

Introduction
We attempted to generate cyclooxygenase-1 and cyclooxygenase-2 deficient (COX-1−/−COX-2−/−) mice on a C57BL/6J background by crossing male COX-1+/−COX-2−/− with female COX-1+/−COX-2+/− and female COX-1−/−COX-2+/− mice. This approach did not yield viable COX-1−/−COX-2−/− mice but we noted that many of the COX-1+/− COX-2−/− mice died by 2–3 months of age. Live COX-1+/−COX-2−/− mice submitted for necropsy were found to have thrombotic events including clots in the heart, liver and lung. We sought to define the mechanism of the thrombotic events associated with this genotype. Cyclooxygenase products have been closely tied to coagulation. Thromboxane A2 (TxA2) promotes thrombosis through platelet aggregation and vasoconstriction [1, 2]. In contrast, prostacyclin, (PGI2) inhibits thrombosis through vasodilation and inhibition of platelet aggregation [3,4,5]. Selective COX-2 inhibitors and NSAIDs, which inhibit COX-1 and COX-2, are associated with an increased incidence of myocardial infarction and other thrombotic events [6, 7]. Meta-analyses show an increased risk of cardiovascular events with one selective COX-2 inhibitor, rofecoxib, and perhaps with another, celecoxib (8, 9). There appears to be an increased risk of cardiovascular events with some NSAIDs including diclofenac but not with naprosyn (10 ). The mechanism of the increased incidence of cardiovascular events is not clear but appears to relate to decreased production of PGI2 (11).
Myocardial infarctions and other thrombotic events can be the result of either enhanced coagulation or impaired clot resolution. The contribution of COX-1 and COX-2 products to thrombosis is complex. TxA2, which promotes platelet aggregation, is produced in platelets through COX-1 [12]. PGI2, which is produced in blood vessels in both endothelial cells and vascular smooth muscle cells, is a product of prostacyclin synthase (PGI synthase) which can be downstream of either COX-1 or COX-2 [13]. At baseline COX-2 expression in vascular tissue is low and most vascular PGI2 is produced through COX-1 [14, 15]. Some investigators have been unable to identify COX-2 in vascular tissue in the absence of injury (16); however, others find that in response to shear forces associated with blood flow COX-2 is induced in vascular tissues and more vascular PGI2 is produced through COX-2 [16, 17]. These findings led to the suggestion that the increase in myocardial infarctions and other thrombotic events seen in patients taking selective COX-2 inhibitors is the product of the inhibition of vascular PGI2 in the absence of any inhibition of platelet thromboxane production [11].
Although the role of cyclooxygenase products in coagulation has been extensively investigated, their role in clot resolution or fibrinolysis is largely unexplored. Incubation of stable PGI2 analogs with vascular smooth muscle cells in culture results in a decreased expression of plasminogen activator inhibitor 1 (PAI-1), a protein that decreases fibrinolysis by inhibiting both tissue-type plasminogen activator and urokinase-type plasminogen activator and thus plasmin formation [18, 19]. Treatment of diabetic patients with iloprost, a stable PGI2 analog, results in decreased plasma PAI-1 activity [20]. Mice deficient in PAI-1 have increased fibrinolysis and a prolonged time to thrombosis in a carotid artery injury model [21]. There is experimental and epidemiological evidence that PAI-1 contributes to the development of cardiovascular disease [22,23]. PAI-1 excess causes spontaneous coronary artery occlusion in mice (24). ApoE−/− mice fed a high fat diet develop severe hypercholesterolemia and have elevated plasma PAI-1 levels (25). These mice exhibit a prothrombotic phenotype with shortened times to thrombotic occlusion in carotid artery injury models. The deletion of the PAI-1 gene in these mice reverses the prothrombotic tendency. Vitronectin (VN), an abundant plasma and matrix glycoprotein, binds PAI-1 and regulates its activity by stabilizing the active PAI-1 conformation [21]. Deletion of either PAI-1 or VN in wild type mice delays thrombotic occlusion following arterial injury [21]. Complete deficiency of PAI-1 in man results in a mild bleeding disorder (26).
In this study we explored the relationship between PAI-1 and cyclooxygenase genotypes using euglobulin lysis times and a carotid artery occlusion model in the mouse. We found that the COX-1+/− COX-2−/− genotype was associated with increased plasma and aortic levels of PAI-1 and with both a prolonged euglobulin lysis time and a shortened time to occlusion in the carotid artery injury model. Moreover, we found that the shortened time to occlusion was normalized by a neutralizing antibody to PAI-1.
Material and Methods
Animals
Except where noted, mice are on a C57BL/6J background. Knockout mice on a C57BL/6J background were COX-1−/−COX-2+/+, COX-1+/−COX-2+/+, COX-1−/−COX-2+/−, COX-1+/+COX-2−/−, and COX-1+/−COX-2−/− where the COX-2 gene was disrupted in exon 8 [278]. COX-1+/+COX-2+/− mice on the B6;129 background were a gift from Garret FitzGerald, University of Pennsylvania. In these mice the COX-2 gene is disrupted in exon 1 [289]. We bred them to produce wild type and COX-2−/− mice. Mice were maintained on a 12 hour light/dark cycle in a pathogen-free barrier facility and fed standard laboratory chow ad libitum. All experiments using mice were approved by the Washington University School of Medicine Animal Studies Committee.
Clot Lysis Assay
A euglobalin clot lysis assay for global fibrinolysis was performed according to a protocol modified from Smith et al (31). See Supplementary Data for detailed protocol.
Arterial Thrombosis Model
Carotid artery thrombosis was induced as described previously [29]. Male and female wild type and knockout C57BL/6J and B6;129 mice (6–9 weeks of age; 16–19 g body weight) were anesthetized with intraperitoneal sodium pentobarbital, secured in a supine position, and placed under a dissecting microscope. The right carotid artery was isolated through a midline cervical incision, and an ultrasonic flow probe (Model 0.5 VB; Transonic Systems, Ithaca, NY) was applied. A 1.5 mW, 540 nm laser beam (Melles Griot, Carlsbad, CA) was applied to the artery from a distance of 6 cm. Rose Bengal dye (3′, 4′, 5′, 6′ – tetrachloro-2, 4, 5, 7-tetraiodofluorescein; (Fisher Scientific, Fair Lawn, NJ) at a dose of 50 mg/kg body weight was then injected into the lateral tail vein, and blood flow was monitored continuously. The occlusion time was taken as the interval between the injection of Rose Bengal dye and complete and stable (>5 minutes) cessation of flow.
