

Abstract
Aims
Adenosine is a potent vasodilator contributing to cerebral blood flow regulation during metabolic stress. However, the distribution of adenosine receptor subtypes and underlying signalling mechanisms for dilation of pial arterioles remain unclear. The present study aimed at addressing these issues.
Methods and results
Isolated porcine pial arterioles were subjected to study of vasomotor function, localization of adenosine receptors, and production of nitric oxide (NO). Concentration-dependent vasodilation to adenosine was inhibited by A2A receptor antagonist ZM241385 but not by A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine. A2A receptors were detected in endothelium and smooth muscle of pial arterioles via immunohistochemistry. Adenosine significantly increased arteriolar production of NO, and the induced dilation was insensitive to KATP channel blocker glibenclamide but was attenuated by endothelial denudation, NO synthase inhibitor l-NAME, or guanylyl cyclase inhibitor ODQ in a similar manner. Both inward rectifier potassium (Kir) channel inhibitor barium and cAMP signalling inhibitor Rp-8-Br-cAMPS attenuated adenosine-induced dilation. In the presence of l-NAME or the absence of endothelium, addition of Rp-8-Br-cAMPS but not barium further reduced adenosine-induced responses. Barium diminished endothelium-independent vasodilation to NO donor sodium nitroprusside. Comparable to the adenosine-induced response, vasodilation to A2A receptor agonist CGS21680 was attenuated by endothelial removal, ZM241385, l-NAME, barium, or Rp-8-Br-cAMPS, but not by glibenclamide.
Conclusion
Adenosine evokes dilation of porcine pial arterioles via parallel activation of endothelial and smooth muscle A2A receptors. Stimulation of endothelial NO production activates smooth muscle guanylyl cyclase for vasodilation by opening Kir channels. Adenosine also activates smooth muscle cAMP signalling leading to vasodilation.
Keywords: Adenosine, Microcirculation, Nitric oxide, Potassium channels, Vasodilation
1. Introduction
The cerebral microvascular network, especially at the level of arterioles, plays an important role in regulating blood flow to the surrounding cerebral tissue for proper nutrition and function.1 Since the seminal studies of Roy and Sherrington,2 cerebral blood flow has been shown to be closely coupled to brain activity through a metabolic regulation mechanism.3 In particular, the tissue metabolite adenosine is a potent vasodilator4 that has been suggested to play a prominent role in the regulation of cerebral blood flow during metabolic stress such as hypotension, neural activation, hypoxia/ischaemia, and hypercapnia/acidosis.5 Interestingly, recent experimental evidence has shown that dilation of pial arterioles on the surface of the cerebral cortex in response to adenosine is attenuated following subarachnoid haemorrhage in rats.6 Although this vasomotor dysfunction could be mediated in part via scavenging of nitric oxide (NO) by haemoglobin as reported for large cerebral arteries,7 the underlying mechanism for arteriolar dysregulation in response to adenosine remains unclear. A better understanding of the fundamental signalling pathways responsible for adenosine-induced dilation of pial arterioles may help to shed light on how cerebral arterioles dilate to metabolic stress and on the possible mechanisms contributing to vasomotor dysfunction in disease states such as cerebral ischaemia in patients following subarachnoid haemorrhage.8
Previous evidence from in vivo studies suggests that activation of adenosine A2A receptors mediates dilation of pial arterioles in response to adenosine.9,10 However, the cellular distribution of the A2A receptors and the underlying signalling pathway responsible for the adenosine-induced dilation at the arteriolar level remain unclear. In vivo studies in the newborn pig have shown that the increased cerebral blood flow in response to adenosine is partly inhibited by pharmacological blockade of NO synthase (NOS) and cAMP signalling,11 suggesting the putative roles of NO and the activation of the cAMP pathway in this process. Notably, the source of NO and the role of cyclic nucleotide signalling remain unclear because the potential neuronal and glial cell influences in this vasodilator response have not been established. Additionally, adenosine-induced activation of potassium channels has been implicated in contributing to the dilation of pial vessels in vivo,10–13 but inconsistent results have been reported for the type of potassium channel involved. Because haemodynamic changes and neural–glial interactions are known to influence vascular function and to produce confounding effects on vasomotor responses to agonists in vivo, we focused on identifying the cellular mechanism for adenosine-induced dilation under a defined environment using an isolated and pressurized vessel preparation, along with immunohistochemical and biochemical approaches. The relative roles of adenosine receptor subtypes, endothelial NOS, smooth muscle potassium channels, and cyclic nucleotide signalling (cAMP and cGMP) in vasodilation to adenosine were determined.
2. Methods
Please see Supplementary material online for expanded Methods section.
2.1. Functional assessment of isolated pial arterioles
All animal procedures were approved by the Scott & White Institutional Animal Care and Use Committee and conform to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health). Pigs were sedated with Telazol (4.4 mg/kg, i.m.) and anaesthetized with 2–4% isoflurane. After a left thoracotomy was performed, the heart was excised for exsanguination. Following exsanguination, the skull was removed and the brain was carefully isolated and placed in ice-cold saline. A segment of brain tissue on the surface of the cerebral cortex was removed and placed in a cooled dissection chamber (∼8°C) containing a physiological salt solution (PSS) with 1% bovine serum albumin. Single pial arteriolar segments (20–60 µm in internal diameter in situ, 0.6–1.0 mm in length without branches) were isolated under a dissection microscope from the surrounding neural/connective tissues. Vessels were cannulated and pressurized to 60 cmH2O intraluminal pressure without flow for measurement of the internal diameter using videomicroscopic techniques.
