

Abstract
Aims
Atrial fibrillation (AF) is increased in patients with heart failure resulting from myocardial infarction (MI). We aimed to determine the effects of chronic ventricular MI in rabbits on the susceptibility to AF, and underlying atrial electrophysiological and Ca2+-handling mechanisms.
Methods and results
In Langendorff-perfused rabbit hearts, under β-adrenergic stimulation with isoproterenol (ISO; 1 µM), 8 weeks MI decreased AF threshold, indicating increased AF susceptibility. This was associated with increased atrial action potential duration (APD)-alternans at 90% repolarization, by 147%, and no significant change in the mean APD or atrial global conduction velocity (CV; n = 6–13 non-MI hearts, 5–12 MI). In atrial isolated myocytes, also under β-stimulation, L-type Ca2+ current (ICaL) density and intracellular Ca2+-transient amplitude were decreased by MI, by 35 and 41%, respectively, and the frequency of spontaneous depolarizations (SDs) was substantially increased. MI increased atrial myocyte size and capacity, and markedly decreased transverse-tubule density. In non-MI hearts perfused with ISO, the ICaL-blocker nifedipine, at a concentration (0.02 µM) causing an equivalent ICaL reduction (35%) to that from the MI, did not affect AF susceptibility, and decreased APD.
Conclusion
Chronic MI in rabbits remodels atrial structure, electrophysiology, and intracellular Ca2+ handling. Increased susceptibility to AF by MI, under β-adrenergic stimulation, may result from associated production of atrial APD alternans and SDs, since steady-state APD and global CV were unchanged under these conditions, and may be unrelated to the associated reduction in whole-cell ICaL. Future studies may clarify potential contributions of local conduction changes, and cellular and subcellular mechanisms of alternans, to the increased AF susceptibility.
Keywords: Atrial fibrillation, Myocardial infarction, Action potential alternans, Afterdepolarization, β-Adrenergic stimulation, T-tubule, Calcium
1. Introduction
Atrial fibrillation (AF), often co-existing with heart failure (HF) as a result of myocardial infarction (MI), is the most common sustained cardiac arrhythmia. It causes substantial morbidity and mortality, and is associated with electrical, structural, and contractile remodelling of the atrial myocardium.1,2 Both re-entrant and non-re-entrant arrhythmias underlie AF initiation and maintenance, and which predominates depends on underlying cardiac pathophysiology and the pattern of associated atrial remodelling.3
Chronic ventricular tachypacing (VTP) models of HF-induced AF have been shown in numerous studies to cause atrial structural and electrophysiological remodelling, with a variety of changes in ion currents, action potentials (APs), and intracellular Ca2+(Ca2+i) handling. For example, AP duration (APD) is either increased,4,5 unchanged,6 or shortened;7 the Ca2+i-transient amplitude is increased5 (in contrast with a decrease, in the ventricle),8 and delayed afterdepolarizations (DADs) also occur.4
However, there are few studies of effects of chronic ventricular MI-induced HF on atrial electrophysiology: in rats9 and dogs,10 MI promoted AF, and in dogs produced an alternating AP morphology, AP alternans.10 Moreover, there are no reports, to our knowledge, of effects of chronic MI on atrial Ca2+i handling. Ventricular electrophysiological and Ca2+i-handling remodelling from chronic MI were studied in rabbits11,12 and, although atrial enlargement occurred, no atrial electrophysiological or Ca2+-handling parameters were measured.
HF, in patients, is associated with increased adrenergic tone and elevated circulating levels of catecholamines.13 Catecholamines promote atrial arrhythmias, mainly by increasing L-type Ca2+ current (ICaL) and Ca2+i-transient amplitude.14 A reduced contractile response to β-adrenoceptor stimulation is well documented in failing ventricle, and ICaL and Ca2+i-transient responses are also blunted,8 but corresponding atrial data are sparse. In the rat atrium, chronic MI reduced ICaL, and potentiated the ICaL increase from β-stimulation.15 β-Stimulation may also affect APD alternans.16 APD alternans precede AF episodes in patients,17 and alternans are associated with Ca2+i-handling abnormalities.18 β-Stimulation enhances Ca2+i cycling and excitation–contraction coupling, and either favours19 or protects against16,20 alternans, depending on whether Ca2+ sequestration or fractional release of Ca2+ from the sarcoplasmic reticulum (SR) predominates.
Atrial conduction disturbances associated with structural changes, including fibrosis, chamber enlargement, and hypertrophy are well established, leading to increased vulnerability to AF.1,21,22 Decreased conduction velocity (CV) from fibrosis could promote re-entry by decreasing re-entry wavelength (λ), since λ = CV × ERP (effective refractory period). Altered atrial gap junction organization and connexin expression could also disturb intercellular connectivity, with the potential to decrease or increase CV.23 Furthermore, disruption of atrial transverse-tubules (t-tubules), as shown in a sheep VTP model of AF,24 may contribute to hypocontractility, Ca2+i-handling changes, and an increased propensity to arrhythmia in these diseased hearts. However, the effects of MI on atrial CV or the t-tubular network are also unknown.
We hypothesized that chronic MI would promote AF under β-stimulation, associated with any or all of the following: decreased atrial λ, increased APD alternans, increased DADs, altered Ca2+i cycling, and disrupted t-tubules. Our aim, therefore, was two-fold: first, to characterize the atrial structural, electrophysiological, and Ca2+i-handling remodelling caused by chronic ventricular MI in rabbits; secondly, to investigate potential mechanisms of AF promotion by MI, by associating changes in propensity to AF with atrial APD, CV, APD alternans, DADs, ICaL, [Ca2+]i, and t-tubule density.
