

Abstract
Background and Purpose
N-arachidonoyl glycine (NAGly) is a lipoamino acid with vasorelaxant properties. We aimed to explore the mechanisms of NAGly's action on unstimulated and agonist-stimulated endothelial cells.
Experimental Approach
The effects of NAGly on endothelial electrical signalling were studied in combination with vascular reactivity.
Key Results
In EA.hy926 cells, the sustained hyperpolarization to histamine was inhibited by the non-selective Na+/Ca2+ exchanger (NCX) inhibitor bepridil and by an inhibitor of reversed mode NCX, KB-R7943. In cells dialysed with Cs+-based Na+-containing solution, the outwardly rectifying current with typical characteristics of NCX was augmented following histamine exposure, further increased upon external Na+ withdrawal and inhibited by bepridil. NAGly (0.3–30 μM) suppressed NCX currents in a URB597- and guanosine 5′-O-(2-thiodiphosphate) (GDPβS)-insensitive manner, [Ca2+]i elevation evoked by Na+ removal and the hyperpolarization to histamine. In rat aorta, NAGly opposed the endothelial hyperpolarization and relaxation response to ACh. In unstimulated EA.hy926 cells, NAGly potentiated the whole-cell current attributable to large-conductance Ca2+-activated K+ (BKCa) channels in a GDPβS-insensitive, paxilline-sensitive manner and produced a sustained hyperpolarization. In cell-free inside-out patches, NAGly stimulated single BKCa channel activity.
Conclusion and Implications
Our data showed that NCX is a Ca2+ entry pathway in endothelial cells and that NAGly is a potent G-protein-independent modulator of endothelial electrical signalling and has a dual effect on endothelial electrical responses. In agonist pre-stimulated cells, NAGly opposes hyperpolarization and relaxation via inhibition of NCX-mediated Ca2+ entry, while in unstimulated cells, it promotes hyperpolarization via receptor-independent activation of BKCa channels.
Keywords: N-arachidonoyl glycine, endothelial, Na+/Ca2+ exchanger, membrane potential, acetylcholine, histamine, BKCa channels
Introduction
The plasma membrane Na+/Ca2+ exchanger (NCXpm) is a bidirectional electrogenic transporter that, depending on the transmembrane gradient of substrate ions and membrane potential, transports Ca2+ either out of cells or into cells in exchange for three ions of Na+. In many cell types, this transporter is the principle mechanism of extrusion of Ca2+ (forward mode) under conditions of stimulated Ca2+ entry. In addition, Ca2+ entry via NCXpm operating in reverse mode contributes to intracellular Ca2+ overload under pathological conditions such as myocardial ischaemia and reperfusion (Lee et al., 2005). Experimentally, such conditions can be mimicked in other cell types by exposure to hypoxic, acidic, ion-shifted Ringer followed by return to normal solution. In primary astrocyte cultures, such manoeuvres result in widespread cell loss (Bondarenko and Chesler, 2001) due to massive Ca2+ influx at the onset of reperfusion (Bondarenko et al., 2005).
NCXpm is also present in endothelial cells, and reports regarding its functional role vary. In experiments using the excised rat aorta, it was found that the Na+ influx into endothelial cells induced by stimulation with ACh was sufficient to trigger Ca2+ inflow via NCXpm (Bondarenko, 2004). Further, the reversed mode of NCXpm following ACh exposure was shown to sustain endothelium-dependent relaxation and NO release (Schneider et al., 2002), indicating that in the vascular endothelium the NCXpm may have an important physiological and pathophysiological role. Given the functional importance of Ca2+ entry into endothelial cells, exogenous compounds and endogenous signalling molecules that affect the activity of the NCXpm may have a profound impact on endothelial cell signalling and function.
Over the past years, a number of endogenous compounds consisting of arachidonic acid conjugated with amino acids, including N-arachidonyl glycine (NAGly), have emerged as powerful modulators of pain (Vuong et al., 2008), inflammation (Burstein et al., 2011) and vascular function (O'Sullivan et al., 2005; Milman et al., 2006; Parmar and Ho, 2010). NAGly is an endogenous, enzymatically-oxygenated metabolite of the endocannabinoid anandamide (N-arachidonoyl ethanolamide) (Bradshaw et al., 2009; McHugh, 2012). NAGly is produced from anandamide via two distinct pathways including oxidative metabolism of the ethanolamine moiety of anandamide and conjugation of glycine to arachidonic acid, which is released during anandamide hydrolysis by fatty acid amide hydrolase (FAAH; Bradshaw et al., 2009). Unlike anandamide, NAGly has no effects on cannabinoid receptors, CB1 and CB2 (Huang et al., 2001) and has low affinity for the vanniloid transient receptor potential cation channel V1, TRPV1 (Sheskin et al., 1997). Recently, NAGly was demonstrated to be an endogenous ligand for GPCR 18 (GPR18; Kohno et al., 2006; McHugh et al., 2010; Takenouchi et al., 2012) and GPCR 92 (GPR92; Oh et al., 2008). In a microglial cell line (BV-2), human endometrial cell line (HEC-1B) and GPR18-transfected HEK-293 cells, NAGly was shown to control cell migration in a GPR18-dependent manner (McHugh et al., 2010; McHugh, 2012; McHugh et al., 2012). Consequently, GPR18 has been proposed to represent the putative abnormal cannabidiol receptor (McHugh et al., 2010; McHugh, 2012). However, given the discrepancies between the levels of expression of GPR18 and the levels of NAGly in different tissues (Alexander, 2012), the association between GPR18 and NAGly is not straightforward. Indeed, activation of GPR18 by NAGly was not observed in a recent study employing high-throughput beta-arrestin-based screen (Yin et al., 2009). In addition, the concept that NAGly acts as an agonist for GPR18 was further challenged recently when it was found that, in GPR18-expressing rat sympathetic neurons, NAGly failed to induce any GPR18-mediated inhibition of N-type (Cav2.2) Ca2+ channel, a primary downstream effector of Gi/o (Lu et al., 2013). The authors concluded that NAGly is either not an agonist for GPR18 or that GPR18 signalling involves a non-canonical pathway.
Importantly, endocannabinoids and cannabinoid-like lipid compounds are able to modulate the activity of a number of ion channels independently of G-proteins (Oz, 2006; Barana et al., 2010; Bondarenko et al., 2011a,b). In rat odontoblasts, cannabinoid-induced Ca2+ influx through TRPV1 was recently shown to be functionally coupled to NCXpm-mediated Ca2+ extrusion (Tsumura et al., 2012). Remarkably, NAGly exhibits a number of G-protein-independent effects, including inhibition of T-type (Cav3.1, Cav3.2 and Cav3.3) Ca2+ channels (Barbara et al., 2009; Ross et al., 2009). NAGly was also shown to enhance inhibitory glycinergic synaptic transmission by blocking glycine uptake and inhibiting excitatory NMDA-mediated synaptic transmission (Jeong et al., 2010). In the vasculature, NAGly (Parmar and Ho, 2010) and its parent molecule anandamide (Jarai et al., 1999; McCollum et al., 2007) act as vasorelaxants, supposedly via stimulation of a putative G-protein coupled non-CB1 or CB2 cannabinoid receptor expressed in vascular endothelium, so-called anandamide or abnormal cannabidiol receptor coupled to NO production (Wagner et al., 1999; Herradon et al., 2007; McCollum et al., 2007). However, whether and how NAGly affects NCXpm-mediated endothelial electrical signalling and, if so, whether GPCRs are required for the observed effects is not clear.
Previously, it was shown that stimulation of a human umbilical vein-derived cell line (EA.hy926) with histamine in rather artificial nominally Ca2+-free conditions (no added Ca2+, no EGTA) induces Ca2+ oscillations due to a coordinated interplay between reversed NCXpm operation and Ca2+ sequestration by sarcoplasmic-endoplasmic reticulum Ca2+ ATPase (Paltauf-Doburzynska et al., 2000). In the present study, we showed that, in the presence of physiological levels of Ca2+, these cells respond to histamine by a sustained hyperpolarization supported by Ca2+ influx via reversed mode NCX. We further demonstrated that NAGly concentration-dependently inhibits this NCXpm-mediated ion current in a G-protein-independent manner and the sustained endothelial hyperpolarization, both in cultured and in situ endothelial cells, an effect that underlies the transient inhibition of endothelium-dependent relaxation to ACh. In contrast, in unstimulated cells, NAGly promoted hyperpolarization via direct stimulation of large-conductance Ca2+-activated K+ (BKCa) channels.
Methods
Cell culture
The human umbilical vein-derived endothelial cell line, EA.hy926 (Edgell et al., 1983) at passage >45 was grown in DMEM containing 10% fetal calf serum and 1% HAT (5 mM hypoxanthine, 20 μM aminopterin, 0.8 mM thymidine) and cells were maintained in an incubator at 37°C in 5% CO2 atmosphere. Cells were plated on either 10 mm (for patch-clamp recordings) or 30 mm glass coverslips (for Ca2+ measurements).
Ca2+ measurements
Cytosolic free Ca2+ was measured using Fura-2/AM as previously described (Paltauf-Doburzynska et al., 2000; Bondarenko et al., 2010). Briefly, cells were loaded with 2 μM Fura-2/AM for 45 min at room temperature. Before the experiments, cells were washed and equilibrated for a further 20 min. Subsequently, cells were illuminated on an inverted microscope (Eclipse 300 TE, Nikon Instruments Inc., Melville, NY, USA) alternatively at 340 and 380 nm (filters: 340HTI15 and 380HTI15; Omega Optical, Brattleboro, VT, USA) and emitted light was collected at 510 nm (510WB40 emission; Omega Optical) using a cooled charge-coupled device camera (−30°C; Quantix KAF 1400G2, Roper Scientific, Acton, MA, USA). All Ca2+ measurements were performed with a 40 × 1.3 N.A. oil-immersion objective (Plan Fluor, Nikon) at room temperature.