In a subset of experiments, mice were given a single intraperitoneal injection of NS-398 (Biomol, Plymouth Meeting, PA) at a dose of 1 mg/kg body weight 2 hours before injecting Rose Bengal. NS-398 was reconstituted in DMSO as a concentrated stock solution at 25 mg/ml. The working solution was made by diluting an aliquot 125 fold (8 μl/ml) in 5% sodium bicarbonate. Control mice received vehicle. In another subset of experiments, mice were given a single tail vein injection of a mouse monoclonal inhibitory antibody, H4B3, against mouse PAI-1 (Molecular Innovations, Novi, MI) (30) at a dose of 0.5 mg/kg body weight 15 minutes before injecting Rose Bengal.
qRT-PCR
Total RNA was extracted from aorta tissues by homogenization in RiboZol RNA extraction reagent (Amresco, Solon, OH). qRT-PCR was used to measure gene expression, with actin expression as a control. The final results were expressed as fold differences in gene expression relative to the actin gene. The Ct for each amplicon was determined as the PCR cycle at which the fluorescence intensity crossed a user-established threshold. The following primers were synthesized by Integrated DNA Technologies (Coralville, IA): COX-1 (5′-AGGAGATGGCTGCTGAGTTGG and 5′-AATCTGACTTTCTGAGTTGCC), COX2 (5′-ACACACTCTATCACTGGCACC and 5′-TTCAGGGCGAAGCGTTTGC), PAI-1 (5′-TCAGAGCAACAAGTTCAACTACACTGAG and 5′CCCACTGTCAAGGCTCCATCACTTGCCCCA ), vitronectin (5′-CATCACGTTCAATCTCGTTCTCTTT and 5′-TTGGGCTGGGAGAAAGAGATGAGG) and actin (5′-CAACGAGCGGTTCGGATG and 5′-GCCACAGGATTCCATACC). Commercially available primers for PGI synthase (PPM-05311A) were purchased from SA Biosciences (Frederick MD).
SDS-PAGE and Western blot analysis
Aortas from C57BL/6J wild type, COX-1+/+COX2−/−, and COX1+/−COX2−/− mice were homogenized in ice cold proteinase inhibitor cocktail containing antipain (25 μg/ml), aprotinin (25 μg/ml), leupeptin (25 μg/ml), chymostatin (25 μg/ml), phenanthroline (50 μmol/liter), phenylmethylsufonyl fluoride (100 μmol/liter), pepstatin A (10 μg/ml), and dithiothreitol (2 nmol/liter) in N-Tris (hydroxymethyl)-methyl-2-aminomethane sulfonic acid (20 mmol/liter), pH 7.4. Equal amounts (40 μg) of protein from all mice were separated by SDS-PAGE on a 10% polyacrylamide gel and transferred to an Immobilon membrane (Millipore, Bedford, MA). Antibodies used for detecting proteins were goat anti-mouse COX-1 (SC-1754, 1:200, Santa Cruz Biothecnology, Santa Cruz, CA), mouse monoclonal anti COX-2 (610204, 1:500, BD Biosciences, San Jose, CA), rabbit anti-human PGI synthase (Santa Cruz SC-20933, 1:200), sheep anti-human PAI-1 (SAPAI-IG, 1:500, Affinity Biologicals, Ontario, Canada). Visualizing protein bands was with HRP-linked secondary antibodies and ECL reagent (GE Healthcare Amersham, Piscataway, NJ) with fluorographic detection on Blue-sensitive autoradiography film (Mid Sci, St. Louis, MO). Determination of protein loading was by reprobing blots with goat anti-human actin (Santa Cruz SC-1616, 1:200).
Plasma PAI-1
Blood was obtained from mice by puncturing the right mandibular vein with a 5.5 mm animal lancet and collecting the blood directly into plasma separator tubes kept on ice and centrifuged at 10,000 xg for 5 min within 10 minutes of collection. Plasma was transferred to cryovials and snap frozen in liquid nitrogen and stored at −80° C until the time of assay. ELISA kits from Molecular Innovations (Novi, MI) were used to measure plasma levels of active PAI-1 (MPAI KT) and total PAI-1 (MPAIKT-TOT) in wild type, COX-1+/+COX-2−/−, and COX-1+/−COX-2−/−mice.
Tail vein bleeding time
Tail vein bleeding time was determined as described by Broze et al [32]. Mice were anesthetized (ketamine 75mg/kg; medetomidine 1 mg/kg intraperitoneally) and placed prone on a warming pad. A transverse incision was made with a #11 scalpel blade over a lateral vein at a position where the diameter of tail was 2.25 to 2.5 mm. The tail was immersed in isotonic normal saline (37° C) in a hand-held test tube. The time from incision to the cessation of bleeding was recorded as the bleeding time.
Analysis of urine for metabolites of PGI2 and TxA2
Urine was collected from mice by lining the bottoms of clean mouse cages with clean sheets of plastic wrap and placing a single mouse in each cage [33]. Once a mouse urinated, the voided urine was transferred to a micro centrifuge tube and snap frozen in liquid nitrogen. Urine samples were stored in −80°C. Urine collected from wild type and knockout C57BL/6J mice was assayed for the prostacyclin metabolite 2,3-dinor-6-keto PGF1α and the thromboxane A2 metabolite 11-dehydro-thromboxane B2 by ELISA (Cayman Chemical, Ann Arbor, MI). Urinary metabolites were normalized to urine creatinine. Urinary creatininewas measured by ELISA (Cayman Chemical, Ann Arbor, MI).