After vessels developed stable basal tone, the concentration–diameter relationships for adenosine (1 nM–10 µM), A1 receptor agonist N6-chloro-cyclopentyladenosine (CCPA; 1 nM–10 µM), and A2A receptor agonist 2-[p-(2-carboxyethyl)]phenylethyl-amino-5′-N-ethylcarboxamidoadenosine (CGS21680; 1 nM–10 µM) were independently established. After completing the control response, the vasodilation elicited by adenosine and by the adenosine receptor agonists was re-examined after a 30 min incubation of vessels with the selective A1 receptor antagonist, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, 0.1 µM)14–16 or the selective A2A receptor antagonist, 4-(2-[7-amino-2-(2-furyl)[1,2,4]-triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol (ZM241385, 1 µM; Tocris).14,17
The following studies were performed to elucidate the possible cellular mechanisms involved in the pial arteriolar dilation to adenosine. First, the role of endothelium in the pial arteriolar dilation to adenosine and other agonists was evaluated in vessels following air bolus injection to remove endothelial cells. Vasodilations to agonists were evaluated and compared in intact and denuded vessels from the same animal. The denuded vessels that exhibited normal basal tone, showed no vasodilation to endothelium-dependent vasodilator bradykinin (1 nM), and showed unaltered response to endothelium-independent vasodilator sodium nitroprusside (SNP, 1 nM–10 µM) were accepted for data analysis. Secondly, the involvement of endothelium-dependent signalling pathways such as activation of NOS, cyclooxygenase, and opening of small and intermediate calcium-activated potassium (SKCa and IKCa) channels in adenosine-induced dilation was examined by incubation of vessels with known effective concentrations of their specific inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 10 µM),18 indomethacin (10 µM),19 and a combination of apamin (0.1 µM, SKCa block) and TRAM-34 (1 µM, IKCa block).20,21 Thirdly, the involvement of various potassium channels in vasodilation to adenosine was examined by treating the vessels with selective inhibitors for large conductance KCa (BKCa) channels (iberiotoxin, 0.1 µM),22 ATP-sensitive potassium (KATP) channels (glibenclamide, 2 µM),12 or inward rectifier potassium (Kir) channels [barium chloride (Ba2+), 100 µM].3,10 Fourthly, the role of cyclic nucleotide signalling was evaluated by incubating the vessels with soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one (ODQ, 0.3 µM)18 and cAMP signalling inhibitor Rp-8-Br-cAMPS (100 µM).18 In some vessels, adenosine-induced vasodilation was examined following exposure to Ba2+ or Rp-8-Br-cAMPS with l-NAME (10 µM). Fifthly, to confirm the efficacy of ZM241385, l-NAME, ODQ, glibenclamide, Rp-8-Br-cAMPS, and Ba2+, vasodilations to adenosine A2A receptor agonist CGS21680 (10 µM),14,23 NO-mediated agonist bradykinin (1 nM), soluble guanylyl cyclase/cGMP signalling activator SNP (1 µM), KATP channel opener pinacidil (10 µM), adenylyl cyclase/cAMP signalling activator forskolin (0.1 µM), and Ba2+-sensitive Kir channel opener K+ (10 mM) were examined, respectively. Sixthly, the mechanism contributing to the vasodilator response to CGS21680 (10 µM) was examined in the absence or the presence of endothelium, l-NAME, Ba2+, Rp-8-Br-cAMPS, or glibenclamide. Finally, to assess the impact of nucleotide signalling on Kir channel activation, the vasodilations to forskolin and SNP were examined in the presence of Ba2+ (100 µM). In some vessels, forskolin-induced vasodilation was repeated following exposure to l-NAME (10 µM) without or with Ba2+.
2.2. NO assay
Basal and adenosine (1 µM)-stimulated production of NO from pial arterioles was evaluated by measuring the levels of nitrite, a major breakdown product of NO, using a chemiluminescence NO analyser (Sievers Instruments) as described previously.18
2.3. Immunohistochemical analysis
Frozen sections (10 µm thick) of pial arterioles were fixed in 4% paraformaldehyde and then immunolabelled with anti-adenosine A2A receptor antibody (1:100, Alpha Diagnostic International) or anti-endothelial NOS (eNOS) antibody (1:100, BD Biosciences). Images were observed using fluorescence microscopy.
2.4. Data analysis
Diameter changes in response to vasodilator agonists were normalized to the maximum diameter change in response to 100 µM SNP in ethylenediaminetetraacetic acid (1 mM)-Ca2+-free PSS and expressed as % maximum dilation. Data are reported as mean ± SEM. Statistical comparisons were performed with Student's t-tests or ANOVA when appropriate and tested with the Bonferroni multiple-range test. A value of P < 0.05 was considered significant.
3. Results
3.1. Vasodilation of pial arterioles to adenosine and adenosine receptor agonists
In this study, all vessels (n = 157) developed a similar level of basal tone (constricted to 43 ± 1% of maximum diameter). The average resting and maximum diameters of the vessel were 36 ± 1 and 83 ± 2 µm, respectively. Adenosine, A1 agonist CCPA, and A2A agonist CGS21680 produced concentration-dependent dilation of pial arterioles (Figure 1). The relative order of potency for these agonists was adenosine = CGS26180 > CCPA.
Figure 1.

Effect of A1 and A2A receptor blockade on dilation of isolated pial arterioles to (A) adenosine, (B) A1 receptor agonist CCPA, and (C) A2A receptor agonist CGS21680. Concentration-dependent vasodilation was examined before (Control) and after incubation with A1 receptor antagonist DPCPX (0.1 µM, n = 6 for adenosine and n = 5 for CCPA) or A2A receptor antagonist ZM241385 (1 µM, n = 6 for adenosine and n = 5 for CCPA and for CGS21680). n represents the number of vessels, one per animal. Data are expressed as mean ± SEM. *P < 0.05 vs. Control.
