

Abstract
Highly active antiretroviral therapy (HAART) can suppress plasma human immunodeficiency virus type 1 (HIV-1) levels to below the detection limit of ultrasensitive clinical assays. However, HIV-1 persists in cellular reservoirs, and in adults, persistent low-level viremia is detected with more sensitive assays. The nature of this viremia is poorly understood, and it is unclear whether viremia persists in children on HAART, particularly those who start therapy shortly after birth. We therefore developed a reverse transcriptase PCR (RT-PCR) assay that allows genotyping of HIV-1 protease even when viremia is present at levels as low as 5 copies of HIV-1 RNA/ml. We demonstrated that viremia persists in children with plasma virus levels below the limit of detection of clinical assays. Viremia was detected even in children who began HAART in early infancy and maintained such strong suppression of viremia that HIV-1-specific antibody responses were absent or minimal. The low-level plasma virus lacked protease inhibitor resistance mutations despite the frequent use of nelfinavir, which has a low mutational barrier to resistance. Protease sequences resembled those of viruses in the latent reservoir in resting CD4+ T cells. Thus, in most children on HAART with clinically undetectable viremia, there is continued virus production without evolution of resistance in the protease gene.
The treatment of human immunodeficiency virus type 1 (HIV-1) infection with highly active antiretroviral therapy (HAART) significantly reduces the levels of viral RNA in plasma and lymphoid tissue (3, 15, 17, 26, 27, 31). In many treated adults and children, free virus becomes undetectable in the plasma when measured by ultrasensitive clinical viral load assays that have a detection limit of 50 copies of HIV-1 RNA/ml. Despite the absence of clinically detectable viremia, however, ongoing viremia remains detectable when more-sensitive reverse transcriptase PCR (RT-PCR) assays are used (8, 20, 28). The nature and clinical significance of ongoing virus production during effective HAART remain elusive.
Several mechanisms may contribute to the persistence of viremia during effective HAART. These include the inability of HAART to completely suppress virus replication because of inadequate potency (14), intermittent nonadherence resulting in suboptimal drug concentrations (30), and the emergence of drug-resistant variants (19). Ongoing cycles of replication in the setting of HAART could lead to the accumulation of drug resistance mutations and treatment failure. Another possible explanation for ongoing low-level viremia is the continued production of HIV-1 by infected cells harbored in viral reservoirs or drug sanctuary sites (1, 20, 34). One such reservoir that is established during acute infection is a small pool of latently infected resting memory CD4+ T cells (5, 6). This latent reservoir has been shown to retain HIV-1 in a replication-competent form despite many years of suppression of viremia to <50 copies/ml (6, 11, 12, 32, 38, 40, 45).
Ongoing viremia is a particular concern with HIV-1-infected children who potentially face a full lifetime of treatment. The extent to which low-level viremia continues in children treated with HAART, particularly those treated from infancy, and the clinical significance of this viremia have not been defined. In an initial cross-sectional study, we detected and sequenced plasma viruses in four of seven children on HAART who had plasma virus levels below 50 copies/ml. These viruses were archival wild-type or pre-HAART drug-resistant variants rather than recently derived drug-resistant mutants (20). To more fully characterize ongoing viremia in children, we have developed a more sensitive RT-PCR assay which we have used to better define the frequency of ongoing viremia and the evolution of drug resistance mutations in the protease gene in children initiating suppressive HAART during acute and chronic HIV-1 infection. Our results provide insight into the nature and clinical significance of low-level viremia in patients on HAART.
MATERIALS AND METHODS
Patients.
We studied low-level viremia in acutely and chronically infected children who had durable suppression of HIV-1 replication on HAART for 1 to 5 years. Study participants were recruited from the pediatric specialty clinics at Johns Hopkins University and the University of Maryland. Written informed consent approved by the institutional review boards was obtained from the parents or guardians of the children. A total of 15 HIV-1-infected children were eligible for study during the period from April 2002 to February 2003.
Six of children (C2, C7, C8, C10, C11, and C22) were from a previously characterized cohort and had participated in longitudinal studies of the latent reservoir in resting CD4+ T cells (20, 32, 38). The inclusion of this well-studied group of children allowed for validation of the novel methodologies used for this study and for the evaluation of the phylogenetic relatedness of plasma virus to replication-competent latent HIV-1 retained in resting CD4+ T cells during years of effective therapy.
HIV-1 RNA isolation, amplification, and sequencing from small blood volumes.