Results
Breeding Schemes to generate COX genotypes
COX-1−/− (gene disrupted in exon 11) and COX-2−/− (gene disrupted in exon 8) mice on a mixed B6;129 genetic background were backcrossed 10 generations to wild type mice on the C57BL/6J genetic background to yield COX-1−/− and COX-2−/− mice on a C57BL/6J genetic background. These mice were bred as in Suppl Table 1. Because COX-1−/− females have delayed parturition, COX-1−/− mice were generated from COX-1+/− females. The percentage of COX-1−/− mice generated was 33% from COX-1+/− × COX-1+/− matings and 45% from male COX-1−/− × female COX-1+/− matings. For generating COX-2−/− mice, females expressing one allele of COX-2 were used, because female COX-2−/− mice are infertile (34). The percentage of COX-2−/− mice generated was less than Mendelian expectations with 6% from COX-2+/− × COX-2+/− matings and 23% from male COX-2−/− × female COX-2+/− matings. Breeding mice for the purpose of generating COX-1+/−COX-2+/−, COX-1−/−COX-2+/−, and COX-1+/−COX-2−/− mice utilized female COX-1+/−COX-2+/− breeders. In contrast to female COX-1+/−COX-2+/− mice on the mixed B6;129 genetic background which have delayed parturition (34), female COX-2+/−COX-2+/− mice on the C57B6/6J genetic background have a normal gestation period and consistently deliver live pups. Male COX-1+/+COX-2−/− × female COX-1+/−COX-2+/− matings were used to produce COX-1+/−COX-2−/− mice. Male COX-1+/−COX-2−/− × female COX-1+/−COX-2+/− matings were also used and these also generated COX-1−/−COX-2−/− mice, which died within 24 hours from patent ductus arteriosus (PDA) (35). Male COX-1−/−COX-2+/+ × female COX-1+/−COX-2+/− matings were used to generate COX-1−/−COX-2+/−mice.
COX-1+/−COX-2−/− genotype is associated with premature death and spontaneous thrombosis
COX-1+/−COX-2−/− mice died prematurely with 50% survival at 3 months and 10% survival at one year (Figure 1A). All the other genotypes generated (COX-1−/−, COX-2−/−, COX-1+/−COX-2+/−, COX-1−/−COX-2+/−) had survival curves similar to wild type mice. Neonate survival for the various COX-1 and COX-2 knockout genotypes on the C57BL/6J genetic background was greater than in mice on the mixed B6;129 genetic background (35). The occurrence of PDA on the B6;129 background has been reported to be 35% in COX-2−/−, 79% in COX-1+/−COX-2−/−, and 100% in COX-1−/−COX-2−/− mice (35). In our mice on the C57BL/6J genetic background there was still 100% occurrence of deaths within 24 hours of birth in COX-1−/−COX-2−/− apparently due to PDA, but there was no indication from neonate survival of PDA in the COX-2−/− or in COX-1+/−COX-2−/− mice on the C57BL/6J genetic background.
Fig. 1.

The COX-1+/−COX-2−/− genotype is associated with premature death and spontaneous thrombosis. (A) Survival curve for COX-1+/−COX-2−/− mice on the C57BL/6J genetic background. The dates of birth and death of 140 COX-1+/−COX-2−/− mice were recorded over a 3 year period. The survival curve represents only those mice allowed to live to a natural death, and does not include an additional 47 COX-1+/−COX-2−/− mice removed for experiments and necropsies. (B–E) Photomicrographs of thromboemboli (arrows) in (B) superior vena cava, (C) traversing tricuspid valve from right atrium into right ventricle, (D) pulmonary artery, and (E) liver, with line showing demarcation between normal tissue and infarction. Original magnification B, C, E 40x, D 100x. Abbreviations: av, azygos vein; br, bronchus; tc, tricuspid valve; N, normal tissue; I, infarct.
At about 1–2 months of age the COX-1+/−COX-2−/− mice on the C57BL/6J genetic background began to die. Some died suddenly whereas others first manifested signs of disease including decreased activity and hunched posture. Some of these animals were sacrificed and necropsied. The necropsies demonstrated spontaneous thromboses in a variety of anatomic sites including superior vena cava (Fig 1B), tricuspid valve (Fig 1C), pulmonary artery (Fig 1D), and central vein in the liver (Fig 1E). The liver thrombosis is associated with an area of hepatic infarction. The liver and pulmonary artery thromboses have considerable fibrosis suggesting that they are at least several days old.
COX-1+/− COX-2−/− genotype is associated with a shortened time to occlusion in the Rose Bengal carotid artery injury model
To address the mechanism of the thrombotic deaths in the COX-1+/−COX-2−/− mice we assessed the time to occlusion in a carotid artery injury model. Wild type C57BL/6J mice had a 63 minute time to occlusion in the carotid injury model (Fig 2A). COX-1−/− COX-2+/+, COX-1−/− COX-2+/−, and COX-1+/− COX-2+/+ all had times to occlusion similar to that seen with wild type mice. COX-1+/+ COX-2−/− mice had a markedly prolonged time to occlusion at 100 minutes. COX-1+/− COX-2−/− mice had a markedly shortened carotid occlusion time at 30 minutes.
Fig. 2. COX-1+/−COX-2−/− genotype is associated with a shortened time to occlusion in the Rose Bengal carotid artery injury model.
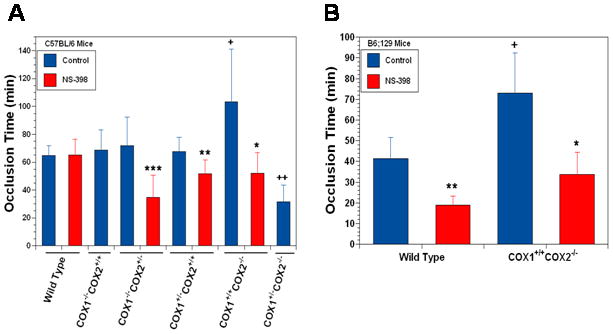
A. C57BL/6J mice. Data are means ± SD of 8–17 mice per group. +p<.01, ++p<.001 compared to wild type control mice, *p<.01, **p<.001, ***p<.001 compared to control mice of the same genotype. B. B6;129 mice. Data are the means ± SD of 5–8 mice per group. +p<.001 compared to wild type, *p<.002, **p<.001 compared to control mice of the same genotype.
The administration of a selective but not specific COX-2 inhibitor, NS-398, to wild type C57BL/6J mice had no effect on the time to occlusion in the carotid injury model. In contrast administration of NS-398 to either the COX-1−/− COX-2+/−, COX-1+/− COX-2+/+, or COX-1+/+ COX2−/− mice resulted in a shortening of the time to occlusion.
Our studies on the carotid injury model used mice on the C57BL/6J genetic background. Other investigators had used mice on a mixed B6;129 background to study the effects of cyclooxygenase products on thrombosis [36]. We therefore repeated some of the carotid artery injury studies using B6;129 mice. Wild type mice on the B6;129 genetic background had a shorter time to occlusion, 41 minutes, than did the wild type on the C57BL/6J genetic background, 63 minutes (Fig 2B and Fig 2A). In contrast to the C57BL/6J mice, the administration of NS-398 to wild type B6;129 mice resulted in a marked diminution in the time to occlusion from 41 minutes to 20 minutes. In COX-1+/+ COX-2−/− mice on the B6;129 genetic background the time to occlusion was markedly prolonged just as it was in the COX-1+/+ COX-2−/− mice on the C57BL/6J genetic background. Similarly administration of NS-398 resulted in a marked shortening of the time of occlusion in the COX-1+/+ COX-2−/− mice on the B6;129 genetic background just as it had in the mice with the similar genotype on the C57BL/6J genetic background.
