

Abstract
Background and Purpose
Emerging evidence indicates that the balance between pro-inflammatory cytokines (PICs) and anti-inflammatory cytokines (AICs) within the brain is an important determinant in the outcome of hypertension. However, the mechanism by which this dysregulation occurs is not known. We aimed to investigate whether AngII induces imbalance between PIC and AIC by modulating downstream transcription factors, NFκB and cyclic AMP response element-binding protein (CREB), and whether AngII-induced effects are mediated by glycogen synthase kinase-3β (GSK-3β).
Experimental Approach
CATH.a neurons were exposed to AngII (10 nM–1 μM) over a preset time course. In another set of experiments, GSK-3β was knock down by using lentivirus containing short hairpin RNA targeting GSK-3β (L-sh-GSK3β) before AngII exposure. Cell extracts were subjected to RT-PCR, immunoblot and immunoprecipitation.
Key Results
AngII caused time-dependent increase in PICs (TNF-α and IL-1β) and reduction in AIC (IL-10). AngII exposure caused reduced phosphorylated CREB(Ser-133) and increased p-NFκB(Ser-276) levels, leading to reduced CREB-CBP and increased NFκB-CBP binding. These results were accompanied by increased activation of GSK-3β, as indicated by increased p-GSK3(Tyr-216) to p-GSK3(Ser-9) ratio. In a subsequent study, pretreatment with L-sh-GSK3β attenuated AngII-induced alterations in PICs and IL-10 by augmenting CREB-CBP and attenuating NFκB-CBP binding.
Conclusions and Implications
Collectively, these findings are the first to provide direct evidence that AngII-induced dysregulation in cytokines is mediated by GSK-3β-mediated alterations in downstream transcription factors in neuronal cells. Our data also reveal that AngII-induced effects could be alleviated by GSK-3β inhibition, suggesting GSK-3β as an important therapeutic target for hypertension that is characterized by increased PICs and NFκB activation.
Keywords: cytokines, angiotensin, hypertension, GSK-3, CATH.a, anti-inflammatory cytokines, brain, PVN
Introduction
Cardiovascular diseases (CVDs) are the leading cause of death in the United States, and of all the CVD conditions, hypertension has the highest prevalence. According to the most recent report from the American Heart Association, an estimated 76.4 million adults ≥20 years of age have high BP (Roger et al., 2011). Despite the success of anti-hypertensive medications such as angiotensin converting enzyme inhibitors, angiotensin receptor blockers and β-adrenergic receptor blockers in reducing BP, the incidence and prevalence of hypertension continues to rise. These statistics clearly suggest the need for novel therapeutic strategies for the treatment of hypertension.
Inflammation is a well-known risk factor for various CVDs including hypertension (Agarwal et al., 2011; Guggilam et al., 2011). Pro-inflammatory cytokines (PICs), such as TNF-α (Dorffel et al., 1999), IL-1β (Dorffel et al., 1999; Peeters et al., 2001) and IL-6 (Chae et al., 2001; Peeters et al., 2001), have been reported to increase with the severity of hypertension and to be of prognostic significance. Besides circulating cytokines, brain cytokines have also been implicated in the pathogenesis of the disease (Guggilam et al., 2011; Kang et al. 2011). Recent discoveries indicate that in addition to elevated levels of circulating and brain PICs (Peeters et al., 2001; Shi et al., 2010), anti-inflammatory cytokines (AICs) such as IL-10 have a significant impact on arterial pressure and cardiac remodelling in experimental models of hypertension (Shi et al., 2010). Additionally, an overactivation of the renin-angiotensin system (RAS) directly or indirectly through PIC plays a vital role in the pathogenesis of hypertension.
The most important transcription factors, viz., NFκB and cyclic AMP response element-binding protein (CREB), are known to play a central role in modulating the gene expression of inflammatory mediators involved in hypertension. However, unlike NFκB, which positively regulates gene expression of PICs (Kang et al. 2011), activation of CREB positively regulates expression of AICs such as IL-10 (Avni et al., 2010). Competition between NFκB and CREB for binding to the co-activator CREB-binding protein (CBP) is important in regulating their transcriptional activity (Grimes and Jope, 2001a; Shenkar et al., 2001). Although angiotensin II (AngII), a major effector molecule of RAS, has been shown to elevate PIC levels in the brain, the effects of overactivation of RAS on AICs are not very well understood. Also, the exact mechanisms underlying AngII-induced effects on inflammatory cytokines are still poorly understood.
Recently, glycogen synthase kinase (GSK)-3 has gained increasing attention from the scientific community due to its role in many biological processes. Research findings in the past several years have now established that GSK-3 acts as a regulatory switch that determines the output of numerous signalling pathways initiated by diverse stimuli (Frame and Cohen, 2001; Grimes and Jope, 2001b; Woodgett, 2001). Of the two isoforms (-α and -β), GSK-3β is particularly abundant in the CNS and is neuron-specific (Leroy and Brion, 1999). Recently, GSK-3β has been reported to modulate the production of inflammatory cytokines in an NFκB-dependent manner (Martin et al., 2005; Steinbrecher et al., 2005; Vines et al., 2006; Beurel and Jope, 2009). However, the role of GSK-3β in AngII-induced dysregulation of inflammatory molecules within the brain has not yet been explored.
Therefore, the present series of studies were undertaken to investigate the novel role of GSK-3β in AngII-induced dysregulation of inflammatory cytokines in neuronal cells. We hypothesized that (i) AngII causes an imbalance between PIC and AIC; (ii) AngII-induced imbalance in PIC and AIC is modulated by downstream transcription factors, NFκB and CREB; and (iii) dysregulation in PIC and AIC are mediated by GSK-3β. To address these, we used CATH.a neurons (a hybridoma derived from mouse locus coeruleus), a catecholaminergic cell line that expresses AT1R and AT2R and has been identified as a reliable cell culture model for investigating AngII intra-neuronal signalling (Sun et al., 2002; 2003; Li et al., 2007). In this study, we constructed highly efficient lentiviral short hairpin RNA (shRNA) targeting GSK-3β to examine the role of GSK-3β in AngII-mediated effects. The results of this study will help us develop newer therapeutics targets for the treatment of hypertension.
Materials and methods
Neuronal cell culture
The CATH.a neurons (stock no. CRL-11179, American Type Culture Collection, Manassas, VA, USA) were grown in RPMI 1640 media containing 4% (v/v) FBS, 8% horse serum and 100 IU·mL−1 penicillin, at 37°C in a humidified atmosphere of 95% air and 5% CO2. Unless otherwise stated, cells were plated at a density of 4 × 106 cells per 60 mm dish or 1 × 107 cells per 100 mm plate. All experiments were performed when the cultures were 70–80% confluent. Before treatment, the cells were allowed to differentiate in serum-free media for 48–72 h (h). To investigate the effects of AngII on inflammatory cytokines, CATH.a cells were exposed to AngII or vehicle. We first performed a pilot experiment to validate the best concentration and time point for AngII stimulation of CATH.a cells, using a concentration range from 10 nM to 1 μM of AngII in culture medium over a stipulated time course (0–24 h). At a defined time point, cells were harvested for real-time RT-PCR for mRNA analysis of TNF-α, we chose TNF-α mRNA levels as the end point because of the focus of the present study. As depicted in Figure 1, we observed that 100 nM is the lowest concentration of AngII that exerts maximum effect on TNF-α mRNA expression in CATH.a neurons. AngII at 1 μM did not cause any additional increase in TNF-α expression and the lowest dose (10 nM) did not produce sufficient increase. The optimal AngII concentration of 100 nM has also been utilized in several previous studies investigating the intra-neuronal AngII signalling in CATH.a cells (Sun et al., 2002; Zimmerman et al., 2004; Mitra et al., 2010; Haack et al., 2012). In all subsequent experiments, cells were exposed to AngII (100 nM) for 6 h, a time point that induces maximal changes in the levels of inflammatory cytokines (Figure 3). In another set of experiments, cells were transfected with lentiviral short hairpin RNA targeting GSK-3β (L-sh-GSK3β) for 48 h before AngII exposure for 6 h. Following exposure to agonists, cells were harvested for real-time RT-PCR, Western blot, immunoprecipitation (IP) and immunoflurorescence analysis. Results are presented as the means ± SD and represent set of three independent experiments in CATH.a cells. In each experiment, n = 6 per treatment groups were used.