Virus particles were pelleted from 3 to 4 ml of plasma by ultracentrifugation at 17,000 × g for 2 h at 4°C in a Heraeus centrifuge. Virus particles were then lysed, and the RNA was isolated using a Qiagen column purification method according to the manufacturer's directions. Isolated RNA was treated with DNase (Invitrogen Corp, Carlsbad, Calif.) and divided into a total of seven to nine reaction tubes. For the first step, the RNA was reverse transcribed and amplified by PCR using a one-step RT-PCR protocol with primers Prot 3′out (nucleotides 2620 to 2647; 5′GCTTTTATTTTCTCTTCTGTCAATGGCC3′) and 5′ outer pol (nucleotides 2008 to 2031; 5′GCCCCTAGGAAAAAGGGCTGTTGG3′). A nested PCR was then carried out with a high-fidelity proof-reading polymerase on 10 μl of the first round product (diluted 1:40) with the following primers: 5′ inner pol (nucleotides 2057 to 2080; 5′TGAAAGATTGTACTGAGAGACAGG3′) and Prot3 in (nucleotides 2569 to 2593; 5′CCTGGCTTTA-ATTTTACTGGTACAG3′). The positions of the oligonucleotide primers are numbered according to the pol gene of the HXB2 isolate (18). In each case, two control RT-PCRs were set up without the RT to exclude contaminating DNA as a source for the amplified sequences. PCR products were cloned into PCR-BluntII-TOPO vector (Invitrogen Corp, Carlsbad, Calif.) and sequenced using a fluorescent dideoxy termination method of cycle sequencing on a 373A automated DNA sequencer (Applied Biosystems, Foster City, Calif.), following Applied Biosystems protocols.
HIV-1 serology.
HIV-1 immunoglobulin G levels were determined using a commercial enzyme-linked immunosorbent assay (Vironostika HIV-1 Micorelisa system; Organon-Tek, Durham, N.C.). Antibody specificity was confirmed by Western blotting (Calyptebiomedical, Rockville, Md.).
Sequence validation and statistical considerations.
Sequence validation was carried out according to the methods recommended by Learn et al. (24). Algorithms were used to distinguish PCR errors from polymorphisms and resistance mutations and to establish the independence of HIV-1 variants obtained from the same patient. Using an estimation procedure for the frequency of artifactual misincorporations (41) combined with the manufacturer's statement of polymerase fidelity, the expected frequency of sporadic misincorporations was calculated at 1 per 104 residues. To avoid overestimation of diversity, substitutions that occurred only once in this data set were removed prior to analysis. With sporadic substitutions removed, identical sequences derived from the same PCR were considered redundant and likely to have been generated by resampling (25). These “sanitized” sequences were used for the remainder of the analysis. Clones obtained from different PCRs were also considered independent. Clones obtained from the same PCR were only considered independent when they differed by drug resistance mutations or by a number of mutations that exceeded the estimated rates of artifactual misincorporation described above. Basic local alignment search tool (BLAST) searches of GenBank (http://www.ncbi.nlm.nih.gov/GenBank/GenbankOverview.html) revealed that none of the sequences matched those of laboratory strains or other patient isolates.
Phylogenetic trees were inferred from nucleotide sequences through the use of PAUP* version 4.0 (Sinauer Associates Inc., Sunderland, Mass) (43). The HKY-85 model of evolution was suggested by MODELTEST analysis (35). Trees were initially inferred using minimum evolution with 100 random addition sequence replicates and tree bisection-reconnection branch swapping, and the shortest trees were used as input for a maximum likelihood estimation using the HKY85+G model. Most-recent common ancestor sequences were obtained during maximum likelihood analysis as the sequence inferred for the node ancestral to each patient-specific clade. Larger data sets were examined, using the highly efficient neighbor-joining method (39) with testing of internal node support using the bootstrap method (10) with 1,000 replicates, for concordant clustering (e.g., analysis of all 221 plasma and cell sequences plus reference strains).
Plasma sequences that were previously published (20) and had been obtained from three of the children (C2, C11, and C22) were included to allow a complete assessment of the phylogenetic relatedness of samples obtained longitudinally. Previously published (20, 32, 38) and recently determined latent reservoir sequences were also used to validate the patient-specific character of the plasma sequences and to compare the HIV-1 protease of plasma variants to those harbored in the latent reservoir in resting CD4+ T cells. Reference sequences (and their accession numbers) included strains A_SE.SE8131 (AF107771), A1_UG.U455 (M62320), A2_CD.CDKFE4 (AF286240), B_FR.HXB2R (K03455), C_IN.IN21068 (AF067155), C_ET.ETH2200 (U46016), and D_ZR.Z2Z6 (M22639).