COX1+/−COX2−/− genotype is associated with decreased urinary prostacyclin metabolites
The cyclooxygenase products most frequently associated with thrombosis are TxA2, which promotes thrombosis through vasoconstriction and platelet aggregation, and PGI2 which impairs thrombosis through vasodilation and inhibition of platelet aggregation. Platelet TxA2 is produced only by COX-1 whereas TxA2 synthesized in other cell types can be the product of either COX-1 or COX-2. Vascular PGI2 can be produced by either COX-1 or COX-2. We next sought to correlate the COX genotypes with urinary levels of metabolites of TxA2 and PGI2 (Fig 3). COX-1−/−, COX-1−/−COX-2+/−, COX-2−/− and COX-1+/−COX-2−/− mice all had lower levels of 2,3-dinor -6-keto PGF1α, a metabolite of PGI2, when compared with wild type (Fig 3A). Although all four of these genotypes had diminished levels of the PGI2 metabolite, the lowest levels were in the COX-1+/−COX-2−/− mice. Urinary levels of the TxA2 metabolite, 11 dehydro thromboxane B2, were also diminished in the COX-1−/−, COX-1−/−COX-2+/−, and COX-2−/− but not in COX-1+/−COX-2−/− mice.(Fig 3B). Although the levels of the thromboxane metabolite were diminished in several genotypes, the lowest levels were in the mice completely deficient in COX-1. Other investigators demonstrated an association of thrombotic events with the ratio of PGI2 urinary metabolites to TxA2 urinary metabolites; the higher the ratio the less likely a thrombotic event [37, 38]. COX-1−/−, COX-1−/−COX-2+/− and COX-2−/− mice all had ratios of 0.6 to 0.7 compared to the ratio of 1.2 seen in wild type mice (Fig 3C). In contrast COX-1+/−COX-2−/−mice had a much lower ratio of 0.25. Thus the COX-1+/−COX-2−/− mice had a PGI2/TxA2 metabolite ratio associated with an increased risk of thrombosis.
Fig. 3. COX-1+/−COX-2−/− genotype is associated with decreased urinary levels of a metabolite of PGI2.

A. Urinary levels of 2,3-dinor-6-keto PGF1α, a PGI2 metabolite. B. Urinary levels of 11-dehydro-thromboxane B2, a TxA2 metabolite. C. Ratio of 2,3-dinor-6- keto PGF1α to 11-dehydro-thromboxane B2. Urinary metabolites were normalized to urine creatinine. Data are means ± SD of 8 mice per group. *p<.03 compared to wild type (WT), **p< .003 compared to wild type, ***p<.0001 compared to wild type.
COX-1+/− COX-2−/− genotype is associated with increased aortic levels of VN and PAI-1
One determinant of the time to thrombosis in the carotid artery injury model is the rate of fibrinolysis. PAI-1 blocks fibrinolysis by inhibiting plasminogen activator and VN stabilizes the active form of PAI-1 [18, 19]. Deletion of PAI-1 or VN in wild type mice delays thrombotic occlusion in arterial injury [21]. We next sought to determine if the shortened time to occlusion in the carotid artery seen with COX-1+/−COX-2−/− mice was associated with an increase in PAI-1. Total mRNA was isolated from the aortas of wild type, COX-1+/+ COX-2−/− and COX-1+/− COX-2−/− mice all on the C57BL/6J genetic background. Levels of VN and PAI-1 mRNA were markedly increased in the aortas of COX-1+/+ COX-2−/− mice and were further increased in the aortas of COX-1+/− COX-2−/− mice (Fig 4A). The levels of PGI synthase mRNA were reduced in the COX-1+/+ COX-2−/− mice and further reduced in the COX-1+/− COX-2−/− mice. We also extracted protein from the aortas of wild type, COX-1+/+ COX-2−/− and COX-1+/− COX-2−/− mice. Western blots for PGI synthase, and PAI-1, confirmed the results of the qRT-PCR data (Fig 4B). There was an eight fold increase in PAI-1 in the COX-1+/− COX-2−/− aortas compared to wild type. There was also a marked decrease in the expression of PGI synthase in the COX-1+/− COX-2−/− mouse aortas. COX-1 levels were diminished in the COX-1+/− COX-2−/− mice compared to wild type. As expected COX-2 was not detectable in either the COX-1+/− COX-2−/− and COX-1+/+ COX-2−/− mice compared to wild type.
Fig. 4. COX-1+/− COX-2−/− genotype is associated with increased aortic levels of PAI-1 and VN.
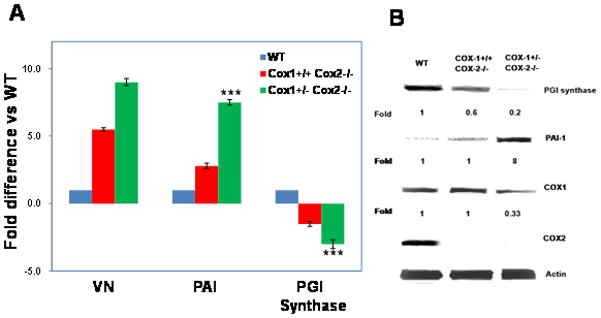
A. qRT-PCR using mRNA extracted from the aortas of C57BL/6J mice. n=3 mice per group *p<0.01; **p<0.004, ***p<0.001 compared to wild type (WT) mice. B. Western blots for PGI synthase, PAI-1, COX-1, COX-2 and actin using protein extracts of aorta from wild type, COX-1+/+ COX-2−/− and COX-1+/−COX-2−/− mice on a C57BL/6J genetic background. Numbers are fold change compared with the expression of the protein in wild type mice.
Plasma levels of PAI-1 are significantly higher in COX-1+/− COX-2−/− mice compared with wild type or COX-1+/+ COX-2−/− mice
Having found increased levels of PAI-1 in the aortas of COX-1+/−COX-2−/− mice we next sought to determine if there were increased plasma levels of PAI-1 in these mice. We measured both total PAI-1 and active PAI-1 levels in the plasma of wild type, COX-1+/+ COX-2−/− and COX-1+/− COX-2−/− mice. There was a fivefold increase in both total PAI-1 and active PAI-1 in the plasma of COX-1+/− COX-2−/− mice compared with the levels in the plasma of either wild type or COX-1+/+ COX-2−/− mice (Fig 5).