Figure 1.
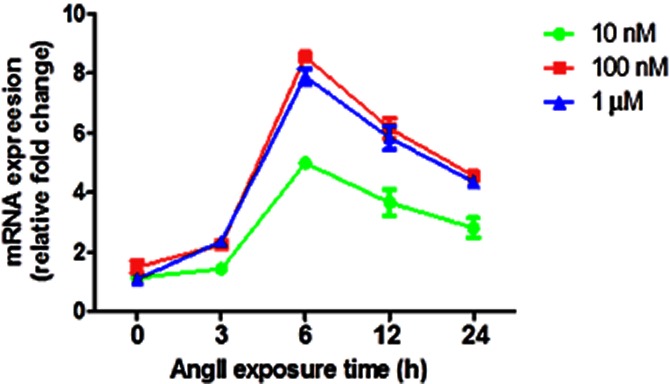
Dose–response relationship between AngII concentration in the culture media and mRNA expression of TNF-α in whole cell lysate in CATH.a cell culture. A pilot experiment was performed to validate the best concentration and time-point for Ang II stimulation of CATH.a cells, using a concentration range from 10 nM to 1 μM of Ang II in culture medium over a stipulated time course (0–24 h). Results showed that 100 nM (at 6 h) is the lowest concentration of Ang II that exerts maximum effect on TNF-α mRNA expression in CATH.a neurons. The results are means ± SD of three independent experiments (n = 6 per treatment groups in each experiment).
Figure 3.
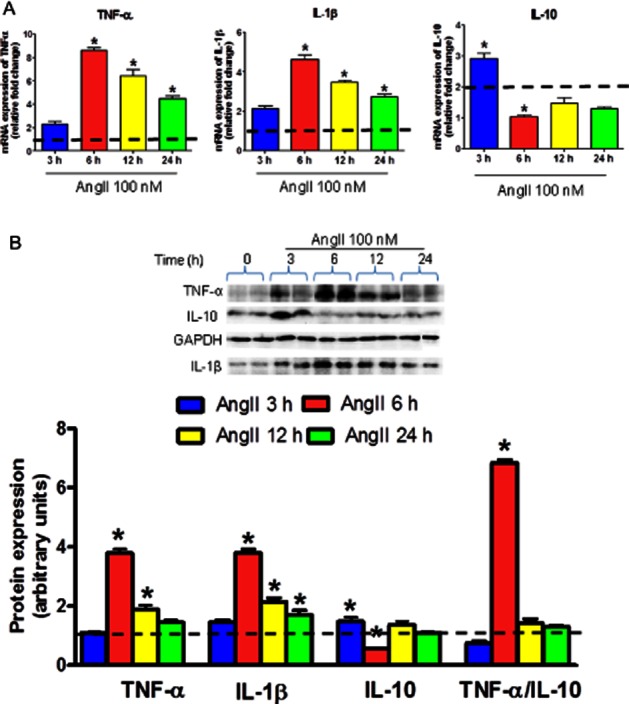
Effects of AngII treatment on TNF-α, IL-1β and IL-10 expression levels in neuronal cells. Serum-starved CATH.a cells were stimulated with 100 nM AngII for the indicated time. (A) mRNA expression of TNF-α, IL-1β and IL-10. (B) A representative Western blot and densitometric analysis of protein expression of TNF-α, IL-1β and IL-10. Quantitative Western blot analysis is shown as the ratio of intensities of the protein of interest and GAPDH, relative to unstimulated control cells (represented by the dashed line). AngII resulted in increased TNF-α and IL-1β, and reduced IL-10 levels indicating an imbalance between PIC and AIC in the CATH.a cells in time-dependent manner with maximum alterations at 6 h of AngII treatment. The results are means ± SD of three independent experiments (n = 6 per treatment groups in each experiment). *P < 0.05 compared to their respective vehicle-treated groups.
Lentiviral construction and transduction
We explored the effects of inhibition of GSK-3β by using gene knock-down approach: RNA interference (RNAi) through delivery of a small hairpin RNA (shRNA) against GSK-3β using a lentiviral vector (L-sh-GSK3β) containing the target sequence 5′-CATGAAAGTTAGCAGAGATAA-3′. L-sh-GSK3β was commercially obtained (NitAn Biotech LLC, Columbus, OH, USA) and these vectors were tagged with eGFP. A scrambled sequence of the same length was used as a control (mentioned as L-scrambled in text). Twenty-four hours after plating, CATH.a cells were transduced (in triplicate) separately in 6-well laminin coated plates with 30 MOI (multiplicity of infection, which is equal to ratio of infectious viral particles to cell) of L-sh-GSK3β and scrambled sequence (L-scrambled) viral particles in the presence of 8 μg·mL−1 of polybrene. We use 2 mL of viral supernatant, which contain 2 × 107–108 viral particles for each transduction experiment. After 48 h, Western blotting was performed to assess the silencing effects of L-sh-GSK3β. Cells were stimulated with AngII 48 h after transduction. Cells were also transduced with L-scrambled separately in presence of AngII. Densitometric analysis of immunoblot showed that cells transduced with L-sh-GSK3β (MOI 30) had significantly lower (more than 60% reduction) protein expression of GSK-3β when compared to cells transduced with scrambled sequence (Figure 2). These results confirmed efficient suppression of GSK-3β by L-sh-GSK3β in neuronal cells.
Figure 2.
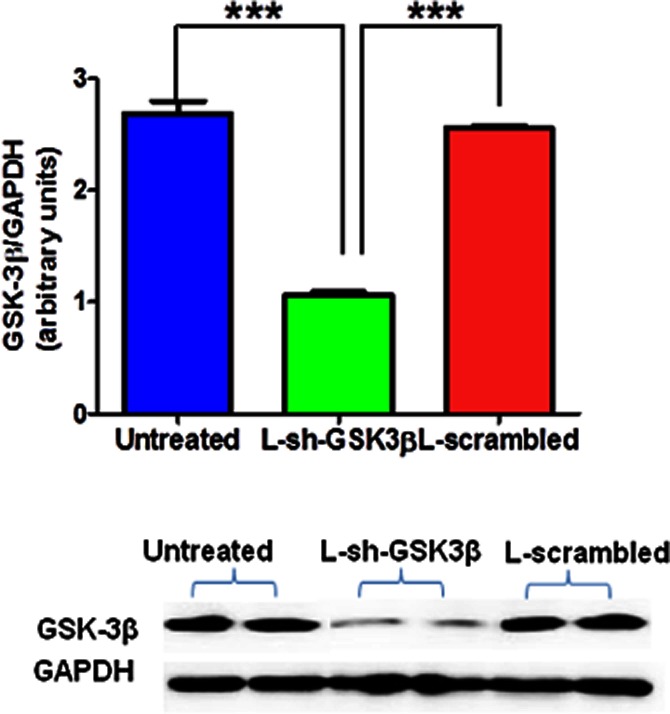
Transduction efficiency of lentiviral shRNA targeting GSK3β (L-sh-GSK3β) in CATH.a cells. Serum-starved CATH.a cells were transduced with L-sh-GSK3β at a multiplicity of infection (MOI) of 30 for 48 h. An immunoblot analysis (upper panel) and a representative blot (lower panel) showing efficient suppression of GSK-3β protein expression by shRNA. The results are means ± SD of three independent experiments (n = 6 per treatment groups in each experiment). ***P < 0.001.