2. Methods
2.1. Animal model of MI
Procedures conformed to the Guide for the Care and Use of Laboratory Animals published by US National Institutes of Health. Project license: 60/4206. Adult male New Zealand White rabbits (3.0–4.0 kg) were pre-medicated with intramuscular Hypnorm (0.3 mg/kg) and anaesthetized with midazolam (0.17–0.3 mg/kg) via the marginal ear vein. Ventilation was with N2O and O2 (1:1) containing 1% isofluorane. A thoracotomy was performed and the left descending coronary artery ligated to produce an ischaemic area of 30–40% of the left ventricle (LV) and subsequent apical infarction (Figure 1Ai). Post-surgery analgaesia: intramuscular Vetergesic (0.04 mg/kg). Sham-operated rabbits underwent thoracotomy without coronary artery ligation. LV function was assessed by echocardiography, under sedation with Hypnorm (0.3 mg/kg). Coronary artery-ligated animals with left ventricle ejection fraction (LVEF) >45% were excluded from the MI group. Rabbits were humanely killed at the point of experimentation (8 weeks post-procedure) by sodium pentobarbitone (100 mg/kg) with 1000 IU heparin intravenously into the left marginal ear vein and removal of the heart.
Figure 1.

LA cellular hypertrophy and t-tubule loss resulting from ventricular MI. (A) Rabbit model of MI (i), LVEF (ii), LV end-diastolic dimension (LVEDD) (iii) n = 13 non-MI hearts, 32 MI. (B) Cardiomyocyte capacitance (i); n = 52 cells (13 non-MI hearts) and 56 cells (15 MI), width (ii), and length (iii); n = 110 cells (6 non-MI hearts) and 191 (10 MI). (C) T-tubule (TT) density. Representative confocal images (i) LV non-MI cell (a), LA non-MI (b), LA MI (c); TT density (ii); n = 14 LV cells (six hearts), 18 LA cells (seven non-MI hearts), 30 LA cells (five MI). *P < 0.05, **P < 0.01, ***P < 0.001.
2.2. Atrial cardiomyocyte isolation
Atrial cells were enzymatically dissociated as previously described.25 Briefly, the left atrium (LA) was dissected from the collagenase-digested heart, finely chopped, and gently shaken in enzyme, at 37°C. Tissues were re-suspended in enzyme and agitated, the supernatant centrifuged, and the cardiomyocyte pellet re-suspended in KB solution comprising (mM): KOH (70), KCl (40), l-glutamic acid (50), taurine (20), KH2PO4 (20), MgCl2 (3), glucose (10), HEPES (10), EGTA (0.5), pH 7.2 (37°C). This was repeated, twice. Cardiomyocyte length and width were measured using a crossed-scale eyepiece graticule (10 µm/division).
2.3. T-tubule density measurement
T-tubule density quantification was based on a previous report.26 Briefly, cardiomyocytes were suspended in Krebs–Henseleit solution containing di-4-ANEPPS (10 µM) for 10 min. Confocal fluorescence Z-stack images were obtained with 1 µm inter-segment spacing, using an LSM510 laser-scanner. From a binary image of internal membranes, pixels with intensity ≥mean pixel value of the peripheral sarcolemma were attributed to t-tubule, and t-tubule density calculated relative to the total number of pixels occupied by the cell width in central segments, using custom-made software.
2.4. Intact heart atrial electrophysiological and optical recording
Hearts were Langendorff perfused with Tyrode's solution comprising (mM): NaCl (120.9), KCl (5), MgSO4 (1), KH2PO4 (0.7), NaHCO3 (24.8), CaCl2 (1.8), glucose (15), 95% O2/5% CO2, with blebbistatin (mechanical uncoupler, 10 µM), with or without the β-adrenoceptor-agonist isoproterenol (ISO), 1 µM or the ICaL-blocker nifedipine (0.3 nM−3 µM), at 37°C, 35 mL/min. Drugs were protected from light during preparation and perfusion. Hearts were either unpaced, or paced (as specified) at 5 Hz with 2 ms pulses of twice diastolic threshold via bipolar electrodes hooked onto the right atrial appendage. Atrial vulnerability to AF induction was tested with a standard AF threshold (AFT) protocol: after 5 Hz pacing, short (1 s) bursts of high frequency (100 Hz) current pulses were delivered to the LA, increasing in 5 mA steps from 5 to 100 mA, until AF was induced. AFT was measured as the amplitude of the smallest current inducing AF lasting >4 s, and a median value from five induction attempts calculated. For optical AP recording, hearts were loaded with di-4 ANEPPS (2 mM, 100 µL bolus), and APs measured from the LA epicardial surface using a 3 mm diameter light guide system. AP shape was assessed, including APD measurement at 50, 75, and 90% repolarization (APD50,75,90), and alternans magnitude was calculated as the mean of the absolute difference between consecutive APDs. LA CV was measured, in paced hearts, with a custom-made electrode comprising bipolar pacing electrodes and two electrogram-recording electrodes (1 mm apart), rotatable to allow CV measurement in the direction of fastest signal propagation, giving an estimate of atrial global CV. Data were acquired using custom-written programs (FL Burton), and analysed using those, MATLAB, or Origin 6.1.