Fig. 5. Plasma levels of PAI-1 are higher in COX-1+/− COX-2−/− mice compared with wild type (WT) or COX-1+/+ COX-2−/− mice.

Total PAI-1 and active PAI-1 levels were measured in the plasma of C57BL/6J mice. Data are the means +/−SD of 12 mice for active PAI-1, and 20 mice for total PAI-1. *P<.01 compared with wild type mice active PAI-1; **P<.001 compared with wild typemice total PAI-1.
The shortened occlusion time seen in the COX-1+/− COX-2−/− mice was normalized by the administration of an inhibitory antibody to PAI-1
Wild type and COX-1+/− COX-2−/− mice were given an inhibitory antibody to PAI-1 prior to the administration of Rose Bengal in the carotid artery injury model. Administration of the antibody to PAI-1 had no effect on the occlusion time in the wild type mice but reversed the shortening of the time of occlusion seen in the COX-1+/− COX-2−/− mice (Fig 6).
Fig. 6. The shortened time to occlusion in COX-1+/− COX-2−/− mice is normalized by the administration of an inhibitory antibody to mouse PAI-1.

Wild type (WT) and COX-1+/− COX-2−/− mice were given a inhibitory antibody to mouse PAI-1 prior to the administration of Rose Bengal in the carotid artery injury model. The time to occlusion was measured. *P<.001 compared with wild typecontrol mice; **P<.001 compared with COX-1+/− COX-2−/− control mice. Data are means ± SD of 13 mice per group for wild type control, 6 for wild type + anti PAI-1, 13 for COX-1+/− COX-2−/− control, 9 for COX-1+/− COX-2−/− anti-PAI-1 mice.
COX-1+/−COX-2−/− genotype is associated with impaired fibrinolysis
We peformed euglobulin lysis times using platelet-poor plasma (Fig 7). The euglobulin lysis time in the COX-1+/−COX-2−/− mice was significantly prolonged in comparison with wild type mice. Addition of an inhibitory antibody to mouse PAI-1 to the wild type plasma had no effect on the euglobulin lysis time; whereas addition of an inhibitory antibody to mouse PAI-1 to COX-1+/−COX-2−/−plasma resulted in a trend to a shorter euglobulin lysis time although this trend did not reach statistical significance. Addition of exogenous mouse PAI-1 to the wild type serum resulted in a marked prolongation of the lysis time such that the optical density never fell below 0.50 (data not shown).
Fig. 7. The COX-1+/−COX-2−/− genotype is associated with a prolonged euglobulin lysis time.

(A) Graphic representation of average time courses for euglobulin clot lysis assays in plasma from wild type C57BL/6J and COX-1+/−COX-2−/− mice with and without the addition of an inhibitory mouse monoclonal antibody against mouse PAI-1. (B) The clot lysis time for COX-1+/−COX-2−/− mice is significantly longer than for wild type C57BL/6J mice. The lysis time in wild type mouse plasma is not affected by the addition of anti-PAI-1, but there is a trend toward shorter lysis times in anti-PAI-1 treated plasma from COX-1+/−COX-2−/− mice compared to untreated controls, although it does not reach statistical significance. Lysis times are measured from the time of peak absorbance to the time the absorbance decreases back to 0.30au, and the area under the curve for this time period is also presented. Two tailed t-test was used to compare lysis times and the area under the curve for different treatment groups. *p<.004 for COX-1+/−COX-2−/− control compared with wild type control. n=6 for all control plasma, and n=4 for all anti PAI-1 treated plasma.
Tail vein bleeding time in COX-1+/− COX-2−/− mice is the same as that in wild type mice
The tail vein bleeding time in the COX-1+/− COX-2−/− mice was the same as the wild type mice (Fig 8). The normal tail vein bleeding time in the COX-1+/−COX-2−/− mice suggests that there is no defect in platelet function. As expected the tail vein bleeding time in COX-1−/− COX-2+/+ mice was markedly prolonged.
Fig. 8. Tail vein bleeding time is normal in COX-1+/− COX-2−/− mice.

*P<.001 compared with wild type. Data are means ± SD of 10 mice per group.
Discussion
In attempting to explain the premature deaths and spontaneous thromboses we saw in COX-1+/−COX-2−/− mice we found that this genotype is associated with a shortened time to occlusion in the carotid artery injury model, with increased levels of aortic and plasma PAI-1 and with a prolonged euglobulin lysis time. Moreover the shortened time to occlusion seen in the COX-1+/−COX-2−/− genotype was normalized by an inhibitory antibody to mouse PAI-1. This suggests that the increased risk of thrombosis seen in this genotype is the result of decreased fibrinolysis mediated by an increase in PAI-1.
The association of elevated levels of PAI-1 with spontaneous thrombosis and a prolonged euglobulin lysis time is consistent with previous studies. Mice deficient in PAI-1 have increased fibrinolysis and a prolonged time to thrombosis in a carotid artery injury model (21) and PAI-1 excess is associated with spontaneous coronary artery occlusions. (24). The spontaneous thromboses seen in the PAI-1 overexpressing mice is consistent with the spontaneous thromboses seen in the COX-1+/−COX-2−/− mice. The COX-1+/− COX-2−/− genotype was also associated with an increase in VN which stabilizes the active form of PAI-1. There is some evidence for regulation of VN expression by cyclooxygenase products (40).
We attempted to determine if specific cyclooxygenase products were involved in mediating the decreased time of occlusion or the increased PAI-1 expression seen in COX-1+/− COX-2−/− mice by measuring urinary metabolites of PGI2 and TxA2. The analysis of the urinary metabolites demonstrated that both PGI2 and TxA2can be produced through either COX-1 or COX-2 although COX-1 is the major pathway for TxA2 production. The urinary metabolites studies also demonstrated that the COX-1+/− COX-2−/− genotype was associated with the lowest levels of PGI2 metabolites and the lowest ratio of PGI2 metabolites to TxA2 metabolites. There are several other lines of evidence that support the suggestion that decreased PGI2 production mediates the elevated PAI-1 levels and the shortened time to occlusion associated with the COX-1+/− COX-2−/− genotype. Stable analogs of PGI2 decrease PAI-1 expression in vascular smooth muscle cells (18, 19). Moreover, the administration of iloprost, a stable analog of PGI2, to diabetic patients resulted in diminished PAI-1 activity (20). Administration of PGI2 to patients undergoing hemodialysis resulted in effective anti-coagulation with laboratory evidence for increased fibrinolysis although PAI-1 levels were not measured (39). Therefore, a genotype associated with low levels of PGI2 production might be expected to be associated with increased PAI-1 and decreased fibrinolysis.