RNA extraction and real-time RT-PCR
Semi-quantitative real-time RT-PCR was used to determine the mRNA levels of TNF-α, IL-1β and IL-10 in CATH.a neurons by using specific primers (Table 1). Total RNA isolation, cDNA synthesis and RT-PCR were performed as previously described (Agarwal et al., 2009). Semi-log amplification curves were evaluated by the comparative quantification method (2−ΔΔCt), and GAPDH was used for normalization of all reported gene expression levels. The data are presented as the fold change of the gene of interest relative to that of control group.
Table 1.
Primer sequences used for PCR and amplified product sizes
| Gene | Primer | Sequence (5′ to 3′) | Size (bp) |
|---|---|---|---|
| GAPDH | Forward | TGAATGACATCAAGAAGGTGGTGGAG | 239 |
| Reverse | TCCTTGGAGGCCATGTAGGCCAT | ||
| IL-1β | Forward | CTGTGTCTTTCCCGTGGACC | 200 |
| Reverse | CAGCTCATATGGGTCCGACA | ||
| IL-10 | Forward | CCAGTTTTACCTGGTAGAAGTGATG | 324 |
| Reverse | TGTCTAGGTCCTGGAGTCCAGCAGACTC | ||
| TNF-α | Forward | CCTCCCTCTCATCAGTTCTA | 501 |
| Reverse | GCAATGACTCTAAAGTAGACCTG |
Immunoblot analysis
For whole cell extracts, cells were washed twice with ice-cold PBS and were scraped into in100 μL per dish of cell lysis buffer (Cell Signaling Technology, Inc., Danvers, MA, USA) containing protease and phosphatase inhibitors. Samples were incubated on ice for 10 min and then centrifuged (10 000× g, 5 min, 4°C). The supernatants were retained. Protein concentrations were determined by the Bradford method (Bradford, 1976). The lysates were stored at −80°C until used for immunoblotting.
Cell lysates were mixed with Laemmli sample buffer (Bio-Rad Laboratories, Berkeley, CA, USA) and placed in a boiling water bath for 5 min. Proteins (30 μg) were separated by SDS-PAGE using 10–15% (w/v) resolving gels and 6% (w/v) stacking gels, and then transferred to nitrocellulose membrane. Non-specific binding sites were blocked with 1% (w/v) casein (for non-phosphorylated antibodies) in PBS or 1% (w/v) BSA (for phosphorylated antibodies) in TBST [20 mM Tris–HCl (pH 7.5), 137 mM NaCl, 0.1% (v/v) Tween 20]. Blots were probed (overnight, 4°C) with the primary antibodies. Specific antibodies used included TNF-α, IL-1β, IL-10, GSK-3β, p-GSK3β(Ser-9), p-GSK3β(Tyr-216), p-CREB(Ser-133) and CBP at 1:1000 dilution. Antibodies were commercially obtained: TNF-α (Abcam Inc., Cambridge, MA, USA); IL-1β (AbD Serotec, Oxon, UK), IL-10 (Abbiotec, San Diego, CA, USA); GSK-3β (BD Transduction Laboratories, San Jose, CA, USA), p-GSK3β(Ser-9), p-GSK3β(Tyr-216), p-CREB(Ser-133) and p-p65(Ser-276) (Cell Signaling Technology, Inc.); and CBP (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Blots were washed in TBST, incubated (60 min, room temperature) with HRP-conjugated secondary antibodies (1:10 000) in blocking solution. Immunoreactive bands were visualized using enhanced chemiluminescence (ECL Plus, Amersham, Piscataway, NJ, USA);, band intensities were quantified using VersaDoc MP 5000 imaging system (Bio-Rad Laboratories) and were normalized with GAPDH.
Immunoprecipitation
To determine the role of GSK-3β in regulating downstream transcription factors, we used the catch and release IP system (Upstate Biotechnology, Billerica, MA, USA) as described previously (Martin et al., 2005). For these experiments, protein CBP was immunoprecipitated by incubating cell lysates with 2 μg of CBP monoclonal antibody (Pharmingen, San Diego, CA, USA) overnight at 4°C. Samples were incubated with 60 μL of protein G sepharose beads (Amersham) for 1 h at 4°C with gentle agitation. The immune complexes were washed three times with lysis buffer. Samples in Laemmli buffer were placed in a boiling water bath, proteins were separated by SDS-PAGE, and samples were immunoblotted with anti-p-CREB(Ser-133) or anti-p-p65(Ser-276). The membranes were re-probed with an anti-CBP antibody to confirm the efficiency and specificity of IP.
Statistical analysis
Statistical analysis was completed by either unpaired t-test or one-way anova with Bonferroni post hoc test using Graph Pad Prism software (version 5.0) (GraphPad Software, Inc., La Jolla, CA, USA). Data are presented as the fold change of each gene of interest relative to controls. Results were considered significant when P < 0.05.
Results
AngII causes an imbalance between pro- and anti-inflammatory cytokines in neuronal cells
To investigate the influence of AngII on PICs and AIC in the neuronal cells, CATH.a cells were exposed to AngII (100 nM) for indicated time and then we examined the mRNA (Figure 3A) and protein (Figure 3B) levels of TNF-α, IL-1β, and IL-10 in whole cell extracts. We observed that AngII-treated cells exhibited time-dependent increase in TNF-α and IL-1β levels with maximal effects at 6 h of exposure. At mRNA level, AngII exposure (6 h) resulted in eightfold increase in TNF-α and about fourfold increase in IL-1β expression in CATH.a neurons (Figure 3A). TNF-α and IL-1β levels in AngII-exposed cells were reduced after 12 and 24 h of exposure when compared with 6 h; however, it remains elevated in comparison with vehicle-treated cells. On the contrary, IL-10 levels in cells treated with AngII for 6 h were significantly lower when compared to vehicle-treated cells. At mRNA level, AngII exposure (6 h) resulted in more than twofold decrease in IL-10 expression (Figure 3A). At 12 and 24 h, IL-10 levels remained lower in comparison with vehicle groups, although the differences were not significant. Noteworthy, AngII exposure for 1 h significantly up-regulated IL-10 levels, whereas TNF-α level was slightly higher at this time point.
To further confirm that AngII causes an imbalance between PIC and AIC in neuronal cells, we determined the alterations in ratio of TNF-α to IL-10 protein levels in cells treated with AngII or vehicle. A significant increase of more than sixfold in TNF-α /IL-10 protein ratio was observed upon 6 h of AngII exposure when compared to all other groups (Figure 3B). These data provide evidence that AngII exposure results in an imbalance between PIC and AIC in favour of PICs in CATH.a neurons.