3.2. Role of adenosine receptors
Blockade of A1 receptors by DPCPX had no effect on vasodilation to adenosine (Figure 1A). In contrast, the A2A receptor antagonist ZM241385 nearly abolished vasodilation induced by the lower concentrations of adenosine (<1 µM) and reduced the response to the highest concentration of adenosine (10 µM) from 97% (Control) to 35% (Figure 1A). The dilation of arterioles to CCPA was not altered by DPCPX, but was almost completely inhibited by ZM241385 (Figure 1B). Pial arteriolar dilation to CGS21680 was nearly abolished by ZM241385 (Figure 1C). Notably, ZM241385 did not alter resting basal tone (Control: 47 ± 6% vs. ZM241385: 51 ± 6%, n = 6, P = 0.08) or receptor-mediated vasodilation to 1 nM bradykinin (84 ± 11% dilation vs. 89 ± 6% dilation with ZM241385 treatment, n = 5, P = 0.39).
3.3. Localization of adenosine A2A receptors
For cellular localization of proteins, we performed immunohistochemical analysis of A2A receptors and eNOS in isolated pial arterioles. The A2A receptor staining was detected in both smooth muscle and endothelial layers with the latter showing overlap with eNOS staining in the endothelium (yellow staining in merged image) (Figure 2).
Figure 2.
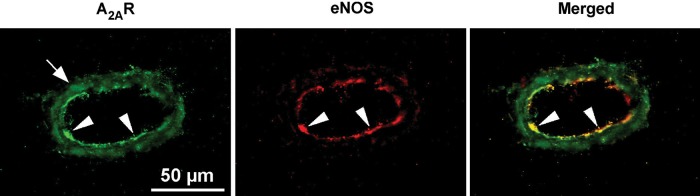
Immunohistochemical detection of adenosine A2A receptors in isolated pial arterioles. In the presence of anti-A2A receptor (green) or anti-eNOS (red) antibodies, high levels of immunostaining were detected in the endothelium and smooth muscle for A2A receptors (A2AR) and in the endothelium for eNOS. The merged image shows overlap staining (yellow) in the endothelial layer. White arrowheads denote endothelial cells and arrows denote vascular smooth muscle cells. Data shown are representative of four separate experiments.
3.4. Roles of endothelium, NOS, and soluble guanylyl cyclase
Disruption of the endothelium attenuated adenosine-induced dilation (Figure 3A) and abolished bradykinin (1 nM)-induced dilation (88 ± 4% dilation vs. 4 ± 2% dilation following denudation, n = 8) of pial arterioles without significantly altering resting basal tone (Control: 41 ± 2% vs. Denudation: 48 ± 3%, n = 8, P = 0.07) or dilation to SNP (Supplementary material online, Figure S1). In a similar manner, exposure to NOS inhibitor l-NAME did not significantly affect basal tone (Control: 37 ± 3% vs. l-NAME: 38 ± 3%, n = 7, P = 0.65) but shifted the vasodilator response curve of adenosine to the right (Figure 3B). The l-NAME concentration was effective for blocking NOS because it significantly reduced the pial arteriolar dilation to endothelium-dependent NO-mediated agonist bradykinin (83 ± 4% dilation vs. 10 ± 2% with l-NAME treatment, n = 7). Furthermore, soluble guanylyl cyclase inhibitor ODQ significantly reduced the pial arteriolar dilation to adenosine (Figure 3C) without altering basal tone (Control: 38 ± 2% vs. ODQ: 33 ± 3%, n = 5; P = 0.34). The efficacy of ODQ for soluble guanylyl cyclase inhibition was confirmed by significantly attenuating vasodilation in response to 1 µM SNP (68 ± 2% dilation vs. 26 ± 4% dilation with ODQ treatment, n = 8). The cyclooxygenase-derived prostaglandins and SKCa/IKCa-dependent EDHF20,21 were not involved in the adenosine-induced vasodilation because indomethacin and apamin plus TRAM-34 treatments did not alter the response (Supplementary material online, Figure S2).
Figure 3.

Roles of endothelium, NOS, and soluble guanylyl cyclase in dilation of isolated pial arterioles to adenosine. Concentration-dependent vasodilation to adenosine was examined (A) in the presence (Control, n = 8) or absence (Denudation, n = 8) of endothelium. The contributions of NO and soluble guanylyl cyclase to adenosine-elicited vasodilation was assessed in endothelium-intact vessels before (Control) and after incubation with their specific inhibitor (B) l-NAME (10 µM, n = 7) and (C) ODQ (0.3 µM, n = 5), respectively. n represents the number of vessels, one per animal. Data are expressed as mean ± SEM. *P < 0.05 vs. Control.
3.5. Adenosine-stimulated NO production
In the absence of adenosine, the NO production from pial arterioles was 72 ± 10 nmol/g protein. Adding adenosine (1 µM) to the vessels increased NO production by more than four-fold (Figure 4). In the presence of l-NAME, the adenosine-stimulated NO production was reduced (Figure 4).
Figure 4.

Adenosine-stimulated NO production in isolated pial arterioles. Chemiluminescence detection of NO was measured in pial arterioles treated with vehicle (Control, n = 8), adenosine (1 µM, n = 8), or adenosine and NOS inhibitor l-NAME (10 µM, n = 4) for 30 min. n represents the number of experiments, one per animal. Data are expressed as mean ± SEM. *P < 0.05 vs. Control.