Animals and tissue preparation
All animal studies were performed in accordance with the recommendations of the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes and approved by the Biomedical Ethics Committee of the A. A. Bogomoletz Institute of Physiology (Kiev, Ukraine). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Male Wistar rats (250–300 g body weight) were killed by cervical dislocation. The abdomen was opened by a midline incision, and the aorta was carefully excised and placed in ice-cold modified Krebs bicarbonate buffer solution of the following composition (mM): 118.3 NaCl, 25 NaHCO3, 4.7 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 2.5 CaCl2, 10 glucose (pH 7.4). All connective and perivascular adipose tissues were removed. The aorta was then cut into ring segments, 2–3 mm wide, care was taken not to disrupt the endothelium. The rings were stored in bubbled Krebs solution maintained at room temperature before use in electrophysiological and contractile experiments.
Contractile recording experiments
Vascular rings were mounted isometrically in a tissue bath between a stationary stainless steel hook and an isometric transducer coupled to a polygraph. The rings were mounted with a resting tension of 1 g. Throughout the experiment, vascular preparations were superfused with modified Krebs bicarbonate buffer solution gassed with 5% CO2 in O2. After the equilibration period, arteries were precontracted with noradrenaline (NA, 10 μM) and when a stable level of contraction was established, 2 μM ACh was applied to observe relaxation. 5 μM NAGly was applied in addition to ACh when sustained relaxation was developed. Contractile experiments were conducted at 37°C. In some experiments, NAGly was applied to precontracted de-endothelized vessels. Denudation was achieved by an infusion of distilled water for 5–7 min and was confirmed by the inability of ACh to produce relaxation in the presence of NA.
Electrophysiological recordings
The membrane potential of endothelial cells from excised rat aorta was recorded using a perforated patch-clamp technique as described previously (Bondarenko, 2004). Briefly, ring segments were cut open and a strip was pinned to the rubber bottom of the chamber, which was perfused with modified Krebs bicarbonate buffer solution at a rate of 1 mL·min−1. The membrane potential of EA.hy926 cells was recorded using a nystatin-perforated patch clamp technique. For membrane potential recordings from EA.hy926 cells, the standard bath solution contained (in mM) 140 NaCl, 5 KCl, 1.2 MgCl2, 10 HEPES, 10 glucose, 2.4 CaCl2. For membrane potential recordings from both in situ endothelial cells and EA.hy926 cells, patch pipettes were filled with a solution containing (in mM) 140 KCl; 10 NaCl; 0.3 EGTA; 10 HEPES (pH adjusted to 7.2 using KOH). The resistance of the pipettes was 3–5 MΩ. Pharmacological agents were applied to the preparation by bath perfusion. Experiments were conducted at room temperature.
Whole-cell INCX was recorded from single EA.hy926 cells 24–36 h after plating using a conventional whole cell patch-clamp technique. Membrane currents and potential were recorded using a List EPC7 amplifier (List, Darmstadt, Germany) and pClamp 8.2 software (Axon Instruments from Molecular Devices, Sunnyvale, CA, USA). For recording the reverse (outward) NCXpm current obtained after the addition of Ca2+ to a Na+ free solution, the pipette solution contained (in mM) 125 NaCl, 10 CsCl, 2 MgCl2, 5 EGTA, 4.28 CaCl2 (1 μM free Ca2+), 10 HEPES and the bath solution contained 140 N-methyl-d-glucamine (NMDG)-Cl, 1 MgCl2 10 TEA, 10 HEPES, 10 glucose with either 1 EGTA or 2.5 CaCl2. For recording the reverse NCXpm current using voltage ramps, the bath solution contained (in mM) 130 NaCl, 10 CaCl2, 1.2 MgCl2, 10 HEPES, 10 glucose and the pipette solution contained (in mM): 80 Cs-methanesulfonate, 25 NaCl, 20 CsCl, 3 NaATP, 1 MgCl2, 10 HEPES, 5 EGTA and 1.93 Ca2+ to set the free Ca2+ concentration to 100 nM. For recordings of both reversed and forward modes of the exchanger using voltage ramps, the bath solution contained (in mM) 140 NaCl, 5 TEACl, 2.4 CaCl2, 1.2 MgCl2, 10 HEPES, 10 glucose and the pipette solution contained (in mM): 110 Cs-methanesulfonate, 10 NaCl, 20 TEACl, 2 MgATP, 10 HEPES, 5 EGTA, 1.93 CaCl2 (100 nM free Ca2+). Voltage ramps of 1 s duration from −100 mV to +90 mV were delivered every 5 s from the holding potential of −40 mV.
Whole-cell recordings of BKCa channels were obtained in standard bath solution, the pipette solution contained (in mM) 145 KCl, 10 HEPES, 1 MgCl2, 5 EGTA and free Ca2+ concentration was set to 300 nM by adding 3.27 mM CaCl2. The bath and the pipette K+-based solutions with 300 nM free Ca2+ were used for recordings of BKCa single-channel activity in inside-out configuration.
Statistical analysis
Experimental data are expressed as mean ± SEM. Student's t-test was used to compare results, with P < 0.05 taken as the level of significance. In experiments with EA.hy926 cells, n denotes the number of cells studied and in experiments with vascular tissue, n denotes the number of rats used.
The nomenclature used for receptors and ion channels conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Results
Reversed mode of NCXpm contributes to sustained endothelial hyperpolarization to histamine in the human umbilical vein-derived endothelial cell line EA.hy926
In the presence of 2.4 mM bath Ca2+, histamine (10–100 μM) produced a sustained endothelial hyperpolarization with a very low decay (Figure 1A). In order to explore the contribution of NCXpm to endothelial hyperpolarization in response to histamine under physiological, Ca2+-containing conditions, bepridil, a blocker of NCXpm, was administered during the plateau phase of the hyperpolarization. Administration of bepridil (50 μM) in the continued presence of histamine fully and reversibly inhibited (P < 0.05) the hyperpolarizing response (Figure 1B). Because NCXpm operating in a reversed mode may contribute to the endothelial hyperpolarization by two mechanisms, that is net efflux of positive charge per cycle and secondary stimulation of Ca2+-activated K+ channels (Bondarenko, 2004), the sensitivity of the hyperpolarization response to bepridil is indicative of NCXpm operating in its Ca2+ entry mode during histamine-evoked hyperpolarization. To test more specifically that the reversed mode of NCXpm was involved in this response, the drug KB-R7943 was used. Similar to bepridil, KB-R7943 (20 μM), an inhibitor of reversed NCXpm, reversibly inhibited (P < 0.05) the endothelial hyperpolarization response to histamine (Figure 1C).
Figure 1.
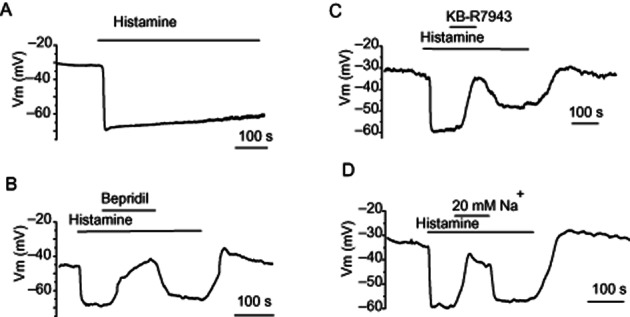
Impact of inhibition of NCXpm on endothelial cell hyperpolarization to histamine. (A) Representative effect of histamine (10 μM) on the membrane potential of EA.hy926 cells. (B) Inhibition of sustained hyperpolarization to histamine (10 μM) by bepridil (50 μM) application during the plateau phase of hyperpolarization. (C) Inhibitory effect of KB-R7943 (20 μM) on sustained endothelial hyperpolarization to histamine. (D) Effect of reduction of external Na+ concentration to 20 mM on endothelial hyperpolarization to histamine. (A–D) Exemplary traces out of 4–7 individual experiments.
The operation of NCXpm in Ca2+ entry mode following histamine stimulation is apparently ensured by a pronounced increase in Na+ influx, which would override the forward mode of the exchanger activated by a rise in intracellular Ca2+ concentration. To explore the role of Na+ entry in the NCXpm-mediated sustained endothelial hyperpolarization to histamine, the Na+ gradient was reduced during the plateau phase of the hyperpolarization. Although the reduction of external Na+ is an intervention that increases the driving force for reversed mode NCXpm under a fixed level of intracellular Na+, this manoeuvre was expected to inhibit NCXpm-mediated Ca2+ entry in confluent cells, when Na+ influx triggers the reversed mode of NCXpm. In these experiments, histamine (10 μM) hyperpolarized endothelial cells from −34.3 ± 2.3 mV to −56 ± 3.6 mV (n = 6). A reduction in extracellular Na+ concentration from 140 mM to 20 mM reversibly inhibited (P < 0.05) the sustained component of the hyperpolarization, driving the membrane potential level to −33.7 ± 2.4 mV (n = 5; Figure 1D). Collectively, these results indicate that cells from the human umbilical vein endothelial cell-derived cell line express a functional NCXpm, the operation of which, in its Ca2+ entry mode, ensures a sustained endothelial hyperpolarization during cell stimulation by histamine.