2.5. Cellular electrophysiology and [Ca2+]i
LA cardiomyocytes were superfused with a Krebs–Henseleit solution comprising (mM): NaCl (140), KCl (4), HEPES (5), MgCl2 (1), CaCl2 (1.8), glucose (11.1), pH 7.4 (37°C), with or without ISO or nifedipine. For recording cellular APs, microelectrodes (∼5–6 MΩ) contained (mM): KCl (20), K-aspartate (110), HEPES (5), MgCl2 (1), Na2ATP (4), disodium creatine phosphate (1), K2EGTA (0.15), pH 7.25. Cells were stimulated with 5 ms, 1 nA current pulses at 1 Hz for 90 s using an Axoclamp-2B amplifier and Clampex 9.2 software. In each cell, APs were recorded both with and without current clamping the resting membrane potential (RMP) (to −80 mV), since some cells in each group had an unclamped RMP more positive than ∼−60 mV and thus an inactivated INa. APD50–90, and the occurrence of inter-AP spontaneous depolarizations (SDs), were monitored. For recording ICaL and [Ca2+]i, microelectrodes contained (mM): KCl (20), K-aspartate (100), TEA-Cl (20), HEPES (10), MgCl2 (4.5), Na2ATP (4), Na2-creatine phosphate (1), K2EGTA (0.01), and Fura-2 (0.1 mM); pH 7.25, pCa ∼7.2. Extracellular 4-AP (5 mM), niflumic acid (0.1 mM) and TTX (5 µM) were used to eliminate contaminating currents. A voltage clamp protocol (Figure 5Ai) was cycled at 1 Hz for 90 s to record steady-state ICaL and Ca2+i transients. Fluorescence was measured at 340 and 380 nm every 15 ms using a Cairn Optoscan. Minimum (Rmin) and maximum (Rmax) fluorescence ratios (340/380 nm) were measured, and cytoplasmic [Ca2+] calculated as 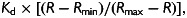 with Kd of 1.2 µM.27 The SR Ca2+ content was assessed by rapid application of 10 mM caffeine [within ∼1 s of the end of the 90 s voltage-pulse train, using a 4 mm diameter perfusion pen (Cell Micro Controls, Norfolk, USA) in close proximity to the cell] to fully release SR-Ca2+ stores, and estimated as the absolute time-integral of the resultant INa/Ca, expressed in coulombs normalized to capacitance.27
with Kd of 1.2 µM.27 The SR Ca2+ content was assessed by rapid application of 10 mM caffeine [within ∼1 s of the end of the 90 s voltage-pulse train, using a 4 mm diameter perfusion pen (Cell Micro Controls, Norfolk, USA) in close proximity to the cell] to fully release SR-Ca2+ stores, and estimated as the absolute time-integral of the resultant INa/Ca, expressed in coulombs normalized to capacitance.27
Figure 5.

Under β-stimulation, ICaL and systolic [Ca2+]i are decreased by MI. Effects of MI and/or ISO on: (A) ICaL stimulated with voltage-pulse (i) in non-MI (ii) and MI (iii) atrial cardiomyocytes. (B) ICaL density (i) and Ca2+ flux via ICaL (ii). (C) [Ca2+]i recordings from non-MI (i) and MI (ii) cardiomyocytes. (D) Diastolic (i) and peak systolic (ii) [Ca2+]i, and Ca2+-transient amplitude (iii), and rate constant (RC) of decay (iv). n =13 non-MI hearts, 12 MI. (E) Caffeine-evoked [Ca2+]i increase (i) and resulting INa/Ca integral (ii), representing SR Ca2+ content. n = 7–10 non-MI hearts, 6–11 MI.
2.6. Statistics
Continuous data are expressed as means ± SEM, and compared using unpaired or paired Student's t-tests, or ordinary or repeated measures ANOVA with appropriate post-tests. Categorical data were compared with a χ2 test. P < 0.05 was considered statistically significant.
3. Results
3.1. MI caused LV dysfunction, and LA cellular hypertrophy and detubulation
Ventricular MI decreased LVEF by 31% (Figure 1Aii) and increased LVEDD by 9% (Figure 1Aiii), with no significant effect on dimensions (mm) of LA (10.6 ± 0.2 in non-MI vs. 11.4 ± 0.3, MI), atrioventricular valve (3.1 ± 0.3 vs. 2.7 ± 0.1), or aorta (9.0 ± 0.3 vs. 8.6 ± 0.2). MI increased atrial cardiomyocyte capacitance by 56% (Figure 1Bi), width by 18% (Figure 1Bii), and length by 14% (Figure 1Biii). T-tubule density was lower in LA cells than LV cells (Figure 1Ci), and MI decreased LA t-tubule density by ∼60% (Figure 1Cii).
3.2. In the presence of β-stimulation, MI promoted AF, SDs, and APD alternans
Figure 2A shows an original electrogram recording of AF induction in an intact non-MI heart. Under β-stimulation, AFT was decreased by MI, by 58% (Figure 2Bi), indicating increased susceptibility to AF, and AF duration was increased by 44% (Figure 2Bii). AF parameters were not altered by MI in the absence of β-stimulation. Atrial global CV was not altered by MI, with or without β-stimulation (Figure 2Biii). During AP-recording, in either paced or un-paced hearts, there was no evidence of atrial SDs in non-MI or MI, with or without β-stimulation. SDs occurred in isolated cardiomyocytes, categorized as threshold: premature APs with normal morphology; or subthreshold: low-amplitude diastolic depolarizations without regenerative phase 0. Under β-stimulation, MI markedly increased the number of threshold SDs (Figure 2Cii). Subthreshold SDs were infrequent (typically averaging <1 per 30 s period of AP stimulation), in non-MI or MI, with or without β-stimulation, suggesting that MI+β-stimulation converted subthreshold SDs to threshold. Figure 3A shows an original recording of APD alternans, from an intact, paced, MI heart, in the absence of β-stimulation. In the unpaced hearts, the mean magnitude of APD alternans was not significantly affected by MI, either in the absence or presence of β-stimulation (Figure 3B). However, in the paced hearts, MI significantly increased APD alternans (Figure 3C), particularly, at APD90, by 147% in the presence of β-stimulation, and by 103% in its absence (Figure 3Ciii).
Figure 2.

Under β-stimulation, atrial arrhythmia is provoked by MI. (A) Original electrogram recording of AF induction in a non-MI heart; pacing protocol shown beneath. (B) AFT (i) and AF duration (ii); n = 7 non-MI hearts, 7 MI. CV (iii); n = 6 non-MI, 5 MI. (C) SDs: original AP trace recorded in an MI atrial cell, showing SDs (*) during stimulation (↑); bar = 250 ms (i); mean number (ii) and incidence (iii) of SDs occurring during AP trains. n = 23–31 cells, 12–13 hearts. ISO = 1 µM ISO.
Figure 3.