3.6. Role of Kir channels
As shown in Figure 5A, Kir channel inhibitor Ba2+ (100 µM) significantly reduced adenosine-induced vasodilation. Ba2+ did not affect basal tone (Control: 42 ± 3% vs. Ba2+: 43 ± 3%, n = 9, P = 0.52) or vasodilation to KATP channel opener pinacidil 10 µM (91 ± 7% dilation vs. 85 ± 10% dilation with Ba2+ treatment, n = 5, P = 0.64), but effectively blocked vasodilation to 10 mM K+ (95 ± 3% dilation vs. 10 ± 5% dilation with Ba2+ treatment, n = 7) and significantly attenuated vasodilation to 1 µM SNP (56 ± 4% dilation vs. 38 ± 1% dilation with Ba2+ treatment, n = 8). Following the combined treatment of vessels with Ba2+ and l-NAME, there was no further reduction of the adenosine-induced dilation than to Ba2+ alone (Figure 5A). Similarly, in the absence of endothelium, vasodilation to adenosine was not further diminished by Ba2+ (Supplementary material online, Figure S3A). In another series of experiments, administration of KATP channel inhibitor glibenclamide or BKCa channel inhibitor iberiotoxin had no effect on the pial arteriolar dilation to adenosine (Supplementary material online, Figure S4) or basal tone (Control: 43 ± 2% vs. Glibenclamide: 46 ± 2%, n = 7, P = 0.08; Control: 38 ± 2% vs. Iberiotoxin: 41 ± 3%, n = 8, P = 0.09). The efficacy of glibenclamide to block KATP channels was confirmed by its ability to inhibit vasodilation to pinacidil (96 ± 2% vs. 12 ± 3% dilation with glibenclamide treatment, n = 6).
Figure 5.

Roles of Kir channels and cAMP signalling in isolated pial arteriolar dilation to adenosine and forskolin. Concentration-dependent vasodilation to adenosine was examined before (Control) and after incubation with (A) Kir channel inhibitor Ba2+ (100 µM, n = 9) or (B) cAMP signalling inhibitor Rp-8-Br-cAMPS (100 µM, n = 8). Vasodilation was also examined following incubation with l-NAME and either Ba2+ (n = 14) or Rp-8-Br-cAMPS (n = 6). (C) Vasodilation to forskolin was examined in endothelium-intact vessels before (Control) and after incubation with Rp-8-Br-cAMPS (Rp, 100 µM, n = 7), Ba2+ (100 µM, n = 6), l-NAME (l-N, 10 µM, n = 6), Ba2+ (100 µM) plus l-NAME (n = 5), and in vessels following removal of endothelial cells (-EC, n = 7). n represents the number of vessels, one per animal. Data are expressed as mean ± SEM. *P < 0.05 vs. Control. †P < 0.05 vs. Rp-8-Br-cAMPS.
3.7. Role of cAMP signalling
The cAMP signalling inhibitor Rp-8-Br-cAMPS significantly reduced adenosine-induced vasodilation (Figure 5B) without altering basal tone (Control: 40 ± 2% vs. Rp-8-Br-cAMPS: 41 ± 2%, n = 8, P = 0.71). Greater reduction in the adenosine-induced dilation was observed following the combined treatment with Rp-8-Br-cAMPS and l-NAME than to Rp-8-Br-cAMPS alone (Figure 5B). The diminished vasodilation to adenosine in the absence of endothelium was further attenuated following incubation with Rp-8-Br-cAMPS (Supplementary material online, Figure S3B). The concentration of Rp-8-Br-cAMPS used in this study effectively inhibited vasodilation in response to the adenylyl cyclase/cAMP signalling activator forskolin (Figure 5C). The forskolin-induced vasodilation was also partially blocked in a similar manner following removal of endothelium or treatment with l-NAME or Ba2+, with no further reduction in the response when exposed to both l-NAME and Ba2+, indicating that cAMP signalling is likely activated in both endothelium (i.e. NO) and smooth muscle by forskolin. Although Rp-8-Br-cAMPS did not alter vasodilation to 10 mM K+ (97 ± 2% dilation vs. 91 ± 4% dilation with Rp-8-Br-cAMPS treatment, n = 5, P = 0.12), it significantly attenuated vasodilation to 1 µM SNP (58 ± 4% dilation vs. 36 ± 3% dilation with Rp-8-Br-cAMPS treatment, n = 5). The combination of Rp-8-Br-cAMPS and Ba2+ treatment almost completely blocked SNP-induced dilation (64 ± 5% dilation vs. 10 ± 3% dilation with Rp-8-Br-cAMPS and Ba2+ treatment, n = 7).
3.8. A2A receptor-mediated vasodilation
The signalling pathway contributing to the vasodilator response to A2A receptor agonist CGS21680 (10 µM) was also examined. Vasodilation to CGS21680 was unaffected by glibenclamide but was significantly attenuated in the absence of endothelium and following incubation with l-NAME, Ba2+, or Rp-8-Br-cAMPS (Figure 6).
Figure 6.

A2A receptor signalling in dilation of isolated pial arterioles to CGS21680. Vasodilation to A2A receptor agonist CGS21680 (10 µM) was examined in the presence (Control, n = 6) or absence of endothelial cells (-EC, n = 6) or in endothelium-intact vessels before (Control) and after incubation with l-NAME (l-N, 10 µM, n = 5), Ba2+ (100 µM, n = 6), Rp-8-Br-cAMPS (Rp, 100 µM, n = 5), or glibenclamide (GB, 2 µM, n = 6). n represents the number of vessels, one per animal. Data are expressed as mean ± SEM. *P < 0.05 vs. Control.
4. Discussion
A considerable volume of evidence has implicated that adenosine is an important mediator in the metabolic regulation of cerebral blood flow.5 However, the mechanism contributing to the adenosine-induced dilation of arterioles in the pial circulation remains largely unclear in view that previous results were obtained from in vivo studies. The salient findings of the present study indicate that adenosine-induced dilation of isolated pial arterioles from the pig is mediated via parallel activation of endothelial and smooth muscle A2A receptors. The endothelial release of NO linking cGMP activation to opening of smooth muscle Kir channels and the cAMP signalling in the smooth muscle are key mechanisms contributing to the vasodilator response.