Analysis of HIV-1 diversity in plasma.
Assessment of HIV-1 diversity in the early-treated cohort versus the late-treated group was performed using protease sequences amplified from the first visit from which multiple viral variants were detected. Diversity was evaluated as the average pairwise genetic distance calculated using the same model and parameters as described above for the phylogenetic analysis. Median values for the early- and late-treated groups were compared using a nonparametric tool (Wilcoxon rank sum test).
Nucleotide sequence accession numbers. Novel sequences have been submitted to GenBank (accession numbers AY429144 to AY429259).
RESULTS
Patients.
Two groups of children who differed by the age of initiation of HAART were studied (Table 1). The members of group 1, the late-treated group, initiated HAART after 1 year of age. Group 1 was subdivided into two subgroups, groups 1A and 1B, on the basis of the length of suppression of viremia on HAART. Before HAART was a standard method of care, six of these children were treated with nonsuppressive regimens consisting of nucleoside analogue RT inhibitors. Group 2, the early-treated group, was comprised of children initiating HAART in the first few months of life with sustained viral suppression resulting from their first HAART regimen. These children were selected to evaluate the impact of early initiation of HAART on residual HIV-1 production in children with perinatally acquired infection.
TABLE 1.
Patient characteristics
| Group and patient | Age (yr) | Sex/ racea | Nonsuppressive therapy
|
Suppressive therapy
|
Plasma HIV RNA at start of sup pressive therapy (copies/ml) | Plasma HIV RNA at first analysis (copies/ml) | |||
|---|---|---|---|---|---|---|---|---|---|
| Regimen(s)b | Duration of therapy (yr) | Age at initiation (yr) | Regimensb | Duration of suppression (yr) | |||||
| 1A | |||||||||
| C2c | 5.5 | F/AA | None | None | 1.0 | ZDV/3TC/RTV | 5.4 | 117,506 | <50 |
| C7c | 12.9 | F/AA | ZDV/DDI | 4.8 | 7.9 | D4T/3TC/NFV | 5.0 | 2,874 | 148 |
| C8c | 11.3 | M/C | ZDV | 2 | 5.6 | ZDV/3TC/RTV | 6.3 | 207,554 | <50 |
| DDC | 0.5 | ||||||||
| ZDV/DDI | 0.2 | ||||||||
| ZDV/3TC | 0.5 | ||||||||
| C10c | 12.6 | F/AA | ZDV/DDI | 4.5 | 7.5 | D4T/3TC/NFV | 5.0 | 12,017 | <50 |
| C22d | 9.3 | F/AA | ZDV | 2.4 | 4.8 | DDI/DLV/RTV/SQV | 4.5 | 246,499 | 124 |
| ZDV/3TC | 0.6 | ||||||||
| ZDV/3TC/RTV | 0.3 | ||||||||
| ZDV/3TC/NFV | 0.8 | ||||||||
| 1B | |||||||||
| C40c | 6.4 | M/AA | ZDV/3TC/NVP | 0.6 | 4.9 | D4T/DDI/NFV | 1.5 | 7,888 | <50 |
| D4T/3TC/NVP | 0.4 | ||||||||
| C42c | 7.5 | F/AA | None | None | 5.4 | D4T/3TC/NFV | 2.2 | 211,254 | <50 |
| C11e | 11.5 | F/AA | ZDV | 4.6 | 10.9 | D4T/EFV/LPVr | 0.7 | 1,602 | <50 |
| ZDV/DDI | 2.4 | ||||||||
| D4T/3TC/NFV | 3.5 | ||||||||
| C45e | 11.4 | M/AA | ZDV/DDI | 0.2 | 9.3 | 3TC/EFV/APV | 1.1 | 128,956 | <50 |
| ZDV | 1.7 | ||||||||
| ZDV/DDI | 0.6 | ||||||||
| ZDV/3TC | 0.6 | ||||||||
| D4T/3TC/RTV | 3.8 | ||||||||
| ZDV/DDI/APV | 1.0 | ||||||||
| 2 | |||||||||
| C101c | 4.6 | M/AA | None | None | 0.15 | D4T/3TC/RTV | 4.4 | >750,000 | <50 |
| D4T/3TC/EFV/RTV | |||||||||
| D4T/DDI/ABC/EFV/RTV | |||||||||
| C102c | 2.8 | F/AA | Nonef | None | 0.13 | ZDV/3TC/NFV | 3.0 | >750,000 | <50 |
| C103c | 5.2 | M/AA | Nonef | None | 0.2 | ZDV/3TC/NFV | 5.2 | 517,384 | <50 |
| 3TC/D4T/NFV | |||||||||
| 3TC/D4T/EFV | |||||||||
| C104c | 4.3 | F/AA | None | None | 0.2 | ZDV/3TC/NFV | 4.1 | >750,000 | <50 |
| C107c | 0.7 | F/AA | Nonef | None | 0.3 | ZDV/3TC/EFV/NFV | 0.5 | >750,000 | <400 |
| ZDV/3TC/EFV | |||||||||
| C108c | 2.2 | M/AA | Nonef | None | 0.2 | D4T/3TC/NVP/NFV | 2.0 | 360,119 | <50 |
F, female; M, male; AA, African-American; C, Caucasian.