The COX-1+/− COX-2−/− genotype would be expected to result in decreased vascular PGI2 production because vascular PGI2 is produced through both COX-1 and COX-2. This is supported by the finding that, of all the genotypes tested, the COX-1+/− COX-2−/− genotype was associated with the lowest level of urinary PGI2 metabolites. Finally the COX-1+/− COX-2−/− genotype is associated with decreased aortic PGI synthase as assessed both by qRT-PCR and Western blot.
How do these data fit with earlier studies of the role of cyclooxygenase products in thrombosis? No previous studies have specifically addressed the effects of cyclooxygenase genotype on PAI-1 levels or fibrinolysis in vivo; however, there have been studies of the role of cyclooxygenase products in the carotid injury model. Cheng et al addressed the role of cyclooxygenases in the Rose Bengal carotid artery injury model used here [36]. Mice deficient in the prostacyclin receptor (IP−/−) had a markedly shortened time to occlusion compared with wild type mice. The time to occlusion in IP+/− mice was intermediate between that seen in wild type and IP−/− mice. These data support the suggestion that decreased PGI2 could lead to increased thrombosis but do not directly address the mechanism. The same investigators found that administration of DFU, a selective COX-2 inhibitor, to wild type mice resulted in a shortening of the time of occlusion. This study was done using B6;129 mice and is consistent with our finding that NS-398 shortened the time to occlusion in wild type mice on the same genetic background. These investigators also developed a mouse (PGHS-1KD) in which COX-1 activity was knocked down by 95%. They found that this genotype is associated with a prolonged time to occlusion. Administration of DFU to these mice shortened the time to occlusion. The 95% knockdown of COX-1 would be expected to decrease platelet thromboxane production and thus lead to a prolonged time to occlusion. In contrast a 50% decrease in COX-1 activity, as is seen in COX-1+/− mice has no effect on platelet aggregation as demonstrated by the normal bleeding time.
One remarkable aspect of the results with the carotid artery injury model was the observation that although COX-1+/− COX-2−/− mice had a shortened time to occlusion neither COX-1+/− COX-2+/+ nor COX-1+/+COX-2−/− mice has a shortened time to occlusion. This suggests that a shortened time to occlusion requires complete, or near complete, loss of COX-2 activity and partial loss of COX-1 activity.
Why is the time to occlusion shorter in the B6;129 wild type mice than in the C57BL/6J wild type mice? Different mouse strains have different baseline values in the carotid artery thrombosis assay [41]. Potential explanations for the differences in time of occlusion between the two strains include events that affect coagulation and events that affect fibrinolysis; these events could include COX- related events.
Why is the time of occlusion prolonged in COX-1+/+ COX-2−/− mice both in the C57BL/6J genetic background and in the B6;129 genetic background? We would have predicted that COX-1+/+COX-2−/− mice would have a time to occlusion similar to or shorter than wild type mice. We were surprised by the prolonged time to occlusion in the COX-1+/+ COX-2−/− mice on the C57BL/6J genetic background, where the COX-2 gene is disrupted in exon 8, and thought this might reflect some non-COX-2-related property in these mice. To confirm this observation we repeated the study in COX-1+/+COX-2−/− mice on the B6;129 genetic background, where the COX-2 gene is disrupted in exon 1; we obtained similar results. Thus the time to occlusion is prolonged in COX-1+/+COX-2−/− mice on different genetic backgrounds with the COX-2 genes disrupted in different exons. The prolonged time to occlusion in the COX-1+/+COX-2−/− is not associated with decreased PAI-1 levels and, in fact, is associated with PAI-1 levels that are increased compared to wild type. Thus the prolonged time to occlusion does not appear to be due to increased fibrinolysis resulting from decreased PAI-1.
The effects of NS-398 on the time to occlusion in the different genotypes fits with the model that a shortened time to occlusion requires a complete, or almost complete, loss of COX-2 activity with partial loss of COX-1 activity. As expected administration of NS-398 resulted in a shortening of the time to occlusion in COX-1−/−COX2+/− and COX-1+/−COX-2+/+ mice. The response of wild type mice to NS-398 varied with the background with NS-398 having no effect on the time to occlusion in C57BL/6J mice but resulting in a shorter time to occlusion in the B6;129 mice. This may reflect the relative contribution of COX-1 and COX-2 to PGI2 synthesis in the two genetic backgrounds. Other investigators using a different selective COX-2 inhibitor in B6;129 mice also found that inhibition of COX-2 results in shortening of the time to occlusion in this model [35]. The prolonged time to occlusion in the COX-1+/+COX2−/− mice is shortened by treatment with NS-398. This effect is seen in COX-1+/+COX-2−/− mice on both backgrounds. NS-398 is not completely specific for COX-2, and it is possible that the shortening of the occlusion time is due to the partial inhibition of COX-1 by NS-398.
Previous studies have demonstrated that cyclooxygenase products have profound effects on thrombosis(1–5). Here we have demonstrated that cyclooxygenase genotypes influence PAI-1 levels and thus occlusion times in carotid injury. The relative importance of these effects in clinical thrombosis is not clear; however, in the carotid artery injury model the short time to occlusion associated with the COX-1+/− COX-2−/− genotype was completely reversed by an inhibitory antibody against mouse PAI-1. In contrast the antibody to PAI-1 had no effect on the time to occlusion in wild type mice.. These findings suggest that PAI-1 levels do not influence the time to occlusion in wild type mice but that increased PAI-1 levels were responsible for the shortened time to occlusion in the COX-1+/− COX-2−/− mice. There is, however, evidence that complete absence of PAI-1 induces a mild fibrinolytic state as is shown by the accelerated clot lysis in PAI-1 deficient mice, (42).
The current study raises the possibility that the role of decreased fibrinolysis in the development of thrombosis may be increased in the face of drugs that inhibit COX-1 and COX-2, and especially drugs that block COX-2 activity completely and block COX-1 activity partially. This study has two potential implications for defining the mechanism of the increased number of myocardial infarctions and the other thrombotic events seen with selective COX-2 inhibitors. The first is that agents that completely inhibit COX-2 activity and partially inhibit COX-1 activity may be more likely to block fibrinolysis and thus result in thrombotic events than agents that completely inhibit COX-2 without affecting COX-1 activity. This suggests that the association of NSAIDs with cardiovascular events may depend on their selectivity for COX-1 and COX-2. The second potential implication is that the administration of a selective COX-2 inhibitor to an individual with diminished COX-1 activity, either on a genetic basis or from taking an NSAID, may result in decreased fibrinolysis and an increased risk of thrombotic events.