Atrial APD alternans is increased by MI. (A) Original optical recording of an AP train (5 Hz pacing) from intact rabbit LA, post-ventricular MI, showing AP alternans. APD alternans magnitude in unpaced (B) and 5 Hz-paced (C) hearts, measured at 50% (i), 75% (ii), and 90% (iii) repolarization. n = 13 non-MI hearts, 12 MI.
3.3. Under β-stimulation, APD was not altered by MI
β-Stimulation increased intrinsic heart rate by 50–60% in non-MI and MI hearts (Figure 4Ai). In unpaced (Figure 4A) and paced (Figure 4B) hearts, under β-stimulation, MI had no significant effect on optically recorded atrial AP morphology or APD50–90. In contrast, in the absence of β-stimulation, MI increased APD75 and APD90, in unpaced (Figure 4A) and paced (Figure 4B) hearts. Figure 4C shows corresponding AP data obtained from isolated cardiomyocytes, comparable except for the lower stimulation rate (1 Hz), which perhaps accounts for the ‘spikier’ AP phase 1 (Figure 4Ci). Neither ISO nor MI affected RMP (Figure 4Cii) and, in agreement with the intact atrial AP data, MI had no significant effect on APD in the presence of β-stimulation (Figure 4Ciii–v), and tended to prolong APD in its absence.
Figure 4.

Under β-stimulation, APD is not changed by MI. (A) Effects of MI and/or 1 µM ISO on rabbit isolated heart rate (i) and, in unpaced hearts, optically recorded atrial APs in non-MI (ii) and MI (iii), and on mean APD50–90 (iv–vi). (B) Corresponding data (i–v) obtained from paced (5 Hz) hearts. n = 13 non-MI, 12 MI. (C) Atrial isolated cardiomyocyte APs (i), at 1 Hz-stimulation, in non-MI (a) and MI (b). Effects of MI and/or ISO on RMP (ii) and, in cells with RMP clamped (−80 mV), APD50–90 (iii–v). n = 33 cells, 14 non-MI hearts, 22 cells, 11 MI.
3.4. Under β-stimulation, ICaL and systolic [Ca2+]i were decreased by MI
Figure 5 shows that in the presence of β-stimulation, but not in its absence, ICaL density and Ca2+ flux through ICaL (Figure 5A and B), and the systolic Ca2+i transient (Figure 5C and D), were each significantly and similarly decreased by MI, while diastolic [Ca2+]i was unaffected (Figure 5Di). Both the rate of decay of the Ca2+i transient (Figure 5Div), and the SR Ca2+ content (Figure 5Eii), were increased by ISO, and not significantly affected by MI.
3.5. Pharmacological inhibition of ICaL, to mimic the ICaL reduction from MI, did not promote AF
In atrial cardiomyocytes from non-MI rabbits, nifedipine inhibited ICaL in a concentration-dependent manner, with the inhibition significantly attenuated by ISO (Figure 6Ai and ii). Using the dose–response curve in the presence of ISO (Figure 6Aii, triangles), two concentrations of nifedipine were chosen, 0.02 and 2 µM (which inhibited ICaL by 35 and 41%, respectively), to test effects of ICaL reduction, in non-MI hearts and cells, on APDs and AFT under β-stimulation. Nifedipine at 0.02 and 2 µM shortened atrial cellular APD90, by 11 and 65%, respectively (Figure 6Aiii), and intact atrial epicardial APD90, by 30 and 42%, respectively (Figure 6Bi). The higher concentration (2 µM) markedly and significantly reduced the AFT, by 85% (Figure 6Bii). However, 0.02 µM nifedipine, which caused an equivalent ICaL reduction to that produced by MI under β-stimulation (35%), did not affect AF susceptibility, despite significantly decreasing APD.
Figure 6.

Under β-stimulation, ICaL reduction to mimic that from MI, did not promote AF. (A) Effects of nifedipine (NIF) on isolated cardiomyocyte (non-MI): (i) ICaL, in the absence (a) and presence (b) of ISO; (ii) nifedipine concentration-dependence of ICaL decrease, +/−ISO (Imax = maximum inhibitory effect; n = 3 hearts); (iii) APDs, under β-stimulation (n = 5 cells, 4 hearts). (B) Effects of ICaL inhibition in intact, non-MI hearts under β-stimulation, on APDs (i) and AFT (ii). n = 13 hearts.
4. Discussion
This is the first study, to our knowledge, to characterize the effects of chronic LV MI, rather than chronic VTP, on atrial Ca2+i handling and electrophysiology. We recorded from intact atria, and isolated cardiomyocytes, with and without a β-agonist. The key findings are that under β-stimulation, MI promoted AF, APD alternans, and cellular SDs, associated with reduced ICaL, Ca2+i transient and t-tubule density. β-Stimulation was used to represent in-vivo sympathetic activation (particularly in HF, with catecholamine elevation13), with consequent phosphorylation/activation of numerous ion channels and Ca2+i-handling proteins.14 We used ISO at supra-maximal concentration to ensure full adrenergic activation, but recognize that this is also supra-physiological.
The APD alternans could contribute to the increased AF susceptibility, by promoting spatial electrical heterogeneity and wavebreak.10 Since the SDs were not observed in intact atrium, this might suggest the alternans as the more likely contributor to the increased AF susceptibility. However, although isolated cardiomyocytes are more prone to afterdepolarizations than tissues, due to the absence of current sink, we cannot exclude the possibility that SDs occurred also in the intact hearts, remote from the myocardial region sensed by the fibre-optic detector. In support, chronic VTP-induced HF promoted DADs in atrial cells5 and triggered-activity characteristic or DADs in intact atria.4 Moreover, since MI increased APD alternans with or without ISO, yet MI increased AF susceptibility only with ISO, this argues against APD alternans as the sole potential electrophysiological mechanism, with the accompanying increase in SDs, as resulted from MI exclusively under β-stimulation, perhaps required.