AngII induces activation of GSK-3β in neuronal cells
To investigate whether AngII exposure for 6 h (maximal effective exposure time) activates GSK-3β, we determined the protein expression levels of p-GSK3β(Ser-9) and p-GSK3β(Tyr-216) by immunoblot analysis in neuronal cells exposed with AngII or vehicle. The activity of GSK-3β is mainly regulated by post-translational phosphorylation with N-terminal phosphorylation of GSK-3β at Ser-9 has an inhibitory effect, whereas phosphorylation of Tyr-216 activates it (Forde and Dale, 2007). Our immunoblot analysis demonstrated that GSK-3β are expressed in CATH.a neurons and there was a slight but significant increase in the phosphorylation of GSK3β(Ser-9) (Figure 4B; quantitation in Figure 4E). Simultaneously, however, AngII dramatically up-regulated phosphorylation of GSK3β(Tyr-216), in CATH.a cells (Figure 4C; quantitation in Figure 4E). Densitometric analysis further revealed that the ratio of protein expression of p-GSK3β(Tyr-216) to p-GSK3β(Ser-9) is significantly up-regulated on AngII exposure (Figure 4D). As phosphorylation of GSK3β(Tyr-216) is essential for their catalytic activity, these results indicate overall activation of GSK-3β upon AngII (100 nM) exposure. The same blots were stripped and re-probed for native GSK-3β showing no significant difference on native GSK-3β expression between the vehicle and AngII-treated groups (Figure 4A). These findings suggest that AngII-induced effects in neuronal cells could be mediated by activation of GSK-3β.
Figure 4.

Effects of AngII treatment on total and phosphorylated GSK-3β expression in neuronal cells. Serum-starved CATH.a cells were stimulated with 100 nM AngII for 6 h and cell extracts were then subjected to protein analysis by Western blot. Densitometric analysis of Western blot results showing protein expression of (A) total GSK-3β, (B) p-GSK3β(Ser-9), (C) p-GSK3β(Tyr-216), (D) p-GSK3β(Tyr-216)/p-GSK3β(Ser-9) ratio, and (E) a representative Western blot. AngII caused significant activation of GSK-3β as indicated by reduced p-GSK3β(Ser-9), increased p-GSK3β(Tyr-216), and increased ratio of p-GSK3β(Tyr-216) to p-GSK3β(Ser-9) protein expression in CATH.a neurons. AngII exposure did not alter total GSK-3β protein levels. The results are means ± SD of three independent experiments. *P < 0.05; ***P < 0.001 compared with cells treated with vehicle.
AngII exposure resulted in altered binding of CBP with CREB and NFκB in neuronal cells
To investigate whether AngII induced imbalance in PIC and AIC is mediated by alterations in downstream transcription factors NFκB subunit p65 and CREB, we assessed the binding of CBP (co-activator protein) with p65 and CREB by IP analysis of vehicle- and AngII-treated groups. As phosphorylation of CREB at Ser-133 and p65 at Ser-276 have been shown to be essential for their binding with CBP, we also determined the protein levels of p-CREB(Ser-133) and p-p65(Ser-276). AngII exposure resulted in significant reduction in p-CREB(Ser-133) expression and increased p-p65(Ser-276) (Figure 5A), leading to decreased CREB-CBP binding and increased NFκB-CBP binding as confirmed by IP analysis. As demonstrated in Figure 5B, in rat neuronal cells, binding between CBP and p65 has been increased and binding between CBP and CREB has been decreased, as reflected by increased presence of p-p65(Ser-276) and decreased presence of p-CREB(Ser-133) in CBP immunoprecipitates of AngII-exposed cells when compared to cells treated with vehicle. These data suggest that AngII causes an imbalance in cytokine levels by modulating the downstream transcription factors.
Figure 5.

Effects of AngII treatment on CBP to NFκB and CREB binding in neuronal cells. The serum-starved (24 h) CATH.a cells were treated without or with AngII (100 nM; 6 h). (A) Densitometric analysis and a representative immunoblot showing increased expression levels of p-p65(Ser-276) and decreased p-CREB(Ser-133) in AngII-treated cells when compared to vehicle. (B) Densitometric analysis and a representative immunoblot showing increased CBP to p-p65(Ser-276) binding and decreased CBP to p-CREB(Ser-133) binding in AngII stimulated cells as measured by immunoprecipitation analysis. The results are means ± SD of three independent experiments. **P < 0.01; **P < 0.01, compared with cells treated with vehicle.
Inhibition of GSK-3β by lentivirus reversed AngII-mediated imbalance in PIC and AIC in neuronal cells
As shown in Figure 6, AngII-exposed cells had significantly increased levels of TNF-α and IL-1β, and decreased levels of IL-10 when compared to vehicle-treated cells. Interestingly, pretreatment of cells with L-sh-GSK3β resulted in significant reduction in mRNA and protein levels of TNF-α and IL-1β when compared to AngII-treated cells. In addition, IL-10 mRNA and protein levels were significantly higher in AngII + L-sh-GSK3β compared with AngII groups. There were no significant differences between vehicle-treated and L-sh-GSK3β + AngII-treated groups. Furthermore, densitometric analysis showed that TNF-α /IL-10 protein ratio was significantly higher in AngII groups in comparison with vehicle, whereas significant reduction in TNF-α /IL-10 ratio was observed in L-sh-GSK3β +AngII when compared to AngII-treated cells. Cells transduced with scrambled sequences (L-scrambled) in presence of AngII did not show any effect and there was no difference in these levels between AngII and L-scrambled + AngII-exposed cells. These results demonstrate that pretreatment of cells of lentiviral silencing GSK-3β causes reversal of AngII-induced imbalance between PIC and AIC in neuronal cells.
Figure 6.

Inhibitory effects of L-sh-GSK3β on AngII-induced imbalance between PIC and AIC in neuronal cells. Serum-starved CATH.a cells were transduced with L-sh-GSK3β at a multiplicity of infection (MOI) of 30 for 48 h and were stimulated with 100 nM AngII for 6 h. (A) mRNA expression of TNF-α, IL-1β and IL-10. (B) Densitometric analysis and a representative immunoblot showing protein expression of TNF-α, IL-1β and IL-10. Pretreatment with L-sh-GSK3β resulted in reversal of AngII-induced increase in TNF-α and IL-1β, and attenuation in IL-10 levels indicating improved balance between PIC and AIC by GSK-3β inhibition. Cells transduced with scrambled sequences separately in presence of AngII did not show any effect. The results are means ± SD of three independent experiments. *P < 0.05 AngII versus vehicle-treated cells; #P < 0.05 AngII versus L-sh-GSK3β + AngII-treated cells; $P < 0.05 L-scrambled + AngII versus vehicle-treated cells.
Inhibition of GSK-3β by lentivirus reversed AngII-mediated altered binding of CBP to NFκB or CREB in neuronal cells
As shown in Figure 7, AngII exposure resulted in increased CBP to p65 binding and decreased CBP to CREB binding in CATH.a cells. Interestingly, pretreatment of cells with L-sh-GSK3β caused significant reduction in AngII-induced elevation in CBP to p65 binding. In addition, CBP-CREB binding was found to be significantly higher in L-sh-GSK3β + AngII group when compared to AngII-exposed cells. Furthermore, L-sh-GSK3β + AngII-treated cells exhibited significantly elevated levels of p-CREB(Ser-133) and reduced levels of p-p65(Ser-276), in comparison with AngII group. There were no significant differences between vehicle-treated and L-sh-GSK3β + AngII-treated groups. Moreover, cells transduced with L-scrambled did not affect AngII-induced changes in CBP to NFκB or CREB binding. These results indicate that AngII-induced imbalance in cytokine levels and transcription factors are mediated by GSK-3β in neuronal cells.
Figure 7.