Supplementary Material
Acknowledgments
We thank George Broze for help with the tail vein bleeding times, and Aubrey Morrison for reviewing the manuscript.
This project supported by DK33165 (WFS), DK55753 (WFS), and HL55520 (DMT)
Footnotes
Contribution: Terrence E. Riehl helped experiments design, performed experiments, analyzed data and contributed to the manuscript; Li He performed carotid artery occlusion studies; Ling Zheng performed experiments; Suellen Greco performed the necropsies, Douglas M. Tollefsen participated in the design of the study, interpreted data and contributed to the manuscript; William F. Stenson designed the overall study, interpreted data and finalized the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci USA. 1975;72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M, Samuelsson B. Identification of an enzyme in platelet microsomes which generates Thromboxane A2 from prostaglandin endoperoxides. Nature. 1976;261:558–560. doi: 10.1038/261558a0. [DOI] [PubMed] [Google Scholar]
- 3.Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263:663–665. doi: 10.1038/263663a0. [DOI] [PubMed] [Google Scholar]
- 4.Moncada S, Gryglewski R, Bunting S, Vane JR. Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potential inhibitor of platelet aggregation. Lancet. 1977;1:18–20. doi: 10.1016/s0140-6736(77)91655-5. [DOI] [PubMed] [Google Scholar]
- 5.Baenziger NL, Becherer PR, Majerus PW. Characterization of prostacyclin synthesis in cultured human arterial smooth muscle cells, venous endothelial cells and skin fibroblasts. Cell. 1979;16:967–974. doi: 10.1016/0092-8674(79)90111-9. [DOI] [PubMed] [Google Scholar]
- 6.Hippisley-Cox J, Coupland C. Risk of myocardial infarction in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population based nested case-control analysis. Br Med J. 2005;330:1366–1373. doi: 10.1136/bmj.330.7504.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fosbøl EL, Gislason GH, Jacobsen S, Folke F, Hansen ML, Schramm TK, Sørensen R, Rasmussen JN, Andersen SS, Abildstrom SZ, Traerup J, Poulsen HE, Rasmussen S, Køber L, Torp-Pedersen C. Risk of myocardial infarction and death associated with the use of nonsteroidal anti-inflammatory drugs (NSAIDs) among healthy individuals: a nationwide cohort study. Clin Pharmacol Ther. 2009;85:190–197. doi: 10.1038/clpt.2008.204. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee D, Nissen SE, Topol EJ. Risk of Cardiovascular Events Associated With Selective COX-2 Inhibitors. JAMA. 2001;286(8):954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 9.Solomon SD, Wittes J, Finn PV, Fowler R, Viner J, Bertagnolli MM, Arber N, Levin B, Meinert CL, Martin B, Pater JL, Goss PE, Lance P, Obara S, Chew EY, Kim J, Arndt G, Hawk E. Cardiovascular Risk of Celecoxib in Randomized Placebo-Controlled Trials: The Cross Trial Safety analysis. Circulation. 2008;117:2104–2113. doi: 10.1161/CIRCULATIONAHA.108.764530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGettigan P, Henry D. Cardiovascular Risk and Inhibition of Cyclooxygenase. JAMA. 2006;296:1633–1644. doi: 10.1001/jama.296.13.jrv60011. [DOI] [PubMed] [Google Scholar]
- 11.Grosser T, Fries S, FitzGerald GA. Biologic basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. The Journal of Clinical Investigation. 2006;116:4–16. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patrignani P, Sciulli MG, Manarini S, Santini G, Cerletti C, Evangelista V. COX-2 is not involved in Thromboxane biosynthesis by activated human platelets. J Physiol Pharmacol. 1999;50:661–667. [PubMed] [Google Scholar]
- 13.Belton O, Byrne D, Kearney D, Leahy A, FitzGerald DJ. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. 2000;02:840–845. doi: 10.1161/01.cir.102.8.840. [DOI] [PubMed] [Google Scholar]
- 14.Wong E, Huang J, Tagari P, Riendeau D. Effects of COX-2 inhibitors on aortic prostacyclin production in cholesterol-fed rabbits. Atherosclerosis. 2001;157:393–402. doi: 10.1016/s0021-9150(00)00756-5. [DOI] [PubMed] [Google Scholar]
- 15.Créminon C, Habib A, Maclouf J, Pradelles P, Grassi J, Frobert Y. Differential measurement of constitutive (COX-1) and inducible (COX-2) cyclooxygenase expression in human umbilical vein endothelial cells using specific immunometic enzyme immunoassays. Biochim Biophys Acta. 1995;1254:341–348. doi: 10.1016/0005-2760(94)00197-7. [DOI] [PubMed] [Google Scholar]
- 16.Rudic RD, Brinster D, Cheng Y, Fries S, Song WL, Austin S, Coffman TM, FitzGerald GA. COX-2-derived prostacyclin modulates vascular remodeling. Circ Res. 2005;96:1240–1247. doi: 10.1161/01.RES.0000170888.11669.28. [DOI] [PubMed] [Google Scholar]
- 17.Topper JN, Cai J, Falb D, Gimbrone MA., Jr Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci USA. 1996;93:10417–10422. doi: 10.1073/pnas.93.19.10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atsuta H, Uchiyama T, Kanai H, Iso T, Tanaka T, Suga T, Maeno T, Arai M, Nagai R, Kurabayashi M. Effects of a stable prostacyclin analogue beraprost sodium on VEGF and PAI-1 gene expression in vascular smooth muscle cells. International Journal of Cardiology. 2009;132:411–418. doi: 10.1016/j.ijcard.2007.12.119. [DOI] [PubMed] [Google Scholar]
- 19.Kirchrath J, Debey S, Glandorff C, Kirchrath L, Schror K. Gene expression profile of the Gs-coupled prostacyclin receptor in human vascular smooth muscle cells. Biochemical Pharmacology. 2004;67:756–765. doi: 10.1016/j.bcp.2003.07.022. [DOI] [PubMed] [Google Scholar]