Reversed mode NCXpm current is enhanced by histamine stimulation
To demonstrate an endogenous NCXpm current in endothelial cells, we drove the transporter backwards by switching the Na+-free external solution from a Ca2+-free to a 2.5 mM Ca2+-containing solution with 120 mM Na+ in the pipette at a holding potential 0 mV. Adding Ca2+ to the bath solution elicited an outward current that was inhibited by 20 μM KB-R7943 (Figure 2A), demonstrating that these cells develop significant NCXpm currents.
Figure 2.
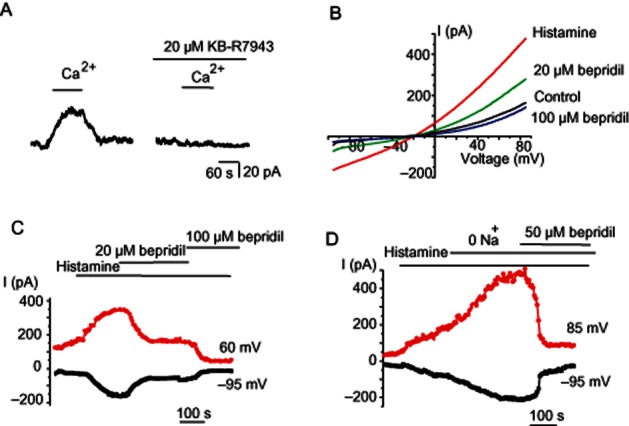
NCXpm is active and is stimulated by histamine. (A) Ca2+ addition produces outward KB-R7943-sensitive current in Na+-loaded endothelial cells. Representative record out of four experiments. Membrane potential was clamped at 0 mV. Pipette was filled with 120 mM NaCl-containing solution. (B) Representative current traces to voltage ramps before (control), during 10 μM histamine exposure (histamine) and in the combined presence of histamine with either 20 μM bepridil or 100 μM bepridil. Internal solution contained 28 mM Na+ and 100 nM free Ca2+. (C) Corresponding time course of the currents shown in (B) taken at −95 and 60 mV in response to 10 μM histamine and the effects of 20 and 100 μM bepridil. Similar results were obtained in 6 cells. (D) Na+ removal amplifies histamine-potentiated current. Representative (n = 5) time course of the current response to histamine application followed by Na+ removal taken at −95 and 85 mV and inhibitory effect of bepridil (50 μM).
We next examined the role of NCXpm in histamine-evoked whole-cell currents. For this purpose, we applied voltage ramps to Na+ loaded (28 mM Na+ in the pipette solution) cells, a condition that favours the NCXpm operating in reversed mode. KCa channels were suppressed by substituting Cs+ for K+ in the internal solution and the internal Ca2+ was buffered at 100 nM. Under these conditions, the voltage ramps produced an outwardly rectifying current with a typical I-V relationship that is characteristic of NCXpm (Figure 2B). Stimulation with 10 μM histamine markedly enhanced the peak currents (by 230%) and subsequent administration of bepridil (20 and 100 μM) strongly suppressed the current amplitude (Figure 2B,C). In another set of experiments, histamine exposure was followed by cell superfusion with Na+-free external solution in the continued presence of Ca2+ and histamine to boost the reversal of the exchanger. This manoeuvre strongly increased both the outward and inward currents (Figure 2D). Because under these experimental ionic conditions, the driving force of the reversed NCXpm is limitless, the inward current observed at negative potentials is highly unlikely to reflect the forward mode of the exchanger. Nevertheless, subsequent addition of 50 μM bepridil suppressed the outwardly rectifying current (Figure 2D). Taken together, these observations indicate that in endothelial cells under our experimental conditions the basal NCXpm currents are enhanced by histamine exposure.
NAGly inhibits NCXpm-mediated ion currents stimulated by histamine
Next, we assessed the effect of NAGly on NCXpm-mediated whole-cell currents. When cells were dialysed with the Cs+-based intracellular solution containing low Ca2+ (100 nM) and high Na+ (28 mM), a condition which favours reversed NCXpm, voltage ramps from −100 to 85 mV revealed an outwardly rectifying current that met the characteristics of NCXpm. This outwardly rectifying current was amplified by histamine and was further potentiated by substitution of external Na+ for NMDG+ (Figure 3A,B). Administration of 10 μM NAGly evoked a gradual suppression of the current (Figure 3A,B), which in some cells was preceded by transient potentiation of current amplitude (Figure 3A). When NAGly (10 μM) was applied to histamine-stimulated cells without prior exposure to Na+-free solution, it abolished the outwardly rectifying current stimulated by histamine and strongly suppressed basal NCXpm currents (Figure 3C,D).
Figure 3.
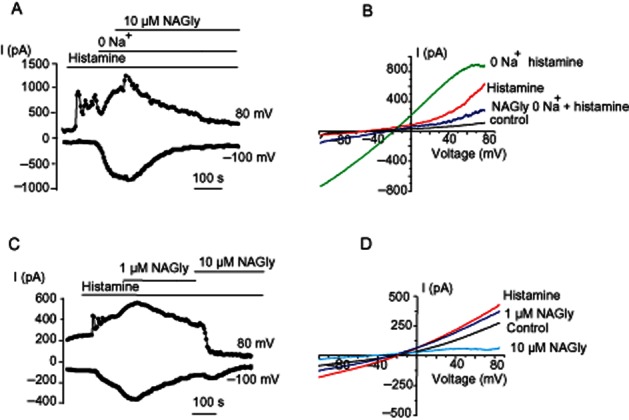
NAGly inhibits NCXpm currents. (A) The time course of inhibition NCXpm currents by 10 μM NAGly. NCX currents were maximally pre-stimulated with histamine and Na+ removal. (B) Representative NCX currents evoked by voltage ramps from the time course shown in (A) before (control), in the presence of 10 μM histamine (histamine) and following Na+ removal (0 Na+ histamine) and the effect of 10 μM NAGly on NCXpm-potentiated currents. Similar results were obtained in six cells. (C, D) The time course of inhibition of NCXpm current amplitude by NAGly (1 and 10 μM) preliminary augmented with histamine (10 μM; C) and corresponding current traces (D) in response to voltage ramps before (control), during 10 μM histamine exposure (histamine) and in the combined presence of histamine with either 1 or 10 μM NAGly. Similar results were obtained in six cells.
NAGly inhibits NCXpm-mediated ion currents and intracellular Ca2+ rise independently of store depletion
The next question we addressed is whether cell stimulation with histamine and the associated store depletion and/or initiation of store-operated or receptor-activated Na+ and Ca2+ influx is required for the inhibitory effect of NAGly on reversed mode NCXpm. To explore the role of the cell signalling cascade initiated by histamine in NAGly-evoked inhibition of INCX, we avoided cell stimulation with histamine and simply drove the transporter backwards by switching to a Na+-free Ca2+-containing solution while delivering voltage ramp pulses. Substitution of external Na+ for NMDG+ strongly augmented the outwardly rectifying current (Figure 4A,B), an observation consistent with increased activity of reversed NCXpm. Again, this outwardly rectifying current was inhibited upon NAGly administration, indicating that cell stimulation with an IP3-generating agonist, such as histamine, is not required for NAGly-induced inhibition of NCXpm. Support for this notion was further obtained by measuring the reversed NCX-driven rise in intracellular Ca2+. Figure 4C shows an exemplary analogous fluorescence trace from a Fura-2-loaded EA.hy926 cell. Replacement of external Na+ with an equimolar amount of NMDG+ induced an increase in [Ca2+]i, which was largely (by 42%) suppressed by subsequent exposure to 3 μM NAGly. Similar results were obtained in a total of 18 cells from four coverslips. These findings indicate that NAGly inhibits the reversed NCXpm independently of endothelial cell stimulation with agonists.
Figure 4.
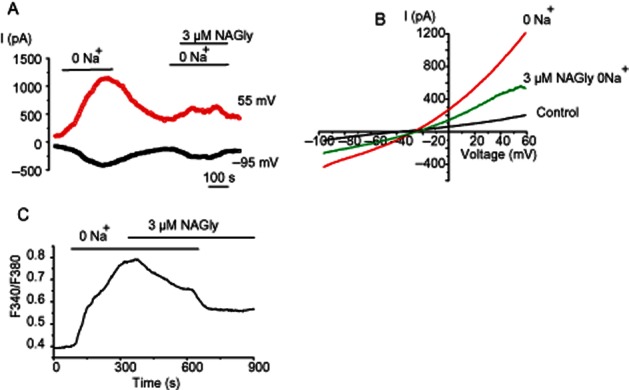
Histamine pre-stimulation is not required for NCXpm inhibition by NAGly. (A, B) Representative time course at voltages indicated (A) and the voltage dependency (B) of NCXpm currents potentiated by Na+ removal in a Ca2+-containing solution and the effect of NAGly (3 μM), (C) Representative effect of 5 μM NAGly on intracellular free Ca2+ rise produced by Na+ removal in Ca2+-containing solution (n = 18).
NAGly reversibly inhibits both reversed and forward mode of NCXpm in a concentration-dependent manner
The above voltage clamp experiments were performed by using pipette solution containing 28 mM Na+ and 100 nM Ca2+, a condition which favours the reverse mode of NCXpm along the membrane potentials tested. To determine whether NAGly exclusively suppresses the reversed mode of the exchanger, in experiments described below cells were dialysed with internal solution containing 10 mM Na+ and 100 nM free Ca2+ in the presence of 2.4 mM bath Ca2+, a condition which allows the recording of both forward and reversed modes of the exchanger depending on the membrane voltage applied. Forward and reverse mode NCXpm currents were estimated at −95 mV and +85 respectively. Representative currents in response to voltage ramps from −100 to 90 mV before and after NAGly administration are shown in Figure 5 A,B. Both outward and inward currents were inhibited by 0.3 μM NAGly with a similar time course (Figure 5A). At a concentration of 0.3 μM, NAGly was slow to exert its full effect on NCXpm currents, the degree of inhibition increased with time and a steady-state value was reached over 200–250 s after initial application (Figure 5A). The effect of NAGly on NCXpm currents was reversible; restoration of INCX occurred within 250–300 s following 0.3 μM NAGly washout. Corresponding I-V curves before application of 0.3 μM NAGly, during the plateau of the response and following washout of the compound are shown in Figure 5B.