MI affected neither atrial APD90 nor CV, under β-stimulation, and thus might not affect re-entry wavelength. However, although APD at late repolarization is a major determinant of ERP, the ERP has yet to be measured in this model. Furthermore, the MI is likely to increase atrial fibrosis, as in rats.9,28 In rabbits, atrial fibrosis and inter-atrial conduction time were increased by VTP,29 and VTP-induced fibrosis in dogs was associated with spatial heterogeneity of CV, and local conduction abnormalities.21 Increased atrial myofibroblast density21 could promote AF via multiple arrhythmia mechanisms since, when coupled to cardiomyocytes, myofibroblasts can act as current sources and/or sinks, depending on myofibroblast density, distribution, and coupling strength, thereby modulating AP characteristics and CV, including production of SDs and alternans.30 Such fibrosis had a greater bearing on atrial CV disturbances and AF than gap junction remodelling (altered connexin subtype expression ratios, phosphorylation status, and cellular distribution), from chronic VTP.31 Nevertheless, whether atrial gap junctions are altered in the present model is unknown and the potential for such remodelling to disturb CV and cause arrhythmias, as proposed in failing ventricle,23 should not be excluded. We recorded CV at a fixed location, in the direction of fastest signal propagation, and acknowledge that extensive spatial, high-resolution analysis would be required to establish the contribution of local changes in CV and its heterogeneity to AF inducibility.
The present lack of effect of MI on atrial APD, under β-stimulation, contrasts with a canine model of chronic MI and sympathetic stimulation by tyramine, in which APD was decreased.32 We found that without β-stimulation, atrial APD was increased by MI. Previous studies of atrial electrophysiological remodelling by HF in rabbits used chronic VTP rather than MI, without β-stimulation, and showed increased APD,33 consistent with the present data, as well as increased ERP33 and altered K+ channel expression.34 The present APD increase is also consistent with several canine VTP studies, e.g.4–6 APD increase may enhance Ca2+i transients as a result of AP plateau elevation from decreased ITO.35 Atrial APD decrease also occurs, after long-term (4 months) VTP in dogs,7 and with LV systolic dysfunction in patients.36 The ionic mechanism of the APD increase from chronic MI in rabbits is unknown, whereas in dogs, VTP decreased atrial ITO, ICaL, and IKs, and increased INa/Ca.1,6 ITO is prominent in the rabbit atrium, and since VTP decreased atrial Kv4.3,34 and ITO decrease prolonged APD90 in rabbit atrial cells,37 ITO reduction might contribute to the APD increase here. However, rabbit atrial ITO is carried primarily by Kv1.4, not measured in the VTP study,34 which confers slow ITO reactivation and could limit the contribution of ITO to APD at physiological and supra-physiological rates.
Since MI provoked AF under β-stimulation, we measured ICaL and the Ca2+i transient under those conditions, and found both to be decreased. The reduced Ca2+i transient likely resulted from the ICaL reduction, since the SR Ca2+ content was unchanged. The mechanism of the ICaL decrease is unclear, but may not involve altered ICaL channel expression, since chronic MI in rats did not affect α1C. 15 Another candidate is down-regulation of atrial β-adrenoceptors. We did not measure β-adrenoceptor density, but previous studies showed reduced atrial β1- and/or β2-expression in HF.38,39 The potential contribution of this ICaL reduction to the AF promotion was tested using nifedipine in intact hearts under β-stimulation. We took into account the effect of ISO to attenuate ICaL block, as observed here and elsewhere.40 MI had decreased ICaL by 35%, under β-stimulation. In non-MI hearts, nifedipine at 0.02 µM, which decreased ICaL by an equivalent degree (35%), did not affect AF susceptibility, and moderately decreased APD. In contrast, maximally effective nifedipine (2 µM), which decreased atrial ICaL by 41%, markedly decreased AFT, but that probably resulted from the accompanying marked decrease in APD; a change which did not result from the MI. Therefore, the whole-cell ICaL reduction alone may not have contributed substantially to the MI-provoked AF. Indeed, ICaL reduction, in the absence of the other atrial electrophysiological and structural consequences of chronic MI, could even suppress AF, since SDs may be provoked by increased ICaL and Ca2+i transient,14 and ICaL inhibition, while inducing Ca2+i alternans in rat ventricular cells,41 suppressed it in human atrial cells.42
However, both APD alternans and SDs result from a complex interaction between membrane potential, ion currents including ICaL and INa/Ca, and Ca2+i cycling, each of which may be altered by myocardial disease as well as stimulation rate. Simultaneous measurement of [Ca2+]i and APs in ventricular myocytes has suggested that APD alternans under rapid pacing is driven by Ca2+i alternans.43 Ca2+i alternans also occurred under tachypacing of cat atrial cells,16,44 although APs were not measured. In rabbit atrial cells, Ca2+i alternans induction depended on refractoriness of SR Ca2+ release.20 Studies of effects of myocardial disease on atrial alternans are sparse. In dogs, chronic MI,10 and chronic atrial tachypacing,45 promoted AF, which was preceded by increased atrial APD alternans. However, in neither report nor the present study was Ca2+i alternans studied. Future studies of alternans in the present rabbit MI model of AF should attempt to measure [Ca2+]i and APs simultaneously, in single cells and intact atrium, under supra-physiological and physiological stimulation rates.