Effects of GSK-3β knock down by lentiviral shRNA (L-sh-GSK3β) on AngII induced alterations in transcription factors in neuronal cells. Serum starved CATH.a cells were transduced with L-sh-GSK3β at a multiplicity of infection (MOI) of 30 for 48 h and were stimulated with 100 nM AngII for 6 h. Cell extracts were then immunoprecipitated with CBP and immunoblotted either with p-p65(Ser-276) or p-CREB(Ser-133). An immunoblot (upper panel) and densitometric analysis (lower panel) showing reversal of AngII-induced increased CBP to p-p65(Ser-276) and reduced CBP to p-CREB(Ser-133) binding in neuronal cells. Cells were also transduced with scrambled sequences in presence of AngII did not show any effect. The results are means ± SD of three independent experiments. ***P < 0.001.
Discussion
The aim of the present study was to investigate the underlying molecular mechanisms by which AngII causes an imbalance between PIC and AIC, and to elucidate the role of GSK-3β in mediating this dysregulation. Three novel findings emerge from this study. First, AngII causes an imbalance between PIC and AIC in neuronal culture by up-regulating binding of CBP to NFκB and down-regulating binding of CBP to CREB. These data explain the increased NFκB-mediated transcription of PIC and decreased CREB-mediated transcription of IL-10 on AngII stimulation. Second, AngII causes significantly increased phosphorylation of GSK-3β at Tyr-216 and increased p-GSK3β(Tyr-216) to p-GSK3β(Ser-9) ratio, indicating increased activation of GSK-3β in neuronal cells. Finally, AngII-induced effects in neuronal cells were reversed by lentiviral-mediated silencing of GSK-3β, suggesting that AngII-induced effects are indeed mediated by GSK-3β in neurons. The results of this study reveal a novel molecular mechanism that AngII-induced increased activation of GSK-3β leads to altered activity of downstream transcription factors, NFκB and CREB, in favour of NFκB-mediated gene transcription, thereby causing an imbalance between PIC and AIC in CATH.a neurons (Figure 8). Our data also suggest that AngII-induced effects could be alleviated by GSK-3β inhibition indicating GSK-3β as potential therapeutic target in various CVDs, particularly hypertension as it is characterized by increased PICs and NFκB activation.
Figure 8.
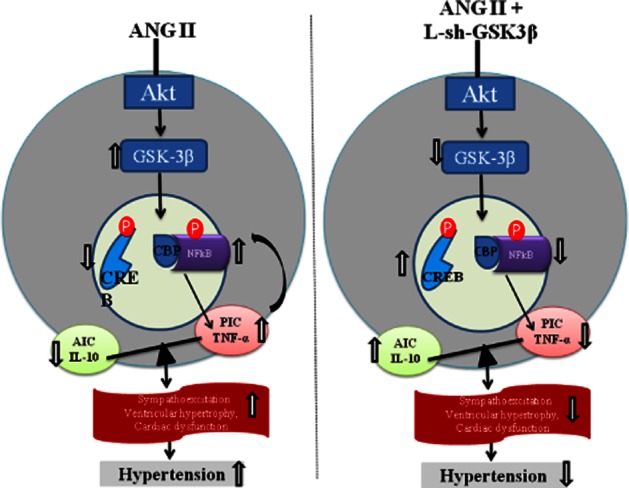
A schematic depicting the proposed pathways (right panel) of AngII-induced dysregulation in inflammatory cytokines in neuronal cells and showing the mechanisms (left panel) by which neuronal cytokines contributes to the pathogenesis of hypertension. It has become clear from the past several years of research that an increased production of PICs in response to overactivated RAS within the cardiovascular regulatory centres of the brain (such as paraventricular nucleus and rostral ventrolateral medulla) causes increased sympathetic outflow leading to increased arterial pressure and cardiac remodelling in experimental models of hypertension. At the cellular level, PICs activate reactive oxygen species which in turn can activate various intracellular signalling pathways, including that of NFκB. Activation of NFκB induces gene transcription of PICs fostering a positive feedback mechanism, and eventually leading to the progression of hypertension. A step further, the results of the present study revealed a novel molecular mechanism that AngII-induced increased phosphorylation of GSK-3β(Tyr-216) and increased p-GSK3β(Tyr-216) to p-GSK-3β(Ser-9) ratio leads to altered activity of downstream transcription factors, NFκB and CREB, in favour of NFκB-mediated gene transcription, thereby, causing an imbalance between PIC and AIC in rat neuronal cells.
Chronic low-grade inflammation is one of the hallmarks of hypertension. PICs, such as TNF-α (Dorffel et al., 1999), IL-1β (Dorffel et al., 1999; Peeters et al., 2001) and IL-6 (Chae et al., 2001; Peeters et al., 2001), have been reported to increase with the severity of hypertension and to be of prognostic significance. Circulating and brain cytokines have both been implicated in the pathogenesis of hypertension. However, emerging evidence indicates that it is not only the PIC (Peeters et al., 2001; Shi et al., 2010) but the balance between pro- and anti-inflammatory cytokines that determines the outcome of the disease, and that these PICs can cross-talk with components of the RAS during the hypertensive response. In the present study, we observed that AngII exposure resulted in upregulation of TNF-α and IL-1β expression in a time-dependent manner in CATH.a neurons with maximal effects at 6 h after AngII exposure. Our results are consistent with those of previous studies from our laboratory which have shown that infusion of AngII in the paraventricular nucleus (PVN), an important cardiovascular regulatory centre in the brain, increases production of TNF-α and IL-1β in rats (Cardinale et al., 2012). Although most of these previous studies have examined the effects of AngII on brain PICs, the effects of overactivation of RAS on anti-inflammatory cytokines are not well understood. In the present study, we observed a significant reduction in IL-10 levels by AngII exposure. Furthermore, the ratio of TNF-α/IL-10 protein expression was found to be dramatically up-regulated in AngII-treated cells. Also, we observed an initial increase in IL-10 mRNA level at 1 h of AngII exposure which could be due to compensatory and protective response to initial increase in TNF-α. Taken together, these results suggest that, at the cellular level, alterations in RAS components not only increases PIC but also causes an imbalance between PIC and AIC in favour of PIC. These results raise another question: what are the exact mechanisms by which alterations in RAS components cause this dysregulation?
Accumulating evidence has suggested that the NFκB-signalling pathway is activated by AngII via the G-protein coupled angiotensin type I receptor (Wolf and Wenzel, 2004). In the unstimulated cells, NFκB is sequestered in the cytoplasm as an inactive complex with inhibitors of NFκB (IκB) (Ghosh and Karin, 2002). Upon stimulation by some inducers such as AngII, IκB is phosphorylated and degraded, leading to translocation of the p65 subunit of NFκB into the nucleus where it activates gene transcription of TNF-α and IL-1β. In addition to the nuclear translocation of NFκB, its transcriptional activity is regulated by a co-activator CREB-binding protein (CBP) that associates with the C-terminal transactivation domain of p65 (Takahashi et al., 2002). Phosphorylation of p65 at Ser-276 has been shown to be required for recruitment of the CBP and transcriptional activity. Besides NFκB, another transcription factor, CREB, has been shown to be involved in the pathogenesis of hypertension. CREB is a 43 kDa phosphoprotein that positively regulates expression of anti-inflammatory cytokines such as IL-10 (Avni et al., 2010). Although activity of CREB is regulated by complex phosphorylation mechanisms that are not yet completely understood, phosphorylation of CREB at Ser-133 has been shown to be required for recruitment of the CBP and transcriptional activity (Chrivia et al., 1993). Due to limited availability of CBP in the nucleus, competition for CBP by diverse transcription factors is inevitable (Yang et al., 2010). As NFκB and CREB are the key transcription factors in the production of cytokines, it is plausible to investigate whether AngII-induced dysregulation in PIC and AIC is mediated by them. In the present study, we observed that AngII exposure resulted in increased phosphorylation of p65 at Ser-276 and reduced phosphorylation of CREB at Ser-133. Moreover, our IP analysis showed that CBP to NFκB binding was increased in AngII (6 h) exposed neuronal cells, whereas CBP to CREB binding was reduced. These results were also associated with elevated levels of TNF-α and IL-1β, and reduced IL-10 levels in AngII (6 h) exposed cells. Collectively, these results suggest that alterations in phosphorylation status of NFκB and CREB by AngII lead to their altered binding with co-activator CBP which in turn leads to an imbalance between PIC and AIC production.