Figure 5.

Inhibitory effect of NAGly on NCXpm currents is reversible, concentration-dependent and non-mode selective. (A, B) Representative time course (A) and NCXpm currents elicited by voltage ramps (B) before (control), during 0.3 μM NAGly administration (NAGly) and following NAGly washout. Similar results were obtained in four cells. (C, D). Representative time course (C) and NCXpm currents elicited by voltage ramps showing concentration-dependent inhibition of current responses by NAGly. Bepridil (50 μM) was administered at the end of the protocol. (D, E) Concentration-dependent inhibition of forward (D) and inward (E) NCXpm currents by NAGly. In parentheses are the numbers of individual cells treated with different concentrations of NAGly.
Inhibition of INCX by NAGly was concentration-dependent within the concentration range tested (0.3–30 μM). Increasing the NAGly concentration from 0.3 to 10 μM produced a stronger inhibitory effect on INCX and reduced the time to reach the steady-state inhibition of INCX to 120–150 s (Figure 5C). Cumulative addition of increasing concentrations of NAGly into the bath solution caused a gradually developing inhibition of the current amplitude (Figure 5D,E). At the end of experiment, the cells were exposed to bepridil (50 μM). The bepridil-sensitive current, which represents only the NCX current, was calculated by subtracting the current remaining after 50 μM bepridil exposure from the total measured current at each NAGly concentration tested. A similar degree of suppression of both the outward and inward current amplitudes was observed at a particular concentration of NAGly that inhibited the reverse and forward modes of NCX activity at an IC50 of 1.22 μM and 1.09 μM respectively (Figure 5F,G). The Hill coefficient was 1.09 for the forward and 1.18 for the reversed mode of the exchanger, suggesting one binding site for NAGly. These experiments indicate that inhibition of NCXpm by NAGly appears to be non-mode selective.
NAGly suppresses NCX currents independently of GPCRs
To examine whether GPCRs mediate the effect of NAGly on INCX, we next tested the effect of the synthetic cannabinoid analogue O-1918 on INCX. It was presumed O-1918 is an antagonist of a putative endothelial abnormal cannabidiol receptor activated by NAGly and anandamide (McHugh et al., 2010; Parmar and Ho, 2010). Exposure to O-1918 (10 μM) gradually suppressed (up to 82%) both the basal INCX (Figure 6A) and that pre-stimulated by low (20 mM) Na+ (switch from 140 mM [Na+]o to 20 mM [Na+]o) (Figure 6B,C). Administration of 10 μM NAGly in the continued presence of 10 μM O-1918 further suppressed the remaining portion of the current. Supplementation of the patch pipette solution with guanosine 5′-O-(2-thiodiphosphate) (GDPβS; 0.5–1 mM), a potent and irreversible G-protein inhibitor, was also tested to examine whether GPCRs are required for suppression of INCX by NAGly. Under these conditions, administration of 10 μM NAGly to the external solution inhibited the currents (n = 7) to the same extent as in the absence of GDPβS (Figure 6D,E). Because NAGly is susceptible to metabolic degradation via FAAH (Bradshaw et al., 2009), we explored whether it is actually NAGly that is causing the effect and not arachidonic acid or glycine generated by NAGly hydrolysis. Pre-incubation and continuous incubation of 1 μM URB597, a FAAH inhibitor (Ho and Randall, 2007; Bradshaw et al., 2009), was unable to prevent inhibition of INCX following NAGly application (Figure 6F,G). When 10 μM NAGly was introduced into the patch pipette, a manoeuvre, which allows NAGly administration to the inner surface of the cell avoiding binding to GPCRs, INCX was gradual suppressed within 200–250 s (Figure 6H,I) However, the mean suppressive effect on INCX induced by intracellular NAGly administration was significantly less than that observed induced by external NAGly application (Figure 6J). The fractional inhibition of INCX by 10 μM of internal NAGly at +85 mV was 55.9 ± 4.2% (n = 4), while external NAGly administration produced a fractional inhibition of INCX of 75.0 ± 7.1% (n = 9). Taken together, these data strongly indicate that the endogenous lipoamino acid NAGly inhibits NCXpm-mediated currents in a GPCR-independent manner.
Figure 6.
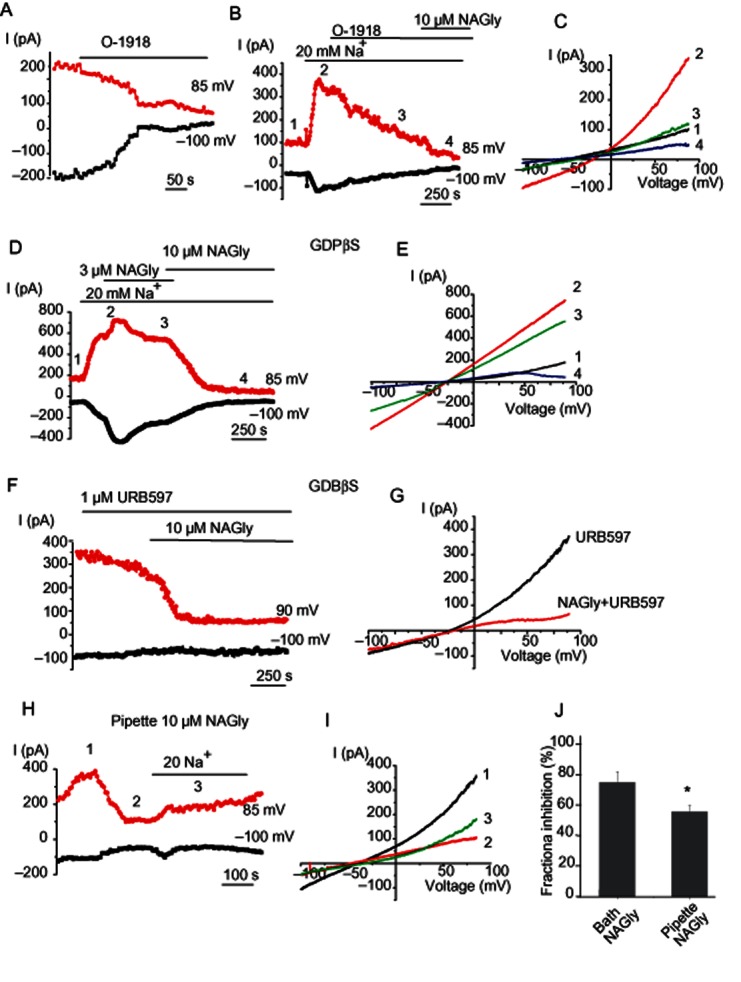
NAGly suppresses NCX currents independently of G-proteins. (A) Representative time course of changes in NCX current amplitudes at −100 and +85 mV during voltage ramps before and during application of 10 μM O-1918. (B) O-1918 (10 μM) suppresses NCXpm current pre-stimulated with a decrease in bath Na+ to 20 mM. The time course of the response. (C) Corresponding current traces elicited by voltage ramps at time points indicated in (B). (D) Representative time course of changes in the NCXpm current amplitudes to voltage ramps at voltages indicated in the presence of GDPβS in the patch pipette under control conditions (1), in the presence of 20 mM Na+ in the bath (2) and in the presence of either 3 μM (3) or 10 μM (4) of bath NAGly in low Na+ solution. (E) Corresponding current traces at time points shown in (D). (F) Representative time course of changes in current amplitudes at −100 and +85 mV during voltage ramps before and during application of 10 μM NAGly in the continued presence of URB597. Pipette solution was supplemented with 0.5 mM GDPβS. (G) Corresponding I-V relationship. (H) Representative time course of the changes in current amplitude during intracellular dialysis of EA.hy926 cell with 10 μM NAGly before and after a decrease in bath Na+, (I) corresponding I-V relationship at time points indicated in (A). (J) Mean fractional inhibition of INCX at +85 mV by extracellular and intracellular NAGly (10 μM). Fractional inhibition was obtained using the function: fractional inhibition = (Icontrol − INAGly)/Icontrol × 100.
Anandamide suppresses NCX currents independently of G-proteins
Unlike NAGly, extracellular administration of anandamide, a parent and structurally similar molecule, at a concentration of 3 μM only marginally suppressed INCX. Increasing the anandamide concentration to 10 μM strongly (up to 80%) inhibited INCX (Figure 7A–C; n = 3), suggesting that anandamide inhibits INCX with the potency lower than that for NAGly. Inclusion of 0.5 mM GDPβS into the patch pipette failed to abolish the anandamide effect on both basal (not shown, n = 3) and pre-stimulated INCX by low (20 mM) Na+ (n = 4; Figure 7D,E).
Figure 7.