- 20.Cozzolino D, Coppola L, Masi S, Salvatore T, Sasso F, Lucia D, Gentile S, Torella R. Short-and long-term treatments with iloprost in diabetic patients with peripheral vascular disease: effects on the cardiovascular risk factor plasminogen activator inhibitor type-1. Eur J Clin Pharmacol. 1999;55:491–4979. doi: 10.1007/s002280050662. [DOI] [PubMed] [Google Scholar]
- 21.Eitzman D, Westrick R, Nabel E, Ginsburg D. Plasminogen activator inhibitor-1 and vitronectin promote vascular thrombosis in mice. Blood. 2000;95:577–580. [PubMed] [Google Scholar]
- 22.Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342:1792–1801. doi: 10.1056/NEJM200006153422406. [DOI] [PubMed] [Google Scholar]
- 23.Hamsten A, de Faire U, Walldius G, Dahlén G, Szamosi A, Landou C, Blombäck M, Wiman B. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet. 1987;2:3. doi: 10.1016/s0140-6736(87)93050-9. [DOI] [PubMed] [Google Scholar]
- 24.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-Dependent Spontaneious Coronary Arterial Thrombosis in Transgenic Mice That Express a Stable Form of Human Plasminogen Activator Inhibitor-1. Circulation. 2002;106:491–496. doi: 10.1161/01.cir.0000023186.60090.fb. [DOI] [PubMed] [Google Scholar]
- 25.Schafer K, Muller K, Hecke A, Mounier E, Goebel J, Loskutoff D, Konstantinides S. Enhanced Thrombosis in Atherosclerosis-Prone Mice Is Associated With Increased Arterial Expression of Plasminogen Activator Inhibitor-1. Arterioscler Thromb Vasc Biol. 2008;23:2097–2103. doi: 10.1161/01.ATV.0000097766.36623.DF. [DOI] [PubMed] [Google Scholar]
- 26.Fay WP, Shapiro AD, Shih JL, Schleef RR, Ginsburg D. Complete Deficiency of Plasminogen-Activator Inhibitor Type 1 Due to a Frame-Shift Mutation. N Engl J Med. 1992;327:1729–1733. doi: 10.1056/NEJM199212103272406. [DOI] [PubMed] [Google Scholar]
- 27.Morhman SJ, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, Smithies O. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 28.Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM, Gorry SA, Trzaskos JM. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 29.He L, Vincente CP, Westnick RJ, Eitzman DT, Tollefsen DM. Heparin cofactor II inhibits arterial thrombosis after endothelial injury. J Clin, Invest. 2002;109:213–219. doi: 10.1172/JCI13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dupont DM, Blouse GE, Hansen M, Mathiasen L, Kjelgaard S, Jensen JK, Christensen A, Gils A, Declerck PJ, Andreasen PA, Wind T. Evidence for a Pre-latent Form of the Serpin Plasminogen Activator Inhibitor-1 with Detached μ-Strand 1C*. Journal of Biological Chemistry. 2006;281:36071–36081. doi: 10.1074/jbc.M606851200. [DOI] [PubMed] [Google Scholar]
- 31.Smith AA, Jacobson LJ, Miller BI, Hathaway WE, Manco-Jonson MJ. A new euglobalin clot lysis assay for global fibrinolysis. Thromobosis Research. 2003;112:329–337. doi: 10.1016/j.thromres.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Broze GJ, Yin ZF, Lasky N. A tail vein bleeding time model and delayed bleeding in hemophiliac mice. Thromb Haemost. 2001;85:747–748. [PubMed] [Google Scholar]
- 33.Kurien BT, Scofield R. Mouse urine collection using clear plastic wrap. Laboratory Animals. 1999;33:83–86. doi: 10.1258/002367799780578525. [DOI] [PubMed] [Google Scholar]
- 34.Davis BJ, Lennard DE, Lee CA, Tiano HF, Morham SG, Wetsel WC, Langenbach R. Anovulation in Cyclooxygenase-2-Deficient Mice Is Restored by Prostaglandin E2 and Interleukin-1μ. Endocrinology. 1999;140:2685–2695. doi: 10.1210/endo.140.6.6715. [DOI] [PubMed] [Google Scholar]
- 35.Loftin CD, Trivedi DB, Tiano HF, Clark JA, Lee CA, Epstein JA, Morham SG, Breyer MD, Nguyen M, Hawkins BM, Goulet JL, Smithies O, Koller BH, Langenbach R. Failure of ductus arteriosus closure and remodeling in neonatal mice deficient in cyclooxygenase-1 and cyclooxygenase-2. PNAS. 2001;98:1059–1064. doi: 10.1073/pnas.031573498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng Y, Wang M, Yu Ying, Lawson J, Funk C, FitzGerald G. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. The Journal of Clinical Investigation. 2006;116:1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.James MJ, Walsh JA. Inter-relationships between Vascular Thomboxane and Prostacyclin Synthesis. Prostaglandins Leukotrienes and Essential Fatty Acids. 1988;31:91–95. doi: 10.1016/0952-3278(88)90081-6. [DOI] [PubMed] [Google Scholar]
- 38.Moncada S, Vane JR. Pharmacology and endogenous roles of prostaglandin endoperoxides, Thromboxane A2. A new group of biologically active compounds derived from prostaglandin endoperoxides. Proceedings of the National Academy of Science USA. 1975;72:2994. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novacek G, Kapiotis S, Jilma B, Quehenberger P, Michitsch A, Traindl O, Speiser W. Enhanced blood coagulation and enhanced fibrinolysis during hemodialysis with prostacyclin. Thrombosis Research. 1997;88:283–290. doi: 10.1016/s0049-3848(97)00255-7. [DOI] [PubMed] [Google Scholar]
- 40.Fernandez-Martinez MR, Bosca A, Martin-Sanz P. Prostaglandin E2 Promotes migration and adhesion in hepatocellular carcinoma cells. Carcinogenesis. 2005;26(4):753–61. doi: 10.1093/carcin/bgi022. [DOI] [PubMed] [Google Scholar]
- 41.Westrick R, Winn M, Eitzman D. Murine Models of Vascular Thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:2079–2093. doi: 10.1161/ATVBAHA.107.142810. [DOI] [PubMed] [Google Scholar]
- 42.Carmeliet P, Stassen JM, Schoonjans L, Ream B, van den Oord JJ, De Mol M, Mulligan RC, Collen D. Plasminogen Activator Inhibitor-1 Gene-deficient mice. J Clin Invest. 1993;92:2756–2760. doi: 10.1172/JCI116893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.