Experimental evidence from in vivo studies in the mouse9 and rat10 indicate that the activation of adenosine A2A receptors mediates pial arteriolar dilation to adenosine. The present study extended these earlier findings to porcine pial arterioles because A2A receptor antagonist ZM241385 but not A1 receptor antagonist DPCPX nearly abolished the dilation to adenosine. Moreover, to the best of our knowledge, we have provided the first immunohistochemical evidence of adenosine A2A receptor expression in both the endothelial and smooth muscle layers of intact pial arterioles. The A2A receptor antagonist ZM241385 is currently the most potent and selective antagonist for high affinity A2A receptors available.17 Ligand binding studies have demonstrated that ZM241385 has 6700-fold selectivity for A2A over A1 binding sites.17 In the present pial microvascular preparation, we also demonstrated that ZM241385 specifically inhibited vasodilation to selective A2A receptor agonist CGS21680, but did not affect vasodilation to bradykinin, indicating that ZM241385 is a specific and selective antagonist for the A2A receptor. It is possible that the residual dilation of porcine pial arterioles to the higher concentrations of adenosine was mediated via low affinity A2B receptors because a potential role of these receptors in residual dilation of pial arterioles to adenosine in the presence of ZM241385 following hypoxia has been suggested in mice.9 However, the direct role of A2B receptors in pial arteriolar dilation to adenosine will have to be addressed when selective A2B agonists and antagonists become available. Additional support for A2A receptors was provided using CCPA and CGS21680, which are the most potent and selective agonists that are commercially available for A124 and A2A15 receptors, respectively. We found that pial arterioles displayed relative order of potency for dilation with adenosine = CGS21680 > CCPA. Notably, pial arteriolar dilation to A1 agonist CCPA was not affected by its antagonist DPCPX, but was effectively inhibited by ZM241385. Although CCPA has binding affinity for A1 receptors in the subnanomolar range, it is able to activate A2A receptors in the micromolar range.24 Therefore, our data suggest that the dilation elicited by 0.1–10 µM CCPA is likely mediated by the activation of A2A receptors and further imply the lack of functional A1 receptors in pial arterioles. Taken together, our results indicate that activation of both endothelial and smooth muscle A2A receptors is responsible in large part for the adenosine-induced dilation of pial arterioles.
The contribution of endothelial adenosine A2A receptors in pial arterioles was supported by the diminished vasodilations to adenosine and CGS21680 in denuded vessels. Activation of the A2A receptors may stimulate the production and release of endothelium-derived factors such as NO,18,25,26 prostaglandins,26 and EDHF,21,26 as suggested from recent reports in other vascular beds. In the present study, it does not appear that prostaglandins or EDHF are involved in pial arteriolar dilation to adenosine because blockade of cyclooxygenase or SKCa and IKCa channels did not alter the response. In newborn pigs, pial arteriolar dilation to topical application of adenosine was found to be reduced by 50% following NOS blockade (NG-nitro-l-arginine, l-NNA),11 suggesting the involvement of NO in this response. However, the exact source of NO for those observed vasomotor responses is uncertain because it is unclear whether the release of NO is a result of secondary changes in local haemodynamics (e.g. shear stress), metabolism, and/or neural-glial activities after adenosine application. In the present study, we report for the first time that pharmacological NOS blockade significantly attenuates adenosine- and CGS21680-induced dilations of isolated pial arterioles, suggesting the direct involvement of vascular NO in mediating the A2A receptor-mediated response. A predominant role of endothelium-derived NO was apparent at the lower concentrations of adenosine from 30 nM to 1 µM because denudation and l-NAME both reduced the vasodilator response by nearly 50–60%. This contention is directly supported by the measurement of adenosine (1 µM)-stimulated NO production from intact pial arterioles. In addition, we found that inhibition of the soluble guanylyl cyclase, a downstream target of NO in the vascular smooth muscle,27 by ODQ attenuated the vasodilation to adenosine similar to that by l-NAME and endothelial denudation (Figure 3). The involvement of endothelium-derived NO/guanylyl cyclase/cGMP pathway in mediating adenosine-induced dilation as reported in the current study may explain the observed inhibition of pial arteriolar dilation11 and cerebral hyperaemic response28 to adenosine by NOS inhibitors, as well as the rise of cGMP level by adenosine in cortical periarachnoid fluid in vivo.11
Another signalling pathway that may be involved in pial arteriolar dilation to adenosine is via the opening of smooth muscle potassium channels. Experimental evidence from in vivo studies has provided disparate results supporting the contribution of KATP,11,12 BKCa13, or Kir channels10 in adenosine-induced dilation of pial arterioles. In contrast, another in vivo study showed no role of KATP channels in the adenosine-induced dilation of pial arterioles.29 In the present study, we found that only Kir channel inhibitor Ba2+ reduced the adenosine-induced dilation of pial arterioles. The ability of Ba2+ to diminish vasodilation to CGS21680 extended support for A2A receptor signalling via Kir channels. Interestingly, in the presence of both Ba2+ and l-NAME, there was no greater reduction of the vasodilation to adenosine than by either Ba2+ or l-NAME alone, suggesting the pivotal role of NO in opening Kir channels in vascular smooth muscle for vasodilation to adenosine. This idea is further supported by the ability of Ba2+ to diminish arteriolar dilation to NO donor SNP and by the lack of further reduction in dilation to adenosine in denuded vessels following exposure to Ba2+. However, the putative role of KATP channels should be considered because it has been shown that Ba2+ inhibits not only Kir channels but also possibly KATP channels in some preparations.30,31 To address this issue, we treated the vessels with Kir channel activator K+ (10 mM) and the KATP channel opener pinacidil, and found that Ba2+ only inhibited K+-induced dilation, suggesting the specific Kir channel inhibition by Ba2+ in the present study. The effectiveness of glibenclamide in blocking vasodilation to pinacidil, but not to adenosine or CGS21680, further suggests the absent role of KATP channels in pial arteriolar dilation to adenosine A2A receptor activation. On the other hand, the early in vivo evidence in newborn pigs shows that glibenclamide diminishes the adenosine-induced dilation of pial arterioles by nearly 50%.11 These disparate results may be related to the indirect influence of the inhibitor on KATP channels in brain neurons in vivo32 and/or to differences in age/maturation (neonatal vs. juvenile) of the animals. Collectively, our data suggest that the activation of endothelial A2A receptors leads to NO-dependent opening of smooth muscle Kir channels, via guanylyl cyclase/cGMP pathway, for pial arteriolar dilation to adenosine.