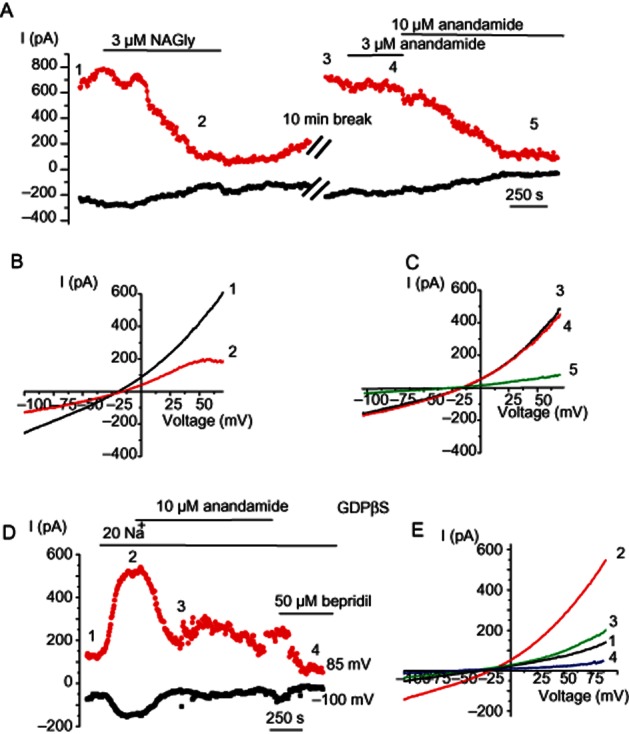
Anandamide suppresses NCX currents independently of G-proteins. (A) Representative time course of changes in current amplitudes at −100 and +85 mV during voltage ramps in response to 3 μM NAGly followed by washout and administration of 3 and 10 μM anandamide. (B, C) Corresponding current traces elicited by voltage ramps at time points indicated in (A). (D) The time course of the changes in NCXpm current amplitude augmented by a decrease in external Na+ concentration to 20 mM followed by administration of 10 μM anandamide and 50 μM bepridil. (E) Corresponding current traces elicited by voltage ramps at time points indicated in (D). Pipette solution was supplemented with 0.5 mM GDPβS.
Dual effect of NAGly on endothelial membrane potential
We examined the effect of NAGly on the membrane potential of unstimulated and histamine-stimulated EA.hy926 cells. NAGly was administered either before or during the plateau phase of the hyperpolarization to histamine. NAGly (3 μM) induced a slowly developing long-lasting hyperpolarization with an amplitude of 8.9 ± 2.1 mV (n = 8). This hyperpolarization was reversed by iberiotoxin (400 nM), an inhibitor of BKCa channels (Figure 8A). When histamine was administered in the continued presence of NAGly, only transient hyperpolarization with a reduced amplitude (7.3 ± 0.9 mV, n = 4, P < 0.05) was produced (Figure 8B). Administration of 3 μM NAGly during the plateau phase of the hyperpolarization to histamine (10 μM) strongly and reversibly inhibited the hyperpolarization within 4 min from 25.2 ± 3.2 mV to 11.0 ± 3.1 mV (n = 4; Figure 8C). Administration of 10 μM NAGly fully inhibited the hyperpolarization to 10 μM histamine (Figure 8D). Upon NAGly washout, the time course of the hyperpolarization was gradually restored within 7–8 min. These results show that NAGly directly affects the endothelial membrane potential.
Figure 8.
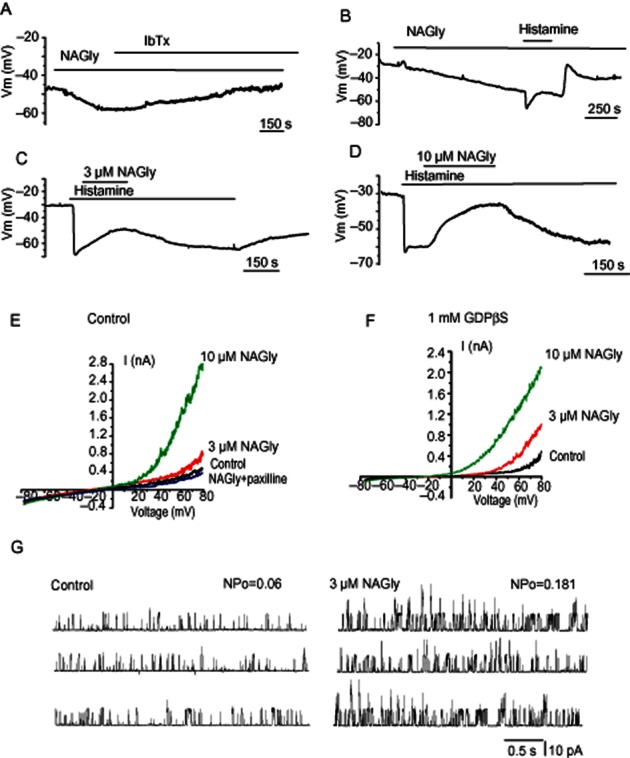
Dual effect of NAGly on the membrane potential of endothelial cells. (A) Representative effect of NAGly (3 μM) and subsequent iberiotoxin (400 nM) administration on the membrane potential of EA.hy926 cells (n = 4). (B) Representative effect of NAGly (3 μM) and subsequent administration of histamine (10 μM) on the membrane potential of EA.hy926 cells (n = 4). (C, D) Effect of 3 μM (C; n = 4) and 10 μM (D) NAGly (n = 6) on histamine (10 μM)-induced sustained hyperpolarization of EA.hy926 cells. (E) Whole cell BKCa currents in response to voltage ramps from −80 to +80 mV in the absence (control), and presence of either 3 or 10 μM NAGly and in the combined presence of 10 μM NAGly and 1 μM paxilline. (F) The same like in (E) but in the presence of 1 mM GDPβS in the pipette solution. (G) Representative recordings of single BKCa channel activity in inside-out configuration in the absence (left) and presence (right) of 3 μM NAGly. Recording were performed at +40 mV and in the presence of 300 nM free Ca2+.
G-proteins are not required for stimulation of BKCa channels by NAGly
Because NAGly produced iberiotoxin-sensitive sustained hyperpolarization in EA.hy926 cells, we next assessed the effect of NAGly on whole-cell potassium currents and whether G-proteins are required for this effect. When the pipettes were filled with KCl-based solution containing 300 nM free Ca2+, voltage ramps from −80 to 80 mV revealed an outwardly rectifying current. External NAGly (3 and 10 μM) strongly potentiated the current with characteristics typical of the BKCa channel present in these cells (Bondarenko et al., 2010; 2011a). The effect of NAGly was completely abolished by 1 μM paxilline (n = 4; Figure 8E), a BKCa channel inhibitor, but not by intracellular dialysis with the irreversible G-protein inhibitor GDPβS (1 mM; n = 4, Figure 8F). To explore whether NAGly affects BKCa channels when applied to the cytosolic face of excised membrane, the BKCa single-channel activity was recorded in inside-out patches under symmetrical K+ conditions and in the presence of 300 nM free bath Ca2+. Under these conditions, 3 μM NAGly potentiated BKCa channel activity (NPo) from 0.046 ± 0.008 to 0.139 ± 0.025 (n = 11), while the single-channel amplitude was unaffected in the presence of NAGly.
Vascular tissue experiments
To determine whether NAGly-induced inhibition of NCXpm and corresponding suppression of the hyperpolarization to histamine was a phenomenon unique to cell culture, experiments were performed on in situ endothelium of excised rat aorta, where NCXpm operating in reverse mode is critical for sustained endothelial cell hyperpolarization (Bondarenko, 2004). When the membrane potential was recorded from in situ endothelium, administration of NAGly (3 μM) in the continued presence of ACh strongly inhibited the hyperpolarization (Figure 9A).
Figure 9.
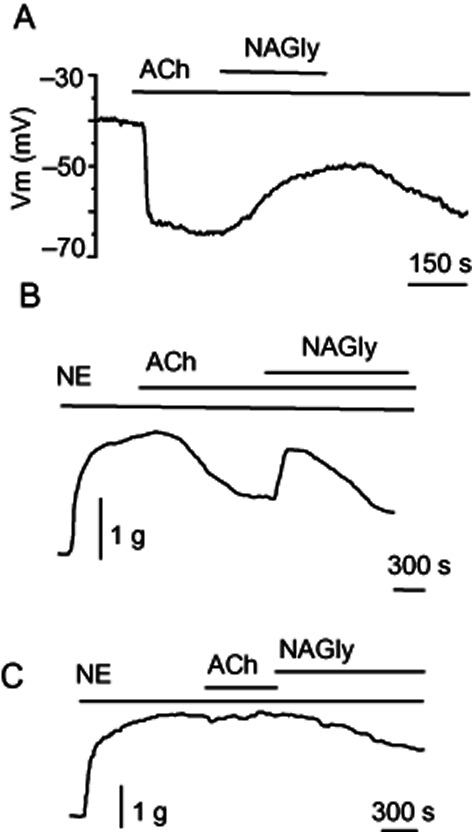
Effects of NAGly on endothelial hyperpolarization and endothelium-dependent relaxation to Ach in excised rat aorta. (A) Representative recording showing the effect of NAGly (3 μM) on the hyperpolarizing response to Ach (2 μM) in rat aortic endothelium (n = 3). (B) Representative recording showing the effect 5 μM NAGly on the relaxation of rat aortic rings produced by Ach (2 μM; n = 4). (C) Representative recording showing the effect of NAGly on norepinephrine-induced contraction of de-endothelized rat aortic ring (n = 4).
To address the functional relevance of the reduction of endothelial hyperpolarization by NAGly, we tested the effect of NAGly on endothelium-dependent relaxation of rat aortic rings produced by ACh (2 μM). Once the relaxation was developed, 5 μM NAGly produced a significant transient reduction of the relaxation (up to 70%) from the pre-NAGly levels (Figure 9B). However, this NAGly-induced inhibition of the relaxation was not persistent and in the continued presence of ACh and NAGly, the relaxation was slowly (within 5–7 min) restored.