Recently, glycogen synthase kinase-3 (GSK-3), an enzyme that was originally discovered for its role in insulin-mediated glycogen metabolism (Embi et al., 1980; Rylatt et al., 1980; Woodgett and Cohen, 1984; Hughes et al., 1993; Ali et al., 2001; Woodgett, 2001; Doble and Woodgett, 2003), has now been shown to regulate the activity of several metabolic, signalling and structural proteins (Frame and Cohen, 2001; Woodgett, 2001; MacAulay and Woodgett, 2008). Not only the activity of GSK3 is regulated by its post-translational phosphorylation, it itself phosphorylates a broad range of substrates and thereby regulates their function (Frame and Cohen, 2001; Woodgett, 2001). Among the signalling proteins regulated by GSK-3β are many transcription factors including CREB and NFκB (Plyte et al., 1992; Grimes and Jope, 2001b). Therefore, we postulated that AngII-induced alterations in phosphorylation status of NFκB and CREB as observed in the present study could be mediated by GSK-3β. To investigate this hypothesis, we first examined whether AngII perhaps had any effect on GSK-3β expression. Interestingly, our immunoblot analysis showed that AngII exposure resulted in a slight increase in p-GSK3β(Ser-9) levels in neuronal cells, at first sight suggesting inactivation of GSK-3β. Surprisingly, however, p-GSK3β(Tyr-216) levels were found to be significantly elevated in AngII-treated cells. Moreover, the ratio of p-GSK3β(Tyr-216) to p-GSK3β (Ser-9) was higher in AngII-exposed cells when compared to vehicle-treated cells. As N-terminal phosphorylation of GSK-3β at Ser-9 has an inhibitory effect, whereas phosphorylation of Tyr-216 activates it (Forde and Dale, 2007), these results clearly suggested activation of GSK-3β upon AngII exposure. Although the role of GSK-3β in CVDs is now becoming a major focus of the scientific community, to the best of our knowledge none of the previous studies have explored the effects of AngII, a key mediator of most of the CVDs, on neuronal GSK-3β. Additionally, most of these previous studies have reported the phosphorylation status of GSK-3β at Ser-9 suggesting inhibition of its activity (Javadov et al., 2009; Tateishi et al., 2010). However, these studies have not investigated the phosphorylation level of GSK-3β(Tyr-216) leaving us with insufficient data to conclude whether those stimuli cause inhibition or activation of GSK-3β. In the present study, we observed that AngII exposure caused up-regulation of p-GSK3β(Ser-9) with concomitant and much higher increase in p-GSK3β(Tyr-216), indicating activation of GSK-3β.
Various upstream kinases, such as PI3K, PKB, MAPK, p70 ribosomal S6 kinase, PKA and PKC, have been reported to be responsible for phosphorylation of GSK-3β at Ser-9 upon stimulation with insulin and other growth factors (Doble and Woodgett, 2003). PKB (also termed Akt), a serine/threonine kinase located downstream of PI3K, has been shown to phosphorylate GSK-3β at Ser-9 in vitro and in vivo (Cross et al., 1994; 1995). AngII is known to exert its cellular effects via activation of several downstream kinases such as PI3K, Akt and MAPK (Wei et al., 2009; Zhang et al., 2012). Therefore, the observed increase in p-GSK3β(Ser-9) levels in AngII-exposed cells in this study could be due to activation of one or more of these kinases. Although which of these kinases is primarily responsible for AngII-induced phosphorylation of GSK-3β(Ser-9) is not clear at this time. However, we found that AngII-exposed neuronal cells had significantly higher levels of p-Akt(Ser-473) (Figure 9), indicating its activation as phosphorylation of Akt at Ser-473 is known to be crucial for its activation (Alessi et al., 1996). However, the upstream kinase or kinases responsible for AngII-induced increased phosphoylation of Tyr-216 is not known at this time and could be a focus of future studies.
Figure 9.
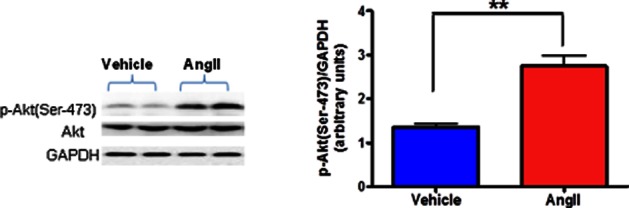
Effects of AngII treatment on phosphorylated Akt expression in neuronal cells. Serum-starved CATH.a cells were stimulated with 100 nM AngII for 6 h and cell extracts were then subjected to protein analysis by Western blot. AngII caused significant activation of Akt as indicated by increased p-Akt(Ser-473) protein expression in CATH.a neurons. AngII exposure did not alter total Akt protein levels. The results are means ± SD of three independent experiments. **P < 0.01 compared with cells treated with vehicle.
As GSK-3β acts as a key regulator of transcription factors NFκB and CREB, it is plausible to speculate that GSK-3β could be the missing link in AngII-induced alterations in inflammatory cytokines. In this study, we observed that suppression of GSK-3β by highly efficient lentiviral shRNA prevented an AngII-induced increase in TNF-α and IL-1β, and a decrease in IL-10 levels in neuronal cells. Furthermore, GSK-3β suppression in AngII-exposed cells led to increased CBP to CREB binding and attenuated CBP to NFκB binding. The altered binding capability of NFκB and CREB to CBP was observed to be due to altered phosphorylation status of both of these transcription factors. Our results showed that GSK-3β silencing caused reduced phosphorylation of NFκB at Ser-276, whereas it increased phosphorylation of CREB at Ser-133. It has been shown previously that phosphorylation of NFκB and CREB at Ser-276 (Reber et al., 2009) and Ser-133 (Chrivia et al., 1993), respectively, is essential for their binding with the CBP and subsequent transactivation. These results suggest that AngII-induced alterations in NFκB and CREB activity are mediated by GSK-3β in neuronal cells. In line with our results, Grimes and Jope, (2001a) showed that inhibition of GSK-3β by lithium facilitates CREB activity in human neuroblastoma SH-SY5Y cells. However, the activity of NFκB is known to be regulated by phosphorylation of IκB and its subsequent nuclear transport. Therefore, the possibility that suppression of GSK-3β affects NFκB regulation at levels other than CBP binding cannot be ignored. Although we have not studied the effects of GSK-3β suppression on phosphorylation of IκB, it has been suggested that GSK-3β does not disrupt NFκB nuclear import in embryonic fibroblasts isolated from GSK3β-null mice (Doble and Woodgett, 2003). Nonetheless, our current results showed that inhibition of GSK-3β in AngII-stimulated neuronal cells altered activity of NFκB and CREB in favour of CREB by modulating their phosphorylation status (Figure 7) and eventually altering their ability to recruit the co-activator CBP. Importantly, these results provide strong evidence that AngII-induced alterations in phosphorylation of NFκB and CREB (Figure 4) in favour of NFκB-mediated transcription of PIC was mediated, at least in part, by GSK-3β.