We found that chronic MI caused atrial t-tubule loss, consistent with previous studies of atrial remodelling from chronic VTP24 and chronic AF.46,47 The concurrent increase in cell capacitance, as with chronic MI in rats,15 infers cell size increase; confirmed here and in studies of chronic VTP24 or AF.46 It is unclear whether t-tubule loss contributes to the development of AF in HF, although t-tubule disorganization may promote atrial Ca2+i alternans,18 with the potential to promote arrhythmia. The consequences of t-tubule loss for Ca2+i cycling have been studied by de-tubulating rat ventricular myocytes using formamide, which decreased the Ca2+i transient and slowed its decay, and de-synchronized SR Ca2+ release.48 MI did not affect the Ca2+i transient or ICaL in the absence of β-stimulation in the present, atrial, cells. No previous studies of rabbit atrial t-tubules and their reduction by myocardial disease could be found. T-tubules contain many of the elements of the β-adrenergic pathway, and the effects of de-tubulation in rat were prevented by β-stimulation, by re-synchronizing Ca2+ release.48 β-Stimulation inhibited atrial alternans,16,20 by facilitating sequestration of Ca2+ from the cytosol.20 The present reduction in APD alternans by β-stimulation (in the paced hearts following MI) could involve a similar mechanism. Mathematical modelling suggests that t-tubule loss promotes Ca2+i alternans by increasing intracellular spatial heterogeneity of Ca2+ diffusion, and steepening the relationship between SR [Ca2+] and cytoplasmic [Ca2+].49 Chronic MI caused frequent failure of AP propagation into subcellular regions which underwent t-tubule remodelling, producing local ‘SDs’.50 Increased SR Ca2+ leak and spontaneous Ca2+i waves also promote SDs.51,52
In conclusion, chronic MI in rabbits remodels atrial structure, electrophysiology and Ca2+i handling. Increased susceptibility to AF by MI, under β-stimulation, may result from associated production of atrial APD alternans and SDs, since steady-state APD and global CV were unchanged under these conditions, and may be unrelated to the associated reduction in whole-cell ICaL. Further studies in this model are warranted, to clarify potential contributions of local conduction changes, and cellular and subcellular mechanisms of alternans, to the increased susceptibility to AF.
Funding
This study was funded by British Heart Foundation (grant no. PG/09/028/27149).
Acknowledgements
Authors thank Aileen Rankin, Michael Dunne, and Karen McGlynn, for excellent technical assistance.
Conflicts of interest: none declared.
References
- 1.Nishida K, Michael G, Dobrev D, Nattel S. Animal models for atrial fibrillation: clinical insights and scientific opportunities. Europace. 2010;12:160–172. doi: 10.1093/europace/eup328. doi:10.1093/europace/eup328. [DOI] [PubMed] [Google Scholar]
- 2.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. doi:10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- 3.Workman AJ, Kane KA, Rankin AC. Cellular bases for human atrial fibrillation. Heart Rhythm. 2008;5:S1–S6. doi: 10.1016/j.hrthm.2008.01.016. doi:10.1016/j.hrthm.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stambler BS, Fenelon G, Shepard RK, Clemo HF, Guiraudon CM. Characterization of sustained atrial tachycardia in dogs with rapid ventricular pacing-induced heart failure. J Cardiovasc Electrophysiol. 2003;14:499–507. doi: 10.1046/j.1540-8167.2003.02519.x. doi:10.1046/j.1540-8167.2003.02519.x. [DOI] [PubMed] [Google Scholar]
- 5.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, et al. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythmia Electrophysiol. 2008;1:93–102. doi: 10.1161/CIRCEP.107.754788. doi:10.1161/CIRCEP.107.754788. [DOI] [PubMed] [Google Scholar]
- 6.Li D, Melnyk P, Feng J, Wang Z, Petrecca K, Shrier A, et al. Effects of experimental heart failure on atrial cellular and ionic electrophysiology. Circulation. 2000;101:2631–2638. doi: 10.1161/01.cir.101.22.2631. doi:10.1161/01.CIR.101.22.2631. [DOI] [PubMed] [Google Scholar]
- 7.Sridhar A, Nishijima Y, Terentyev D, Khan M, Terentyeva R, Hamlin RL, et al. Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc Res. 2009;84:227–236. doi: 10.1093/cvr/cvp216. doi:10.1093/cvr/cvp216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Briston SJ, Caldwell JL, Horn MA, Clarke JD, Richards MA, Greensmith DJ, et al. Impaired β-adrenergic responsiveness accentuates dysfunctional excitation-contraction coupling in an ovine model of tachypacing-induced heart failure. J Physiol. 2011;589:1367–1382. doi: 10.1113/jphysiol.2010.203984. doi:10.1113/jphysiol.2010.203984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardin S, Guasch E, Luo X, Naud P, Le Quang K, Shi Y, et al. Role for microRNA-21 in atrial profibrillatory fibrotic remodeling associated with experimental postinfarction heart failure. Circ Arrhythm Electrophysiol. 2012;5:1027–1035. doi: 10.1161/CIRCEP.112.973214. doi:10.1161/CIRCEP.112.973214. [DOI] [PubMed] [Google Scholar]
- 10.Miyauchi Y, Zhou S, Okuyama Y, Miyauchi M, Hayashi H, Hamabe A, et al. Altered atrial electrical restitution and heterogeneous sympathetic hyperinnervation in hearts with chronic left ventricular myocardial infarction. Implications for atrial fibrillation. Circulation. 2003;108:360–366. doi: 10.1161/01.CIR.0000080327.32573.7C. doi:10.1161/01.CIR.0000080327.32573.7C. [DOI] [PubMed] [Google Scholar]
- 11.McIntosh MA, Cobbe SM, Smith GL. Heterogeneous changes in action potential and intracellular Ca2+ in left ventricular myocyte sub-types from rabbits with heart failure. Cardiovasc Res. 2000;45:397–409. doi: 10.1016/s0008-6363(99)00360-0. doi:10.1016/S0008-6363(99)00360-0. [DOI] [PubMed] [Google Scholar]