Considering that NCX is highly expressed in vascular smooth muscle cells and partially mediates vascular smooth muscle contraction to phenylephrine (Zhang et al., 2010) and noradrenaline (Lagaud et al., 1999), we next explored the effect of NAGly on NA-induced constriction. In de-endothelialized aortic rings, NAGly (5 μM) attenuated the NA-induced contraction by 19 ± 3.8% (n = 4; Figure 9C).
Discussion
In the present study, we demonstrated that in a human umbilical vein endothelial cell-derived cell line, the sustained hyperpolarization to histamine is sensitive to bepridil, the NCXpm blocker, which inhibits both forward and reversed modes of NCXpm, KB-R7943, which preferentially inhibits NCXpm in reversed mode, and to a decrease in Na+ gradient, indicating that in these cells, NCXpm operates in a Ca2+ influx mode during exposure to histamine and this contributes to cell hyperpolarization. Our membrane potential measurements are substantiated by whole-cell current recordings under experimental conditions adjusted to suppress K+ conductance with physiological levels of intracellular Na+. The outwardly rectifying current with typical characteristics of NCXpm observed under basal conditions was potentiated by histamine and further greatly enhanced by Na+ withdrawal. These outwardly rectifying currents were suppressed by bepridil, further demonstrating that the current potentiated by histamine can be attributed to NCXpm. Our electrophysiological data, obtained from ion substitution and pharmacological experiments, together with the typical I-V characteristics of the current indicate that the major mechanism of Ca2+ inflow into EA.hy926 cells following stimulation with histamine is the reversed mode NCXpm. The function of NCXpm as a pathway for Ca2+ entry has also been demonstrated previously in experiments on the endothelium of excised rat aorta exposed to Ach (Bondarenko, 2004), and mechanical injury (Berra-Romani et al., 2012), emphasizing the importance of the transport in regulation of Ca2+-dependent processes.
Recent findings indicate that in endothelium from rat aorta exposed to ATP, NCXpm operates in the forward mode contributing to the restoration of internal Ca2+ (Berra-Romani et al., 2010), suggesting that the mode of NCXpm operation is agonist-specific. Given that NCXpm is an electrogenic transporter, NCXpm operating in the forward mode contributes to a faster decay of the hyperpolarization and intracellular Ca2+ rise. Obviously, NCXpm operation in response to the action of specific agonists not only provides an additional pathway for Ca2+ entry or extrusion, but also shapes the electrical responses ensuring a characteristic ‘fingerprint’ typical for a particular agonist. Thus, the hyperpolarization to ATP (Bondarenko and Sagach, 1996) and ionomycin (Bondarenko and Sagach, 2006) in in situ aortic endothelium is more transient and is followed by the depolarization phase, while ACh produces a long-lasting hyperpolarization, which in addition to reversed NCXpm is supported by Na+ pump stimulation (Bondarenko and Sagach, 2006). The results of the current study show that the EA.hy926 cell line may serve as a convenient model for studying the mechanisms of regulation and modulation of NCXpm-mediated Ca2+ entry in endothelial cells.
Using the same endothelial cell line, it was demonstrated previously that in rather artificial, nominal Ca2+-free conditions (no added Ca2+ no EGTA), histamine evokes oscillations in intracellular Ca2+ concentration due to coordinated interplay between reversed NCXpm operation and Ca2+ sequestration by sarcoplasmic-endoplasmic reticulum Ca2+ ATPase (Paltauf-Doburzynska et al., 2000). The oscillations were not seen in the presence of physiological levels of bath Ca2+. However, it was not clear from that study whether histamine exposure in the presence of physiological levels of Ca2+ results in the NCXpm-mediated Ca2+ entry. In fact, in follow up studies, Ca2+ entry into EA.hy926 cells stimulated with histamine has widely been attributed to the store-operated Ca2+ entry (SOCE; Malli et al., 2003; Naghdi et al., 2010), or receptor-activated Ca2+ entry (RACE; Jousset et al., 2008). In experiments with cytosolic and endoplasmic reticulum (ER) Ca2+ measurements and with the use of 10 μM La3+ as a discriminating compound, the RACE pathway accompanied by very moderate store depletion was suggested to be mainly responsible for histamine-induced Ca2+ entry into EA.hy926 cells (Jousset et al., 2008), which questions the relevance of SOCE.
Like in other non-excitable cells, Ca2+ influx into endothelial cells upon stimulation by vasoactive compounds is believed to be mainly induced by either the SOCE pathway or receptor-activated non-selective Ca2+-permeable cation channels (Nilius and Droogmans, 2001), an influx pathway(s) activated independently of depletion of intracellular Ca2+ stores. A common experimental approach used for SOCE characterization in fluorescent Ca2+ measurements is a classical Ca2+ re-addition protocol. Ca2+ re-addition after a period of exposure to Ca2+-free medium, during which the IP3-generating agonists or inhibitors of sarcoplasmic/endoplasmic reticulum Ca2+ATPase (SERCA) are applied, is associated with a large increase in fluorescence, which is commonly interpreted as a Ca2+ influx via store-operated channels (Malli et al., 2003; Naghdi et al., 2010). However, an increase in intracellular Ca2+ due to store depletion in a Ca2+-free bath solution inevitably results in Ca2+ extrusion in exchange for Na+ and, consequently, to Na+ loading. The accompanying [Ca2+]o and [Na+]i shifts aid the reversal of NCXpm upon Ca2+ re-addition. Hence, the classical Ca2+ re-addition protocol does not allow one to fully discriminate between the SOCE and NCXpm. By using an electrophysiological approach, our results confirm the functional expression of NCXpm in EA.hy926 cells and demonstrate that during stimulation with histamine in the presence of a physiological level of external Ca2+, the exchanger operates in the reverse mode (Girardin et al., 2010) ensuring sustained Ca2+ inflow and membrane hyperpolarization. Remarkably, in endothelial cells, despite a robust increase in Ca2+ following Ca2+ re-addition, whole-cell Ca2+-release-activated inward current (ICRAC) following passive store depletion could only be convincingly shown in the absence of divalent cations (Abdullaev et al., 2008; Beech, 2009; Girardin et al., 2010). This obvious discrepancy may indicate that a mechanism(s) other then SOCE largely support Ca2+ entry following stimulation of endothelial cells by vasoactive agonists. Our results suggest that NCXpm is the Ca2+ influx pathway involved following stimulation of endothelial cells with either histamine or ACh.
Modulation of stimulated Ca2+ entry is an important aspect of endothelial cell (patho)physiology, which influences numerous Ca2+-dependent processes. In the present study, we showed, for the first time, that NAGly, the recently identified endogenous lipoamino acid that acts as a ligand for GPR18/GPR92 (Kohno et al., 2006; Oh et al., 2008), affects endothelial electrical responses in both unstimulated and agonist-stimulated cells. In histamine- and ACh-stimulated cells, NAGly effectively inhibited membrane hyperpolarization via suppression of NCXpm-mediated Ca2+ inflow, while in unstimulated cells NAGly produced hyperpolarization via direct stimulation of BKCa channels. The mechanism of INCX suppression by NAGly appears to be independent of recruitment of GPCRs, since the effect was unaffected by intracellular dialysis with GDPβS, a potent and irreversible inhibitor of all G-proteins. Moreover, in NAGly-dialysed cells, INCX was gradually inhibited, although to a lesser extent than that observed during external NAGly application, further indicating that the action of NAGly involves a receptor-independent mechanism and occurs via a mechanism targeting the external face of the membrane leaflet. The role of GPR18 was addressed and ruled out by investigating the effect of the synthetic cannabinoid antagonist, O-1918, on INCX–induced suppression by NAGly. We found that O-1918 itself acts as an inhibitor of NCXpm eliminating the effect of NAGly. Anandamide, a lipid mediator and a parent molecule for NAGly, also suppressed INCX, although higher concentrations were required to observe the inhibition. INCX inhibition by NAGly was independent of FAAH, indicating that it is actually NAGly that is causing the effect but not arachidonic acids or glycine, the products of NAGly degradation via FAAH. Taken together, these observations indicate that the effect of NAGly on NCXpm is either a consequence of NAGly interacting with the NCX protein itself or modifications of the lipid bilayer, an important regulator of imbedded ion transport proteins (Crowley et al., 2003) including NCX (Kuszczak et al., 2011).
The inhibitory effect of NAGly on endothelial hyperpolarization does not appear to be unique for cell culture since NAGly effectively suppressed the endothelial hyperpolarization response to ACh in excised rat aorta. In this vascular bed, ACh produces a sustained endothelial hyperpolarization supported by Ca2+ inflow via the reverse mode of NCXpm (Bondarenko, 2004) ensuring a sustained NO release and endothelium-dependent relaxation (Schneider et al., 2002). The inhibitory effect of NAGly on endothelial cell hyperpolarization occurs at low micromolar concentrations, which were previously shown to induce endothelium-dependent component of relaxation, presumably by binding to putative non-CB1/non-CB2 endothelial cannabinoid receptors (Parmar and Ho, 2010). In excised rat aorta, the reduced hyperpolarization was accompanied by a transient suppression of the endothelium-dependent relaxation to ACh, demonstrating a functional link between reduced endothelial hyperpolarization and altered endothelium-dependent relaxation induced by NAGly.