In the present study, we found that the dilations of pial arterioles to adenosine, CGS21680, and forskolin were significantly reduced by the cAMP signalling inhibitor Rp-8-Br-cAMPS. It is important to note that treatment of pial arterioles with combined l-NAME and Rp-8-Br-cAMPS (Figure 5B) produced a significantly greater attenuation of the adenosine-induced dilation than treatment with either inhibitor alone, indicating the involvement of two independent vasodilation mechanisms. Because the dilation of pial arterioles to forskolin was inhibited by at least 40–50% following l-NAME treatment or endothelial removal, it appears that this drug can promote cAMP-mediated activation of eNOS in porcine pial arterioles. This conclusion is consistent with previous evidence showing forskolin activates eNOS via cAMP signalling in cultured endothelial cells.33 However, it does not appear that adenosine promotes a cAMP cascade to activate eNOS because in the absence of the endothelium Rp-8-Br-cAMPS further reduced the dilation to adenosine in a comparable manner to the combination treatment with l-NAME and Rp-8-Br-cAMPS. This finding indicates a component of smooth muscle cAMP signalling, independent of eNOS, in adenosine-induced dilation of porcine pial arterioles. Interestingly, we also found that Rp-8-Br-cAMPS had an inhibitory effect on the dilation of pial arterioles to SNP. This result may intuitively suggest the involvement of cAMP in mediating NO signalling for adenosine-induced vasodilation. It should be noted that the observed Rp-8-Br-cAMPS effect on SNP may be explained by a previous report that Rp-diastereomeric analogues of cAMP inhibit both cAMP- and cGMP-induced dilation of mesenteric arteries34 or by evidence showing SNP, which is composed of both NO and iron, can activate cAMP signalling via release of iron rather than NO from its structure.35 Our findings are consistent with the latter finding because the treatment of pial arterioles with Ba2+ or Rp-8-Br-cAMPS caused a comparable 40% reduction in dilation to SNP but the combined treatment with these two blockers nearly abolished the response. These results support the idea that SNP does not act solely as an NO donor in porcine pial arterioles. The activation of dual pathways, guanylyl cyclase/Kir and cAMP, appear to mediate the dilation of pial arterioles to SNP. Further evidence to support the idea that NO and Kir channels are linked in series for mediating dilation of pial arterioles was provided by similar attenuation of forskolin-induced vasodilation following exposure to Ba2+ or l-NAME with no further diminution during their combined treatment (Figure 5C). Collectively, our results indicate that the NO-independent dilation associated with smooth muscle adenosine A2A receptor activation in the porcine pial microvessels is mediated via the adenylyl cyclase/cAMP pathway.
In summary, we found that dilation of porcine pial arterioles to adenosine is mediated by at least two parallel mechanisms. First, adenosine-induced activation of endothelial A2A receptors leads to the production of NO with subsequent activation of smooth muscle soluble guanylyl cyclase and opening of Kir channels for vasodilation. Secondly, activation of smooth muscle A2A receptors evokes vasodilation through cAMP signalling. Notably, we previously found that dilation of porcine coronary18,23 and retinal25 arterioles to adenosine A2A receptor activation is mediated in part via opening of smooth muscle KATP channels independent of cAMP signalling. It appears that the pial arterioles exhibit a unique mechanism for adenosine-induced dilation different from that of other metabolically active organs. Because K+ and adenosine released from neural-glial cells have been implicated as important factors contributing to neurovascular blood-flow regulation in the cerebral circulation,3,5 our present results suggest that the opening of Kir channels plays a central role in the vasodilator response of pial arterioles to both of these metabolites. Furthermore, the NO component of A2A receptor activation of pial arterioles may be a target for impaired adenosine-induced dilation of these vessels following acute subarachnoid haemorrhage.6 The NO scavenging role of haemoglobin36 and/or elevated levels of endogenous eNOS inhibitor asymmetric dimethyl-l-arginine37 may explain the attenuated adenosine response. Identifying the molecular signalling pathways for adenosine-induced dilation of pial arterioles in the present study may help future mechanistic examinations on the role of this critical metabolite under pathophysiological conditions.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Institutes of Health (EY018420 to T.W.H.) and the Kruse Chair Endowment Fund (L.K.).
Supplementary Material
Acknowledgements
The authors gratefully thank Dr Naris Thengchaisri and Dr Guangrong Lu for their expert technical assistance.
Conflict of interest: none declared.