- 12.Ng GA, Cobbe SM, Smith GL. Non-uniform prolongation of intracellular Ca2+ transients recorded from the epicardial surface of isolated hearts from rabbits with heart failure. Cardiovasc Res. 1998;37:489–502. doi: 10.1016/s0008-6363(97)00255-1. doi:10.1016/S0008-6363(97)00255-1. [DOI] [PubMed] [Google Scholar]
- 13.Bolger AP, Sharma R, Li W, Leenarts M, Kalra PR, Kemp M, et al. Neurohormonal activation and the chronic heart failure syndrome in adults with congenital heart disease. Circulation. 2002;106:92–99. doi: 10.1161/01.cir.0000020009.30736.3f. doi:10.1161/01.CIR.0000020009.30736.3F. [DOI] [PubMed] [Google Scholar]
- 14.Workman AJ. Cardiac adrenergic control and atrial fibrillation. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:235–249. doi: 10.1007/s00210-009-0474-0. doi:10.1007/s00210-009-0474-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boixel C, Gonzalez W, Louedec L, Hatem SN. Mechanisms of L-type Ca2+ current downregulation in rat atrial myocytes during heart failure. Circ Res. 2001;89:607–613. doi: 10.1161/hh1901.096702. doi:10.1161/hh1901.096702. [DOI] [PubMed] [Google Scholar]
- 16.Florea SM, Blatter LA. Regulation of cardiac alternans by β-adrenergic signaling pathways. Am J Physiol. 2012;303:H1047–H1056. doi: 10.1152/ajpheart.00384.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narayan SM, Franz MR, Clopton P, Pruvot EJ, Krummen DE. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation. 2011;123:2922–2930. doi: 10.1161/CIRCULATIONAHA.110.977827. doi:10.1161/CIRCULATIONAHA.110.977827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blatter LA, Kockskamper J, Sheehan KA, Zima AV, Huser J, Lipsius SL. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol. 2003;546:19–31. doi: 10.1113/jphysiol.2002.025239. doi:10.1113/jphysiol.2002.025239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng GA, Brack KE, Patel VH, Coote JH. Autonomic modulation of electrical restitution, alternans and ventricular fibrillation initiation in the isolated heart. Cardiovasc Res. 2007;73:750–760. doi: 10.1016/j.cardiores.2006.12.001. doi:10.1016/j.cardiores.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Shkryl VM, Maxwell JT, Domeier TL, Blatter LA. Refractoriness of sarcoplasmic reticulum Ca2+ release determines Ca2+ alternans in atrial myocytes. Am J Physiol. 2012;302:H2310–H2320. doi: 10.1152/ajpheart.00079.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs. Atrial remodeling of a different sort. Circulation. 1999;100:87–95. doi: 10.1161/01.cir.100.1.87. doi:10.1161/01.CIR.100.1.87. [DOI] [PubMed] [Google Scholar]
- 22.Workman AJ, Smith GL, Rankin AC. Mechanisms of termination and prevention of atrial fibrillation by drug therapy. Pharmacol Ther. 2011;131:221–241. doi: 10.1016/j.pharmthera.2011.02.002. doi:10.1016/j.pharmthera.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19. doi: 10.1093/cvr/cvn133. doi:10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dibb KM, Clarke JD, Horn MA, Richards MA, Graham HK, Eisner DA, et al. Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ Heart Fail. 2009;2:482–489. doi: 10.1161/CIRCHEARTFAILURE.109.852228. doi:10.1161/CIRCHEARTFAILURE.109.852228. [DOI] [PubMed] [Google Scholar]
- 25.Workman AJ, Kane KA, Rankin AC. Rate-dependency of action potential duration and refractoriness in isolated myocytes from the rabbit AV node and atrium. J Mol Cell Cardiol. 2000;32:1525–1537. doi: 10.1006/jmcc.2000.1186. doi:10.1006/jmcc.2000.1186. [DOI] [PubMed] [Google Scholar]
- 26.Quinn FR, Currie S, Duncan AM, Miller S, Sayeed R, Cobbe SM, et al. Myocardial infarction causes increased expression but decreased activity of the myocardial Na+-Ca2+ exchanger in the rabbit. J Physiol. 2003;553:229–242. doi: 10.1113/jphysiol.2003.050716. doi:10.1113/jphysiol.2003.050716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seidler T, Miller SLW, Loughrey CM, Kania A, Burow A, Kettlewell S, et al. Effects of adenovirus-mediated sorcin overexpression on excitation-contraction coupling in isolated rabbit cardiomyocytes. Circ Res. 2003;93:132–139. doi: 10.1161/01.RES.0000081596.90205.E2. doi:10.1161/01.RES.0000081596.90205.E2. [DOI] [PubMed] [Google Scholar]
- 28.Boixel C, Fontaine V, Rucker-Martin C, Milliez P, Louedec L, Michel JB, et al. Fibrosis of the left atria during progression of heart failure is associated with increased matrix metalloproteinases in the rat. J Am Coll Cardiol. 2003;42:336–344. doi: 10.1016/s0735-1097(03)00578-3. doi:10.1016/S0735-1097(03)00578-3. [DOI] [PubMed] [Google Scholar]
- 29.Shimano M, Tsuji Y, Inden Y, Kitamura K, Uchikawa T, Harata S, et al. Pioglitazone, a peroxisome proliferator-activated receptor-gamma activator, attenuates atrial fibrosis and atrial fibrillation promotion in rabbits with congestive heart failure. Heart Rhythm. 2008;5:451–459. doi: 10.1016/j.hrthm.2007.12.010. doi:10.1016/j.hrthm.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 30.Zhang P, Su J, Mende U. Cross talk between cardiac myocytes and fibroblasts: from multiscale investigative approaches to mechanisms and functional consequences. Am J Physiol. 2012;303:H1385–H1396. doi: 10.1152/ajpheart.01167.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burstein B, Comtois P, Michael G, Nishida K, Villeneuve L, Yeh YH, et al. Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res. 2009;105:1213–1222. doi: 10.1161/CIRCRESAHA.108.183400. doi:10.1161/CIRCRESAHA.108.183400. [DOI] [PubMed] [Google Scholar]