Abbreviations for drugs; ZDV, zidovudine; 3TC, lamivudine; DDI, didanosine; DDC, zalcitabine; D4T, stavudine; ABC, abacavir; EFV, efavirenz; DLV, delavirdine; NVP, nevirapine; RTV, ritonavir; LPVr, lopinavir-ritonavir; APV, amprenavir; SQV, saquinavir; NFV, nelfinavir.
Suppressed on first PI-based regimen.
Did not take PI component of first two PI-based regimens.
Prior failure on PI-based regimen.
Received neonatal prophylaxis (6 weeks of ZDV after birth).
The majority (11/15; 73%) of the children studied were on standard three-drug HAART regimens with combinations of nucleoside analogue RT inhibitors and protease inhibitors (PIs). The remaining children were either on four (n = 3) or five (n = 1) antiretroviral drugs, including one or more PIs. A total of 60% (9/15) of the children were receiving the PI nelfinavir.
The median ages at the initiation of HAART for groups 1A and 1B were 5.6 and 6.3 years, and the median durations of HAART were 3.3 and 0.6 years, respectively. The median age at the start of HAART for the early-treated group was 2.5 months (range, 1.6 to 3.8 months), and the median duration of HAART was 3.5 years (range, 0.5 to 5.2 years). The geometric mean plasma HIV-1 RNA levels before HAART were 257,833 copies/ml (range, 5,389 to >750,000 copies/ml) for the late-treated group (Table 1) and 840,000 copies/ml (range, 517,384 to >1,500,000 copies/ml) for the early-treated group (Table 1).
Persistence of HIV-1 viremia below 50 copies/ml during suppressive HAART in children.
A novel RT-PCR assay that detects as few as 5 copies of HIV-1 RNA in plasma (Fig. 1A) was used to assess ongoing viremia in 30 plasma samples obtained from the 15 children. With this assay, ongoing HIV-1 viremia was detectable in 26 of 30 plasma samples, including 23 samples obtained while the viral load was <50 copies/ml (Fig. 1B; Table 2). Viremia was detected in five of six children in the early-treated group despite the excellent response to HAART observed in this group. In three samples from group 1, the viral load was >50 copies/ml at the time of analysis and then returned to below 50 copies/ml. All three children with episodes of intermittent detectable viremia of >50 copies/ml continued to have durable suppression of virus replication and required no change in therapy. These episodes were therefore considered to be blips.
FIG. 1.

Nested RT-PCR assay used for genotyping the HIV-1 protease gene from the plasma in patients in whom plasma HIV-1 RNA levels are below 50 copies/ml. (A) The sensitivity of the assay was established using reaction mixtures containing serial dilutions of in vitro-transcribed HIV-1 NL43 RNA in the presence (RT+) or absence (RT−) of RT. Control PCRs for the first (1st) RT-PCR and the second (2nd) nested PCR are also shown. (B) Representative amplifications of protease from 4 ml of plasma from two subjects (subject C11, top panel; subject C40, bottom panel) whose viral loads were <50 copies/ml at the time of analysis.
TABLE 2.
HIV-1 protease sequences amplified from plasma in children with <50 copies of HIV-1 RNA/ml on protease-inhibitor-containing HAART regimensc
 |
 |
Time points with asterisks indicate previously published sequences from the same patient included to provide a complete picture of the evolutionary features of HIV-1 in plasma during HAART.
May represent preexisting polymorphism or early drug resistance to nelfinavir.
Group 1A, late-treated (long-term suppression) group; group 1B, late-treated (short-term suppression) group; group 2, early-treated group.