References
- 1.Kontos HA, Wei EP, Raper AJ, Rosenblum WI, Navari RM, Patterson JL., Jr Role of tissue hypoxia in local regulation of cerebral microcirculation. Am J Physiol. 1978;234:H582–H591. doi: 10.1152/ajpheart.1978.234.5.H582. [DOI] [PubMed] [Google Scholar]
- 2.Roy CS, Sherrington C. On the regulation of the blood supply of the brain. J Physiol. 1890;11:85–108. doi: 10.1113/jphysiol.1890.sp000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nature Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. doi:10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 4.Berne RM, Rubio R, Curnish RR. Release of adenosine from ischemic brain: effect on cerebral vascular resistance and incorporation into cerebral adenine nucleotides. Circ Res. 1974;35:262–271. doi:10.1161/01.RES.35.2.262. [Google Scholar]
- 5.O'Regan M. Adenosine and the regulation of cerebral blood flow. Neurol Res. 2005;27:175–181. doi: 10.1179/016164105X21931. doi:10.1179/016164105X21931. [DOI] [PubMed] [Google Scholar]
- 6.Britz GW, Meno JR, Park IS, Abel TJ, Chowdhary A, Nguyen TS, et al. Time-dependent alterations in functional and pharmacological arteriolar reactivity after subarachnoid hemorrhage. Stroke. 2007;38:1329–1335. doi: 10.1161/01.STR.0000259853.43084.03. doi:10.1161/01.STR.0000259853.43084.03. [DOI] [PubMed] [Google Scholar]
- 7.Macdonald RL, Weir BK. A review of hemoglobin and the pathogenesis of cerebral vasospasm. Stroke. 1991;22:971–982. doi: 10.1161/01.str.22.8.971. doi:10.1161/01.STR.22.8.971. [DOI] [PubMed] [Google Scholar]
- 8.Sehba FA, Bederson JB. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28:381–398. doi: 10.1179/016164106X114991. doi:10.1179/016164106X114991. [DOI] [PubMed] [Google Scholar]
- 9.Miekisiak G, Kulik T, Kusano Y, Kung D, Chen JF, Winn HR. Cerebral blood flow response in adenosine 2a receptor knockout mice during transient hypoxic hypoxia. J Cereb Blood Flow Metab. 2008;28:1656–1664. doi: 10.1038/jcbfm.2008.57. doi:10.1038/jcbfm.2008.57. [DOI] [PubMed] [Google Scholar]
- 10.Paisansathan C, Xu H, Vetri F, Hernandez M, Pelligrino DA. Interactions between adenosine and K+ channel-related pathways in the coupling of somatosensory activation and pial arteriolar dilation. Am J Physiol Heart Circ Physiol. 2010;299:H2009–H2017. doi: 10.1152/ajpheart.00702.2010. doi:10.1152/ajpheart.00702.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armstead WM. Role of nitric oxide, cyclic nucleotides, and the activation of ATP-sensitive K+ channels in the contribution of adenosine to hypoxia-induced pial artery dilation. J Cereb Blood Flow Metab. 1997;17:100–108. doi: 10.1097/00004647-199701000-00013. doi:10.1097/00004647-199701000-00013. [DOI] [PubMed] [Google Scholar]
- 12.Bari F, Louis TM, Busija DW. Effects of ischemia on cerebral arteriolar dilation to arterial hypoxia in piglets. Stroke. 1998;29:222–227. doi: 10.1161/01.str.29.1.222. doi:10.1161/01.STR.29.1.222. [DOI] [PubMed] [Google Scholar]
- 13.Paterno R, Faraci FM, Heistad DD. Role of Ca2+-dependent K+ channels in cerebral vasodilatation induced by increases in cyclic GMP and cyclic AMP in the rat. Stroke. 1996;27:1603–1607. doi: 10.1161/01.str.27.9.1603. doi:10.1161/01.STR.27.9.1603. [DOI] [PubMed] [Google Scholar]
- 14.Hein TW, Belardinelli L, Kuo L. Adenosine A2A receptors mediate coronary microvascular dilation to adenosine: role of nitric oxide and ATP-sensitive potassium channels. J Pharmacol Exp Ther. 1999;291:655–664. [PubMed] [Google Scholar]
- 15.Müller CE, Stein B. Adenosine receptor antagonists: structures and potential therapeutic applications. Curr Pharm Design. 1996;2:501–530. [Google Scholar]
- 16.Alzheimer C, Kargl L, ten Bruggencate G. Adenosinergic inhibition in hippocampus is mediated by adenosine A1 receptors very similar to those of peripheral tissues. Eur J Pharmacol. 1991;196:313–317. doi: 10.1016/0014-2999(91)90445-v. doi:10.1016/0014-2999(91)90445-V. [DOI] [PubMed] [Google Scholar]
- 17.Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett PWR, Jones G, et al. The in vitro pharmacology of ZM 241385, a potent, non-xanthine, A2a selective adenosine receptor antagonist. Br J Pharmacol. 1995;115:1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x. doi:10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hein TW, Kuo L. cAMP-independent dilation of coronary arterioles to adenosine: role of nitric oxide, G proteins, and KATP channels. Circ Res. 1999;85:634–642. doi: 10.1161/01.res.85.7.634. doi:10.1161/01.RES.85.7.634. [DOI] [PubMed] [Google Scholar]
- 19.Hein TW, Liao JC, Kuo L. oxLDL specifically impairs endothelium-dependent, NO-mediated dilation of coronary arterioles. Am J Physiol Heart Circ Physiol. 2000;278:H175–H183. doi: 10.1152/ajpheart.2000.278.1.H175. [DOI] [PubMed] [Google Scholar]