- 32.Numata A, Miyauchi Y, Ono N, Fishbein MC, Mandel WJ, Lin SF, et al. Spontaneous atrial fibrillation initiated by tyramine in canine atria with increased sympathetic nerve sprouting. J Cardiovasc Electrophysiol. 2012;23:415–422. doi: 10.1111/j.1540-8167.2011.02197.x. doi:10.1111/j.1540-8167.2011.02197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frommeyer G, Schmidt M, Clauss C, Kaese S, Stypmann J, Pott C, et al. Further insights into the underlying electrophysiological mechanisms for reduction of atrial fibrillation by ranolazine in an experimental model of chronic heart failure. Eur J Heart Fail. 2012;14:1322–1331. doi: 10.1093/eurjhf/hfs163. doi:10.1093/eurjhf/hfs163. [DOI] [PubMed] [Google Scholar]
- 34.Birner C, Husser O, Jeron A, Rihm M, Fredersdorf S, Resch M, et al. Differential expression of potassium channels and abnormal conduction in experimental tachycardia-induced heart failure. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:473–480. doi: 10.1007/s00210-011-0723-x. doi:10.1007/s00210-011-0723-x. [DOI] [PubMed] [Google Scholar]
- 35.Sah R, Ramirez RJ, Oudit GY, Gidrewicz D, Trivieri MG, Zobel C, et al. Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current (Ito) J Physiol. 2003;546:5–18. doi: 10.1113/jphysiol.2002.026468. doi:10.1113/jphysiol.2002.026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Workman AJ, Pau D, Redpath CJ, Marshall GE, Russell JA, Norrie J, et al. Atrial cellular electrophysiological changes in patients with ventricular dysfunction may predispose to AF. Heart Rhythm. 2009;6:445–451. doi: 10.1016/j.hrthm.2008.12.028. doi:10.1016/j.hrthm.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Workman AJ, Marshall GE, Rankin AC, Smith GL, Dempster J. Transient outward K+ current reduction prolongs action potentials and promotes afterdepolarisations: a dynamic-clamp study in human and rabbit cardiac atrial myocytes. J Physiol. 2012;17:4289–4305. doi: 10.1113/jphysiol.2012.235986. doi:10.1113/jphysiol.2012.235986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roth DA, Urasawa K, Helmer GA, Hammond HK. Downregulation of cardiac guanosine 5′-triphosphate-binding proteins in right atrium and left ventricle in pacing-induced congestive heart failure. J Clin Invest. 1993;91:939–949. doi: 10.1172/JCI116315. doi:10.1172/JCI116315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinfath M, Lavicky J, Schmitz W, Scholz H, Doring V, Kalmar P. Regional distribution of β1- and β2-adrenoceptors in the failing and nonfailing human heart. Eur J Clin Pharmacol. 1992;42:607–611. doi: 10.1007/BF00265923. [DOI] [PubMed] [Google Scholar]
- 40.Legssyer A, Hove-Madsen L, Hoerter J, Fischmeister R. Sympathetic modulation of the effect of nifedipine on myocardial contraction and Ca current in the rat. J Mol Cell Cardiol. 1997;29:579–591. doi: 10.1006/jmcc.1996.0301. doi:10.1006/jmcc.1996.0301. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Diaz ME, Eisner DA, O'Neill S. The effects of membrane potential, SR Ca2+ content and RyR responsiveness on systolic Ca2+ alternans in rat ventricular myocytes. J Physiol. 2009;587:1283–1292. doi: 10.1113/jphysiol.2008.164368. doi:10.1113/jphysiol.2008.164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Llach A, Molina CE, Fernandes J, Padro J, Cinca J, Hove-Madsen L. Sarcoplasmic reticulum and L-type Ca2+ channel activity regulate the beat-to-beat stability of calcium handling in human atrial myocytes. J Physiol. 2011;589:3247–3262. doi: 10.1113/jphysiol.2010.197715. doi:10.1113/jphysiol.2010.197715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wan X, Laurita KR, Pruvot EJ, Rosenbaum DS. Molecular correlates of repolarization alternans in cardiac myocytes. J Mol Cell Cardiol. 2005;39:419–428. doi: 10.1016/j.yjmcc.2005.06.004. doi:10.1016/j.yjmcc.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 44.Kockskamper J, Blatter LA. Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J Physiol. 2002;545:65–79. doi: 10.1113/jphysiol.2002.025502. doi:10.1113/jphysiol.2002.025502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe I, Okumura Y, Nagashima K, Ohkubo K, Ashino S, Kofune M, et al. Electrical remodeling in fibrillating canine atrium: action potential alternans during rapid atrial pacing and late phase 3 early afterdepolarization after cessation of rapid atrial pacing. Int Heart J. 2010;51:354–358. doi: 10.1536/ihj.51.354. doi:10.1536/ihj.51.354. [DOI] [PubMed] [Google Scholar]
- 46.Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D'hooge J, et al. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–885. doi: 10.1161/CIRCRESAHA.109.206276. doi:10.1161/CIRCRESAHA.109.206276. [DOI] [PubMed] [Google Scholar]
- 47.Wakili R, Yeh YH, Qi XY, Greiser M, Chartier D, Nishida K, et al. Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ Arrhythm Electrophysiol. 2010;3:530–541. doi: 10.1161/CIRCEP.109.933036. doi:10.1161/CIRCEP.109.933036. [DOI] [PubMed] [Google Scholar]
- 48.Brette F, Rodriguez P, Komukai K, Colyer J, Orchard CH. β-adrenergic stimulation restores the Ca transient of ventricular myocytes lacking t-tubules. J Mol Cell Cardiol. 2004;36:265–275. doi: 10.1016/j.yjmcc.2003.11.002. doi:10.1016/j.yjmcc.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 49.Li Q, O'Neill SC, Tao T, Li Y, Eisner D, Zhang H. Mechanisms by which cytoplasmic calcium wave propagation and alternans are generated in cardiac atrial myocytes lacking T-tubules-insights from a simulation study. Biophys J. 2012;102:1471–1482. doi: 10.1016/j.bpj.2012.03.007. doi:10.1016/j.bpj.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sacconi L, Ferrantini C, Lotti J, Coppini R, Yan P, Loew LM, et al. Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc Natl Acad Sci USA. 2012;109:5815–5819. doi: 10.1073/pnas.1120188109. doi:10.1073/pnas.1120188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, et al. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. doi:10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- 52.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306. doi:10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]