Past few years of research showed that PICs act as neuromodulators and play a pivotal role in sympathetic regulation of BP (Shi et al., 2010; Agarwal et al., 2011). It is also clear from these studies that increased production of PICs in response to overactivated RAS within the cardiovascular regulatory centres of the brain (such as PVN and rostral ventrolateral medulla) causes increased sympathetic outflow leading to increased arterial pressure and cardiac remodelling in experimental models of hypertension. At the cellular level, PICs activate reactive oxygen species (Zimmerman et al., 2004; Xia et al., 2011), which, in turn, can activate various intracellular signalling pathways, including that of NFκB. Activation of NFκB induces gene transcription of PICs, which leads to further increase in ROS production, fostering a positive feedback mechanism, and eventually leading to the progression of hypertension. A step further, the results of the present study provide evidence of GSK-3β as an important link between RAS, transcription factors and inflammatory cytokines. A summary of the pathway that our data support is depicted in Figure 8.
Limitations
In the present study, we used CATH.a neurons as our neuronal cell model. Clearly, the CATH.a cell line may not exhibit the exact phenotype of neurons located in areas of the brain that regulate BP (e.g. PVN and rostral ventrolateral medulla). However, CATH.a neurons are commonly used to examine AngII-dependent signalling mechanisms, as these cells express the AngII type 1 (AT1R) and type 2 (AT2R) receptors (Sun et al., 2003). For instance, this cell line has been found to be a suitable model for studies involving AngII-induced oxidative stress in hypertension, NFκB (Okada et al., 2006; Mitra et al., 2010), neuronal activation (Sun et al., 2002), expression of AT1R and JNK pathway (Liu et al., 2006), Elk-1(Stefano et al., 2006) and AP-1(Swanson et al., 1998), as well as the effects of AngII on voltage-gated potassium channels (Gao et al., 2010). Therefore, we believe that this cell line is an adequate and widely validated neuronal cell model to determine the AngII signalling pathways. However, future studies involving primary neuronal cell cultures from the cardiovascular regulatory centres of the brain as well as in vivo experiments using animal models of hypertension are still warranted and could be an important perspective of this study.
Another limitation of the present study relates to the involvement of AngII receptors in AngII-induced changes observed in this study. Growing body of evidence suggest that the pro-inflammatory effects of AngII are mediated by angiotensin II type 1 receptor (AT1R). In the context of CNS, previous reports from our laboratory have demonstrated that AngII induces up-regulation of several PICs (e.g. TNF-α, IL-1β and IL-6) in the PVN of Sprague Dawley rats (Kang et al., 2009). More importantly, central (i.c.v.) blockade of AT1R by losartan caused significant down-regulation of PICs in AngII-infused rats (Kang et al., 2009). These findings clearly suggest AT1R as the primary receptor inducing pro-inflammatory effects of AngII in the brain. Therefore, in our opinion, AngII-induced inflammatory response as observed in the present study could be mediated via AT1R activation as well. Nevertheless, future studies could be directed towards dissecting out the role of various AngII receptors in vitro as well as in vivo.
In summary, the results of the present study suggest that AngII exposure causes up-regulation of TNF-α and IL-1β, and down-regulation of IL-10 in neuronal cells by increasing CBP to NFκB binding and attenuating CBP to CREB binding, and AngII-induced dysregulation in inflammatory cytokines is indeed mediated by GSK-3β. The results of this study explain a novel molecular mechanism by which an overactivation of the RAS in the neuronal cells modulates activity of the transcription factors leading to inflammatory alterations. The identification of GSK-3β as a downstream target of AngII in mammalian cells suggests an effector role for GSK-3β in cellular responses to AngII. The results of this study suggest the therapeutic potential of inhibiting GSK-3β in the treatment of CVDs characterized by chronic inflammation. However, in vivo validation of the data presented here could certainly be an important perspective of this study.
Acknowledgments
The work was supported by National Heart, Lung, and Blood Institute Grant HL-80544 to J. F.
Glossary
- AIC
anti-inflammatory cytokines
- AngII
angiotensin II
- CBP
CREB-binding protein
- CREB
cyclic AMP response element-binding protein
- CVDs
cardiovascular diseases
- GSK-3β
glycogen synthase kinase-3β
- L-sh-GSK3β
lentivirus targeting GSK-3β
- PIC
pro-inflammatory cytokines
- RAS
renin-angiotensin system
Conflict of interest
None declared.
References
- Agarwal D, Haque M, Sriramula S, Mariappan N, Pariaut R, Francis J. Role of proinflammatory cytokines and redox homeostasis in exercise-induced delayed progression of hypertension in spontaneously hypertensive rats. Hypertension. 2009;54:1393–1400. doi: 10.1161/HYPERTENSIONAHA.109.135459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal D, Welsch MA, Keller JN, Francis J. Chronic exercise modulates RAS components and improves balance between pro- and anti-inflammatory cytokines in the brain of SHR. Basic Res Cardiol. 2011;106:1069–1085. doi: 10.1007/s00395-011-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101:2527–2540. doi: 10.1021/cr000110o. [DOI] [PubMed] [Google Scholar]
- Avni D, Ernst O, Philosoph A, Zor T. Role of CREB in modulation of TNFalpha and IL-10 expression in LPS-stimulated RAW264.7 macrophages. Mol Immunol. 2010;47:1396–1403. doi: 10.1016/j.molimm.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Beurel E, Jope RS. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J Neuroinflammation. 2009;6:9. doi: 10.1186/1742-2094-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cardinale JP, Sriramula S, Mariappan N, Agarwal D, Francis J. Angiotensin II-Induced Hypertension Is Modulated by nuclear factor-kappaB in the paraventricular nucleus. Hypertension. 2012;59:113–121. doi: 10.1161/HYPERTENSIONAHA.111.182154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001;38:399–403. doi: 10.1161/01.hyp.38.3.399. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS, Cohen P. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem J. 1994;303(Pt 1):21–26. doi: 10.1042/bj3030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116(Pt 7):1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR, et al. Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension. 1999;34:113–117. doi: 10.1161/01.hyp.34.1.113. [DOI] [PubMed] [Google Scholar]
- Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–527. [PubMed] [Google Scholar]
- Forde JE, Dale TC. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci. 2007;64:1930–1944. doi: 10.1007/s00018-007-7045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359(Pt 1):1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Li Y, Schultz HD, Wang WZ, Wang W, Finch M, et al. Downregulated Kv4.3 expression in the RVLM as a potential mechanism for sympathoexcitation in rats with chronic heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H945–H955. doi: 10.1152/ajpheart.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem. 2001a;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001b;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Guggilam A, Cardinale JP, Mariappan N, Sriramula S, Haque M, Francis J. Central TNF inhibition results in attenuated neurohumoral excitation in heart failure: a role for superoxide and nitric oxide. Basic Res Cardiol. 2011;106:273–286. doi: 10.1007/s00395-010-0146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack KK, Engler CW, Papoutsi E, Pipinos II, Patel KP, Zucker IH. Parallel changes in neuronal AT1R and GRK5 expression following exercise training in heart failure. Hypertension. 2012;60:354–361. doi: 10.1161/HYPERTENSIONAHA.112.195693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993;12:803–808. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadov S, Rajapurohitam V, Kilic A, Zeidan A, Choi A, Karmazyn M. Anti-hypertrophic effect of NHE-1 inhibition involves GSK-3beta-dependent attenuation of mitochondrial dysfunction. J Mol Cell Cardiol. 2009;46:998–1007. doi: 10.1016/j.yjmcc.2008.12.023. [DOI] [PubMed] [Google Scholar]
- Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S, Yang ZM, et al. Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovasc Res. 2009;82:503–512. doi: 10.1093/cvr/cvp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YM, Gao F, Li HH, Cardinale JP, Elks C, Zang WJ, et al. NF-kappaB in the paraventricular nucleus modulates neurotransmitters and contributes to sympathoexcitation in heart failure. Basic Res Cardiol. 2011;106:1087–1097. doi: 10.1007/s00395-011-0215-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy K, Brion JP. Developmental expression and localization of glycogen synthase kinase-3beta in rat brain. J Chem Neuroanat. 1999;16:279–293. doi: 10.1016/s0891-0618(99)00012-5. [DOI] [PubMed] [Google Scholar]
- Li Y, Hou LX, Aktiv A, Dahlstrom A. Studies of the central nervous system-derived CAD cell line, a suitable model for intraneuronal transport studies? J Neurosci Res. 2007;85:2601–2609. doi: 10.1002/jnr.21216. [DOI] [PubMed] [Google Scholar]
- Liu D, Gao L, Roy SK, Cornish KG, Zucker IH. Neuronal angiotensin II type 1 receptor upregulation in heart failure: activation of activator protein 1 and Jun N-terminal kinase. Circ Res. 2006;99:1004–1011. doi: 10.1161/01.RES.0000247066.19878.93. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Woodgett JR. Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of type 2 diabetes. Expert Opin Ther Targets. 2008;12:1265–1274. doi: 10.1517/14728222.12.10.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra AK, Gao L, Zucker IH. Angiotensin II-induced upregulation of AT(1) receptor expression: sequential activation of NF-kappaB and Elk-1 in neurons. Am J Physiol Cell Physiol. 2010;299:C561–C569. doi: 10.1152/ajpcell.00127.2010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Okada H, Inoue T, Kikuta T, Watanabe Y, Kanno Y, Ban S, et al. A possible anti-inflammatory role of angiotensin II type 2 receptor in immune-mediated glomerulonephritis during type 1 receptor blockade. Am J Pathol. 2006;169:1577–1589. doi: 10.2353/ajpath.2006.060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters AC, Netea MG, Janssen MC, Kullberg BJ, Van der Meer JW, Thien T. Pro-inflammatory cytokines in patients with essential hypertension. Eur J Clin Invest. 2001;31:31–36. doi: 10.1046/j.1365-2362.2001.00743.x. [DOI] [PubMed] [Google Scholar]
- Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta. 1992;1114:147–162. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- Reber L, Vermeulen L, Haegeman G, Frossard N. Ser276 phosphorylation of NF-kB p65 by MSK1 controls SCF expression in inflammation. Plos One. 2009;4:e4393. doi: 10.1371/journal.pone.0004393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics – 2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylatt DB, Aitken A, Bilham T, Condon GD, Embi N, Cohen P. Glycogen synthase from rabbit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur J Biochem. 1980;107:529–537. [PubMed] [Google Scholar]
- Shenkar R, Yum HK, Arcaroli J, Kupfner J, Abraham E. Interactions between CBP, NF-kappaB, and CREB in the lungs after hemorrhage and endotoxemia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L418–L426. doi: 10.1152/ajplung.2001.281.2.L418. [DOI] [PubMed] [Google Scholar]
- Shi P, Raizada MK, Sumners C. Brain cytokines as neuromodulators in cardiovascular control. Clin Exp Pharmacol Physiol. 2010;37:e52–e57. doi: 10.1111/j.1440-1681.2009.05234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefano L, Al Sarraj J, Rossler OG, Vinson C, Thiel G. Up-regulation of tyrosine hydroxylase gene transcription by tetradecanoylphorbol acetate is mediated by the transcription factors Ets-like protein-1 (Elk-1) and Egr-1. J Neurochem. 2006;97:92–104. doi: 10.1111/j.1471-4159.2006.03749.x. [DOI] [PubMed] [Google Scholar]
- Steinbrecher KA, Wilson W, 3rd, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol. 2005;25:8444–8455. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Sumners C, Raizada MK. Chronotropic action of angiotensin II in neurons via protein kinase C and CaMKII. Hypertension. 2002;39(2 Pt 2):562–566. doi: 10.1161/hy0202.103057. [DOI] [PubMed] [Google Scholar]
- Sun C, Du J, Raizada MK, Sumners C. Modulation of delayed rectifier potassium current by angiotensin II in CATH.a cells. Biochem Biophys Res Commun. 2003;310:710–714. doi: 10.1016/j.bbrc.2003.09.069. [DOI] [PubMed] [Google Scholar]
- Swanson DJ, Zellmer E, Lewis EJ. AP1 proteins mediate the cAMP response of the dopamine beta-hydroxylase gene. J Biol Chem. 1998;273:24065–24074. doi: 10.1074/jbc.273.37.24065. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Tetsuka T, Uranishi H, Okamoto T. Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur J Biochem. 2002;269:4559–4565. doi: 10.1046/j.1432-1033.2002.03157.x. [DOI] [PubMed] [Google Scholar]
- Tateishi A, Matsushita M, Asai T, Masuda Z, Kuriyama M, Kanki K, et al. Effect of inhibition of glycogen synthase kinase-3 on cardiac hypertrophy during acute pressure overload. Gen Thorac Cardiovasc Surg. 2010;58:265–270. doi: 10.1007/s11748-009-0505-2. [DOI] [PubMed] [Google Scholar]
- Vines A, Cahoon S, Goldberg I, Saxena U, Pillarisetti S. Novel anti-inflammatory role for glycogen synthase kinase-3beta in the inhibition of tumor necrosis factor-alpha- and interleukin-1beta-induced inflammatory gene expression. J Biol Chem. 2006;281:16985–16990. doi: 10.1074/jbc.M602446200. [DOI] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Zhang ZH, Felder RB. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol. 2009;296:H1425–H1433. doi: 10.1152/ajpheart.00942.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, Wenzel UO. Angiotensin II and cell cycle regulation. Hypertension. 2004;43:693–698. doi: 10.1161/01.HYP.0000120963.09029.ca. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Judging a protein by more than its name: GSK-3. Sci STKE. 2001;2001:re12. doi: 10.1126/stke.2001.100.re12. [DOI] [PubMed] [Google Scholar]
- Woodgett JR, Cohen P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5) Biochim Biophys Acta. 1984;788:339–347. doi: 10.1016/0167-4838(84)90047-5. [DOI] [PubMed] [Google Scholar]
- Xia H, Suda S, Bindom S, Feng Y, Gurley SB, Seth D, et al. ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS ONE. 2011;6:e22682. doi: 10.1371/journal.pone.0022682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jiang H, Chen SS, Chen J, Xu SK, Li WQ, et al. CBP knockdown inhibits angiotensin II-induced vascular smooth muscle cells proliferation through downregulating NF-kB transcriptional activity. Mol Cell Biochem. 2010;340:55–62. doi: 10.1007/s11010-010-0400-2. [DOI] [PubMed] [Google Scholar]
- Zhang ZH, Yu Y, Wei SG, Felder RB. Aldosterone-induced brain MAPK signaling and sympathetic excitation are angiotensin II type-1 receptor dependent. Am J Physiol Heart Circ Physiol. 2012;302:H742–H751. doi: 10.1152/ajpheart.00856.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]