It is noteworthy that NAGly exhibited an inhibitory effect on both the forward and reversed NCXpm and taking into account that the mode of NCXpm operation in vascular endothelium may depend on incoming stimuli (Berra-Romani et al., 2010) and vary between different vascular beds, it is tempting to speculate that in certain vascular beds, which respond to a specific agonist by stimulation of NCXpm-mediated Ca2+ extrusion from the endothelium, NAGly may promote endothelium-dependent vasodilatation via NCXpm inhibition. Importantly, NAGly appears to have complex effects on different key ion transport systems expressed in endothelial cells. Recently we showed that in cells overexpressing the key molecular constituents of SOCE, the stromal interacting molecule 1 (STIM1) and the pore-forming subunit of SOCE channels, Orai1, NAGly hampers SOCE primarily by uncoupling STIM1 from Orai1 (Deak et al., 2013). Accordingly, that and our current data indicate that endogenous lipoaminoacid NAGly fine-tunes the interplay between crucial molecular players of cellular signalling. Recently, evidence has been obtained indicating that lipoamino acids including NAGly potently inhibit T-type Ca2+ channels (Barbara et al., 2009; Ross et al., 2009). Although endothelial cells commonly lack voltage-gated Ca2+ channels, some reports do suggest the functional presence of these channels in endothelial cells. Hence, we performed additional experiments designed to exclude the possibility that the action of NAGly on NCX in our cell model is influenced by voltage-gated Ca2+ channels. Depolarizing voltage steps failed to induce inward currents in EA.hy926 cells.
Our data indicate that the inhibitory effect of NAGly on NCXpm is not restricted to endothelial cells. Indeed, we showed here that in de-endothelialized NA-precontracted rat aortic segments, NAGly opposes the contraction, which is partially controlled by NCXpm-mediated Ca2+ entry (Lagaud et al., 1999; Zhang et al., 2010). Therefore, it is likely that both NCXpm inhibition and BKCa stimulation may account for the endothelium-independent component of relaxation. Although our findings indicate that the mechanism of the NAGly action on endothelial cells is not dependent on G-proteins and highlight a receptor-independent action, they do not rule out the possible involvement of as yet unidentified GPCRs sensitive to endocannabinoids in the endothelium-dependent component of the vascular relaxation reported earlier in a number of studies (Jarai et al., 1999; Mukhopadhyay et al., 2002; Offertaler et al., 2003; Begg et al., 2005). Because endothelial cells undergo significant changes in culture, some aspects of endothelial cell physiology, including expression of ion channels and receptors may be altered in cultured cells (Marchenko and Sage, 1993; Sandow and Grayson, 2009). These alterations may be relevant for endocannabinoid signalling mechanisms. More extensive electrophysiological studies using in situ vascular endothelium are required to resolve the enigma of the existence of still unidentified cannabinoid receptors in the vascular endothelium. Inasmuch as NCXpm is implicated in ischaemic/reperfusion injury, we hypothesize that this endogenous lipid may exhibit cardio- and neuroprotection against myocardial ischaemia or brain insults. This still unrecognized feature of NAGly and possible alterations of endogenous levels of NAGly in ischaemic tissues warrant further investigation.
Collectively, the results of the present study demonstrate a previously unrecognized inhibitory effect of NAGly on NCXpm activity, which largely controls endothelial cell function. In the vasculature, the action of NAGly does not appear to be limited to promoting endothelial hyperpolarization and vasodilatation, but it may also inhibit agonist-stimulated endothelial hyperpolarization induced by reversed NCXpm. This effect on endothelial electrical signalling is accompanied by a transient reversal of endothelium-dependent vasodilatation. A better understanding of the role of NAGly and related lipid compounds in the mechanisms involved in the regulation and modulation of cardiovascular function in health and disease is important for the development of novel effective approaches for the management of cardiovascular disorders.
Acknowledgments
We are grateful for continuous financial support from the Austrian Science Funds (FWF, P21857-B18 and P22553-B18) and for excellent technical assistance of Dr. R. Rost (Medical University of Graz) and L. Stepanenko (Bogomoletz Institute of Physiology). A. Deak is funded by the PhD programme ‘Neuroscience’ at the Medical University of Graz.
Glossary
- BKCa
high-conductance Ca2+-activated K+ channel
- CB1 receptor
cannabinoid receptor type 1
- CB2 receptor
cannabinoid receptor type 2
- FAAH
fatty acid amide hydrolase
- GDPβS
guanosine 5′-O-(2-thiodiphosphate)
- GPR18
GPCR 18
- GPR92
GPCR 92
- NAGly
N-arachidonoyl glycyne
- NCXpm
plasma membrane Na+-Ca2+ exchanger
- NMDG
N-methyl-d-glucamine
- O-1918
1,3-dimethoxy-5-methyl-2-[(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-benzene
- RACE
receptor-activated Ca2+ entry
- SOCE
store-operated Ca2+ entry
- STIM1
stromal interacting molecule 1
- TRPV1
transient receptor potential cation channel V1
Conflicts of interest
None to declare.
References
- Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. 2008;103:1289–1299. doi: 10.1161/01.RES.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP. So what do we call GPR18 now? Br J Pharmacol. 2012;165:2411–2413. doi: 10.1111/j.1476-5381.2011.01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peter JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (5th edition) 2011;164(Suppl. S1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barana A, Amoros I, Caballero R, Gomez R, Osuna L, Lillo MP, et al. Endocannabinoids and cannabinoid analogues block cardiac hKv1.5 channels in a cannabinoid receptor-independent manner. Cardiovasc Res. 2010;85:56–67. doi: 10.1093/cvr/cvp284. [DOI] [PubMed] [Google Scholar]
- Barbara G, Alloui A, Nargeot J, Lory P, Eschalier A, Bourinet E, et al. T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J Neurosci. 2009;29:13106–13114. doi: 10.1523/JNEUROSCI.2919-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ. Harmony and discord in endothelial calcium entry. Circ Res. 2009;104:e22–e23. doi: 10.1161/CIRCRESAHA.108.191338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg M, Pacher P, Batkai S, Osei-Hyiaman D, Offertaler L, Mo FM, et al. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;106:133–145. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Berra-Romani R, Raqeeb A, Guzman-Silva A, Torres-Jacome J, Tanzi F, Moccia F. Na+-Ca2+ exchanger contributes to Ca2+ extrusion in ATP-stimulated endothelium of intact rat aorta. Biochem Biophys Res Commun. 2010;395:126–130. doi: 10.1016/j.bbrc.2010.03.153. [DOI] [PubMed] [Google Scholar]
- Berra-Romani R, Raqeeb A, Torres-Jacome J, Guzman-Silva A, Guerra G, Tanzi F, et al. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J Vasc Res. 2012;49:65–76. doi: 10.1159/000329618. [DOI] [PubMed] [Google Scholar]
- Bondarenko A. Sodium-calcium exchanger contributes to membrane hyperpolarization of intact endothelial cells from rat aorta during acetylcholine stimulation. Br J Pharmacol. 2004;143:9–18. doi: 10.1038/sj.bjp.0705866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko A, Chesler M. Rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001;34:134–142. doi: 10.1002/glia.1048. [DOI] [PubMed] [Google Scholar]
- Bondarenko A, Sagach V. Na+-K+-ATPase is involved in the sustained ACh-induced hyperpolarization of endothelial cells from rat aorta. Br J Pharmacol. 2006;149:958–965. doi: 10.1038/sj.bjp.0706913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko A, Svichar N, Chesler M. Role of Na+-H+ and Na+-Ca2+ exchange in hypoxia-related acute astrocyte death. Glia. 2005;49:143–152. doi: 10.1002/glia.20107. [DOI] [PubMed] [Google Scholar]
- Bondarenko A, Waldeck-Weiermair M, Naghdi S, Poteser M, Malli R, Graier WF. GPR55-dependent and -independent ion signalling in response to lysophosphatidylinositol in endothelial cells. Br J Pharmacol. 2010;161:308–320. doi: 10.1111/j.1476-5381.2010.00744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko AI, Sagach VF. Modulation of the membrane potential of intact guinea pig aortic endothelium. Neurophysiology. 1996;28:202–207. [Google Scholar]
- Bondarenko AI, Malli R, Graier WF. The GPR55 agonist lysophosphatidylinositol acts as an intracellular messenger and bidirectionally modulates Ca2+-activated large-conductance K+ channels in endothelial cells. Pflugers Arch. 2011a;461:177–189. doi: 10.1007/s00424-010-0898-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko AI, Malli R, Graier WF. The GPR55 agonist lysophosphatidylinositol directly activates intermediate-conductance Ca2+ -activated K+ channels. Pflugers Arch. 2011b;462:245–255. doi: 10.1007/s00424-011-0977-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Hu SS, Benton VM, Stuart JM, Masuda K, et al. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem. 2009;10:14. doi: 10.1186/1471-2091-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein S, McQuain C, Ross A, Salmonsen R, Zurier RE. Resolution of inflammation by N-arachidonoylglycine. J Cell Biochem. 2011;112:3227–3233. doi: 10.1002/jcb.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Treistman SN, Dopico AM. Cholesterol antagonizes ethanol potentiation of human brain BKCa channels reconstituted into phospholipid bilayers. Mol Pharmacol. 2003;64:365–372. doi: 10.1124/mol.64.2.365. [DOI] [PubMed] [Google Scholar]
