;13:147–159. doi: 10.1111/j.1369-1600.2008.00108.x. [DOI] [PubMed] [Google Scholar]
- Simiand J, Keane M, Keane PE, Soubrié P. SR 141716, a CB1 cannabinoid receptor antagonist, selectively reduces sweet food intake in marmoset. Behav Pharmacol. 1998;9:179–181. [PubMed] [Google Scholar]
- Suárez J, Ortíz O, Puente N, Bermúdez-Silva FJ, Blanco E, Fernández-Llebrez P, et al. Distribution of diacylglycerol lipase alpha, an endocannabinoid synthesizing enzyme, in the rat forebrain. Neuroscience. 2011;192:112–131. doi: 10.1016/j.neuroscience.2011.06.062. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Ward SJ, Raffa RB. Rimonabant redux and strategies to improve the future outlook of CB1 receptor neutral-antagonist/inverse-agonist therapies. Obesity (Silver Spring) 2011;19:1325–1334. doi: 10.1038/oby.2011.69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.