- 20.Cipolla MJ, Smith J, Kohlmeyer MM, Godfrey JA. SKCa and IKCa Channels, myogenic tone, and vasodilator responses in middle cerebral arteries and parenchymal arterioles: effect of ischemia and reperfusion. Stroke. 2009;40:1451–1457. doi: 10.1161/STROKEAHA.108.535435. doi:10.1161/STROKEAHA.108.535435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ribeiro AS, Fernandes VS, Orensanz LM, Martinez MP, Recio P, Martinez-Saenz A, et al. Mechanisms involved in the adenosine-induced vasorelaxation to the pig prostatic small arteries. Purinergic Signal. 2011;7:413–425. doi: 10.1007/s11302-011-9238-7. doi:10.1007/s11302-011-9238-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thengchaisri N, Kuo L. Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: role of cyclooxygenase and potassium channels. Am J Physiol Heart Circ Physiol. 2003;285:H2255–H2263. doi: 10.1152/ajpheart.00487.2003. [DOI] [PubMed] [Google Scholar]
- 23.Hein TW, Wang W, Zoghi B, Muthuchamy M, Kuo L. Functional and molecular characterization of receptor subtypes mediating coronary microvascular dilation to adenosine. J Mol Cell Cardiol. 2001;33:271–282. doi: 10.1006/jmcc.2000.1298. doi:10.1006/jmcc.2000.1298. [DOI] [PubMed] [Google Scholar]
- 24.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, et al. Comparative pharmacology of human adenosine receptor subtypes - characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg's Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. doi:10.1007/PL00005131. [DOI] [PubMed] [Google Scholar]
- 25.Hein TW, Yuan Z, Rosa RH, Jr, Kuo L. Requisite roles of A2A receptors, nitric oxide, and KATP channels in retinal arteriolar dilation in response to adenosine. Invest Ophthalmol Vis Sci. 2005;46:2113–2119. doi: 10.1167/iovs.04-1438. doi:10.1167/iovs.04-1438. [DOI] [PubMed] [Google Scholar]
- 26.Radenkovic M, Stojanovic M, Jankovic R, Topalovic M, Stojiljkovic M. Combined contribution of endothelial relaxing autacoides in the rat femoral artery response to CPCA: an adenosine A2 receptor agonist. Sci World J. 2012;2012:143818. doi: 10.1100/2012/143818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA. 1977;74:3203–3207. doi: 10.1073/pnas.74.8.3203. doi:10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dirnagl U, Niwa K, Lindauer U, Villringer A. Coupling of cerebral blood flow to neuronal activation: role of adenosine and nitric oxide. Am J Physiol. 1994;267:H296–H301. doi: 10.1152/ajpheart.1994.267.1.H296. [DOI] [PubMed] [Google Scholar]
- 29.Taguchi H, Heistad DD, Kitazono T, Faraci FM. ATP-sensitive K+ channels mediate dilatation of cerebral arterioles during hypoxia. Circ Res. 1994;74:1005–1008. doi: 10.1161/01.res.74.5.1005. doi:10.1161/01.RES.74.5.1005. [DOI] [PubMed] [Google Scholar]
- 30.Ishizaka H, Kuo L. Endothelial ATP-sensitive potassium channels mediate coronary microvascular dilation to hyperosmolarity. Am J Physiol. 1997;273:H104–H112. doi: 10.1152/ajpheart.1997.273.1.H104. [DOI] [PubMed] [Google Scholar]
- 31.Bonev AD, Nelson MT. ATP-sensitive potassium channels in smooth muscle cells from guinea pig urinary bladder. Am J Physiol. 1993;264:C1190–C1200. doi: 10.1152/ajpcell.1993.264.5.C1190. [DOI] [PubMed] [Google Scholar]
- 32.Li DP, Chen SR, Pan HL. Adenosine inhibits paraventricular pre-sympathetic neurons through ATP-dependent potassium channels. J Neurochem. 2010;113:530–542. doi: 10.1111/j.1471-4159.2010.06618.x. doi:10.1111/j.1471-4159.2010.06618.x. [DOI] [PubMed] [Google Scholar]
- 33.Namkoong S, Kim CK, Cho YL, Kim JH, Lee H, Ha KS, et al. Forskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling. Cell Signal. 2009;21:906–915. doi: 10.1016/j.cellsig.2009.01.038. doi:10.1016/j.cellsig.2009.01.038. [DOI] [PubMed] [Google Scholar]
- 34.Jackson WF. Rp diastereomeric analogs of cAMP inhibit both cAMP- and cGMP-induced dilation of hamster mesenteric small arteries. Pharmacology. 1996;52:226–234. doi: 10.1159/000139387. doi:10.1159/000139387. [DOI] [PubMed] [Google Scholar]
- 35.Kim HJ, Tsoy I, Park MK, Lee YS, Lee JH, Seo HG, et al. Iron released by sodium nitroprusside contributes to heme oxygenase-1 induction via the cAMP-protein kinase A-mitogen-activated protein kinase pathway in RAW 264.7 cells. Mol Pharmacol. 2006;69:1633–1640. doi: 10.1124/mol.105.020487. doi:10.1124/mol.105.020487. [DOI] [PubMed] [Google Scholar]
- 36.Huang KT, Han TH, Hyduke DR, Vaughn MW, Van Herle H, Hein TW, et al. Modulation of nitric oxide bioavailability by erythrocytes. Proc Natl Acad Sci USA. 2001;98:11771–11776. doi: 10.1073/pnas.201276698. doi:10.1073/pnas.201276698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung CS, Iuliano BA, Harvey-White J, Espey MG, Oldfield EH, Pluta RM. Association between cerebrospinal fluid levels of asymmetric dimethyl-L-arginine, an endogenous inhibitor of endothelial nitric oxide synthase, and cerebral vasospasm in a primate model of subarachnoid hemorrhage. J Neurosurg. 2004;101:836–842. doi: 10.3171/jns.2004.101.5.0836. doi:10.3171/jns.2004.101.5.0836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.