- Deak AT, Groschner LN, Alam MR, Seles E, Bondarenko AI, Graier WF, et al. The endocannabinoid N-arachidonoyl glycine (NAGly) inhibits store-operated Ca2+ entry by abrogating STIM1/Orai1 interaction. J Cell Sci. 2013;126:879–888. doi: 10.1242/jcs.118075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardin NC, Antigny F, Frieden M. Electrophysiological characterization of store-operated and agonist-induced Ca2+ entry pathways in endothelial cells. Pflugers Arch. 2010;460:109–120. doi: 10.1007/s00424-010-0825-1. [DOI] [PubMed] [Google Scholar]
- Herradon E, Martin MI, Lopez-Miranda V. Characterization of the vasorelaxant mechanisms of the endocannabinoid anandamide in rat aorta. Br J Pharmacol. 2007;152:699–708. doi: 10.1038/sj.bjp.0707404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Randall MD. Endothelium-dependent metabolism by endocannabinoid hydrolases and cyclooxygenases limits vasorelaxation to anandamide and 2-arachidonoylglycerol. Br J Pharmacol. 2007;150:641–651. doi: 10.1038/sj.bjp.0707141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Petros TJ, Chang SY, Zavitsanos PA, Zipkin RE, et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J Biol Chem. 2001;276:42639–42644. doi: 10.1074/jbc.M107351200. [DOI] [PubMed] [Google Scholar]
- Jarai Z, Wagner JA, Varga K, Lake KD, Compton DR, Martin BR, et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc Natl Acad Sci U S A. 1999;96:14136–14141. doi: 10.1073/pnas.96.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong HJ, Vandenberg RJ, Vaughan CW. N-arachidonyl-glycine modulates synaptic transmission in superficial dorsal horn. Br J Pharmacol. 2010;161:925–935. doi: 10.1111/j.1476-5381.2010.00935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jousset H, Malli R, Girardin N, Graier WF, Demaurex N, Frieden M. Evidence for a receptor-activated Ca2+ entry pathway independent from Ca2+ store depletion in endothelial cells. Cell Calcium. 2008;43:83–94. doi: 10.1016/j.ceca.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno M, Hasegawa H, Inoue A, Muraoka M, Miyazaki T, Oka K, et al. Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem Biophys Res Commun. 2006;347:827–832. doi: 10.1016/j.bbrc.2006.06.175. [DOI] [PubMed] [Google Scholar]
- Kuszczak I, Samson SE, Pande J, Shen DQ, Grover AK. Sodium-calcium exchanger and lipid rafts in pig coronary artery smooth muscle. Biochim Biophys Acta. 2011;1808:589–596. doi: 10.1016/j.bbamem.2010.11.029. [DOI] [PubMed] [Google Scholar]
- Lagaud GJ, Randriamboavonjy V, Roul G, Stoclet JC, Andriantsitohaina R. Mechanism of Ca2+ release and entry during contraction elicited by norepinephrine in rat resistance arteries. Am J Physiol. 1999;276(1 Pt 2):H300–H308. doi: 10.1152/ajpheart.1999.276.1.H300. [DOI] [PubMed] [Google Scholar]
- Lee C, Dhalla NS, Hryshko LV. Therapeutic potential of novel Na+-Ca2+ exchange inhibitors in attenuating ischemia-reperfusion injury. Can J Cardiol. 2005;21:509–516. [PubMed] [Google Scholar]
- Lu VB, Puhl HL, Ikeda SR., 3rd N-Arachidonyl glycine does not activate G protein-coupled receptor 18 signaling via canonical pathways. Mol Pharmacol. 2013;83:267–282. doi: 10.1124/mol.112.081182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollum L, Howlett AC, Mukhopadhyay S. Anandamide-mediated CB1/CB2 cannabinoid receptor – independent nitric oxide production in rabbit aortic endothelial cells. J Pharmacol Exp Ther. 2007;321:930–937. doi: 10.1124/jpet.106.117549. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D. GPR18 in Microglia: implications for the CNS and endocannabinoid system signalling. Br J Pharmacol. 2012;167:1575–1582. doi: 10.1111/j.1476-5381.2012.02019.x. doi: 10.1111/j.1476-5381.2012.02019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Hu SS, Rimmerman N, Juknat A, Vogel Z, Walker JM, et al. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010;11:44. doi: 10.1186/1471-2202-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Page J, Dunn E, Bradshaw HB. Delta(9) -THC and N-arachidonyl glycine are full agonists at GPR18 and cause migration in the human endometrial cell line, HEC-1B. Br J Pharmacol. 2012;165:2414–2424. doi: 10.1111/j.1476-5381.2011.01497.x. doi: 10.1111/j.1476-5381.2011.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malli R, Frieden M, Osibow K, Zoratti C, Mayer M, Demaurex N, et al. Sustained Ca2+ transfer across mitochondria is Essential for mitochondrial Ca2+ buffering, sore-operated Ca2+ entry, and Ca2+ store refilling. J Biol Chem. 2003;278:44769–44779. doi: 10.1074/jbc.M302511200. [DOI] [PubMed] [Google Scholar]
- Marchenko SM, Sage SO. Electrical properties of resting and acetylcholine-stimulated endothelium in intact rat aorta. J Physiol. 1993;462:735–751. doi: 10.1113/jphysiol.1993.sp019579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milman G, Maor Y, Abu-Lafi S, Horowitz M, Gallily R, Batkai S, et al. N-arachidonoyl L-serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc Natl Acad Sci U S A. 2006;103:2428–2433. doi: 10.1073/pnas.0510676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Chapnick BM, Howlett AC. Anandamide-induced vasorelaxation in rabbit aortic rings has two components: G protein dependent and independent. Am J Physiol Heart Circ Physiol. 2002;282:H2046–H2054. doi: 10.1152/ajpheart.00497.2001. [DOI] [PubMed] [Google Scholar]
- Naghdi S, Waldeck-Weiermair M, Fertschai I, Poteser M, Graier WF, Malli R. Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1-dependent store-operated Ca2+ entry. J Cell Sci. 2010;123(Pt 15):2553–2564. doi: 10.1242/jcs.070151. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA, Randall MD. Vascular effects of delta 9-tetrahydrocannabinol (THC), anandamide and N-arachidonoyldopamine (NADA) in the rat isolated aorta. Eur J Pharmacol. 2005;507:211–221. doi: 10.1016/j.ejphar.2004.11.056. [DOI] [PubMed] [Google Scholar]
- Offertaler L, Mo FM, Batkai S, Liu J, Begg M, Razdan RK, et al. Selective ligands and cellular effectors of a G protein-coupled endothelial cannabinoid receptor. Mol Pharmacol. 2003;63:699–705. doi: 10.1124/mol.63.3.699. [DOI] [PubMed] [Google Scholar]
- Oh DY, Yoon JM, Moon MJ, Hwang JI, Choe H, Lee JY, et al. Identification of farnesyl pyrophosphate and N-arachidonylglycine as endogenous ligands for GPR92. J Biol Chem. 2008;283:21054–21064. doi: 10.1074/jbc.M708908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz M. Receptor-independent actions of cannabinoids on cell membranes: focus on endocannabinoids. Pharmacol Ther. 2006;111:114–144. doi: 10.1016/j.pharmthera.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Paltauf-Doburzynska J, Frieden M, Spitaler M, Graier WF. Histamine-induced Ca2+ oscillations in a human endothelial cell line depend on transmembrane ion flux, ryanodine receptors and endoplasmic reticulum Ca2+-ATPase. J Physiol. 2000;524(Pt 3):701–713. doi: 10.1111/j.1469-7793.2000.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmar N, Ho WS. N-arachidonoyl glycine, an endogenous lipid that acts as a vasorelaxant via nitric oxide and large conductance calcium-activated potassium channels. Br J Pharmacol. 2010;160:594–603. doi: 10.1111/j.1476-5381.2009.00622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross HR, Gilmore AJ, Connor M. Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonoyl dopamine. Br J Pharmacol. 2009;156:740–750. doi: 10.1111/j.1476-5381.2008.00072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL, Grayson TH. Limits of isolation and culture: intact vascular endothelium and BKCa. Am J Physiol Heart Circ Physiol. 2009;297:H1–H7. doi: 10.1152/ajpheart.00042.2009. [DOI] [PubMed] [Google Scholar]
- Schneider JC, El Kebir D, Chereau C, Mercier JC, Dall'Ava-Santucci J, Dinh-Xuan AT. Involvement of Na+/Ca2+ exchanger in endothelial NO production and endothelium-dependent relaxation. Am J Physiol Heart Circ Physiol. 2002;283:H837–H844. doi: 10.1152/ajpheart.00789.2001. [DOI] [PubMed] [Google Scholar]
- Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J Med Chem. 1997;40:659–667. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- Takenouchi R, Inoue K, Kambe Y, Miyata A. N-arachidonoyl glycine induces macrophage apoptosis via GPR18. Biochem Biophys Res Commun. 2012;418:366–371. doi: 10.1016/j.bbrc.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Tsumura M, Sobhan U, Muramatsu T, Sato M, Ichikawa H, Sahara Y, et al. TRPV1-mediated calcium signal couples with cannabinoid receptors and sodium-calcium exchangers in rat odontoblasts. Cell Calcium. 2012;52:124–136. doi: 10.1016/j.ceca.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Vuong LA, Mitchell VA, Vaughan CW. Actions of N-arachidonyl-glycine in a rat neuropathic pain model. Neuropharmacology. 2008;54:189–193. doi: 10.1016/j.neuropharm.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Wagner JA, Varga K, Jarai Z, Kunos G. Mesenteric vasodilation mediated by endothelial anandamide receptors. Hypertension. 1999;33(1 Pt 2):429–434. doi: 10.1161/01.hyp.33.1.429. [DOI] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, et al. Lipid G protein-coupled receptor ligand identification using beta-arrestin PathHunter assay. J Biol Chem. 2009;284:12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ren C, Chen L, Navedo MF, Antos LK, Kinsey SP, et al. Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current and lowers blood pressure. Am J Physiol Heart Circ Physiol. 2010;298:H1472–H1483. doi: 10.1152/ajpheart.00964.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]