Seven children from the late-treated group (C2, C7, C8, C10, C11, C22, and C40) had plasma samples analyzed at multiple time points (Fig. 2). Despite suppression of viremia to below the limits of detection by ultrasensitive clinical assays, HIV-1 viremia remained detectable in 9 of the 11 repeat samples obtained from this group. All of the patients continued to have suppression of viral replication to <50 copies/ml for a mean of 8.1 months (range, 0.03 to 21.4 months) from the first analysis. No patients were excluded from the original pediatric cohort for viral rebound. Together with the results of the sequence analysis described below, which provided definitive confirmation of the patient-specific nature of the PCR products obtained, these data demonstrate that HIV-1 viremia persists in children at low levels despite durable suppressive HAART regardless of whether treatment is initiated early and late.
FIG. 2.

Viral load data from study subjects who were monitored longitudinally. Plasma HIV-1 RNA levels are indicated by closed symbols. Open symbols indicate that the level of viremia was below the limit of detection of the assay used (400, 200, or 50 copies/ml); the plotted values serve to indicate the limit of detection. Solid arrows indicate times of sampling. Dashed arrows indicated previously reported time points (20).
Extent of HIV-1 diversity and divergence at plasma RNA levels of <50 copies/ml.
To provide definitive confirmation that the PCR signals detected represent patient-specific sequences that are continuously produced in children whose plasma virus levels are below the limit of detection of clinical assays, we cloned and sequenced the positive PCRs. A total of 181 independent RT-PCR assays were performed on the cohort (median, 6 reactions per time point per patient) (Table 2). A total of 68 amplicons (38% of total reactions) were obtained at the limits of the serial dilution RT-PCR (median, 4 amplicons per patient; range, 0 to 18) (Table 2). A total of 200 clones derived from the PCR amplicons were sequenced and analyzed, yielding 116 distinct HIV-1 variants, as determined by our criteria for clonal independence. As discussed below, phylogenetic analysis demonstrated the expected patient-specific clustering. Nucleotide sequences obtained from the plasma of each patient formed a distinct cluster (Fig. 3), reflecting the patient-specific polymorphisms in the protease gene. These results demonstrate that it is possible to obtain reliable, patient-specific genotypic data even when the starting number of template viral RNA molecules is extremely low.
FIG. 3.

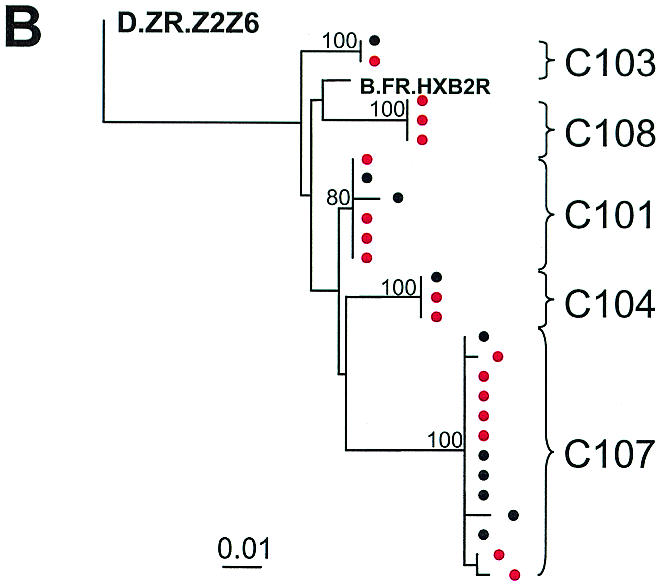
Maximum-likelihood phylogenetic analysis of HIV-1 protease gene sequences from plasma (red circles) of study subjects treated with HAART either later during childhood (A) or from early infancy (B). Sequence information obtained from latently infected CD4+ T cells from the early-treated group (black circles) are included in panel B to confirm the patient-specific nature of the protease sequences amplified at low levels and to emphasize the homogeneity of HIV-1 protease sequences in these individuals. Previously published plasma virus sequences are indicated in open circles (A) and are included to provide a complete picture of the evolutionary profile of HIV-1 in plasma at low levels during prolonged suppressive HAART. Reference sequences from clades A and C are labeled as follows: A*, A.SE.SE8131; A1, A.UG.U455; A2, A2.CD.CDKFE4; C*, C.IN.IN21068; C**, C.ET.ETH2220. Strain B.F.R.HXB2R was used as a reference sequence for clade B.
Because the range of sequences obtained may be affected by both diversity (variability observed within a single specimen) and divergence (increase in genetic distance over time), we performed a temporal analysis of the quasispecies in plasma at viral loads of <50 copies/ml in children. We found that HIV-1 viremia at <50 copies/ml was comprised of diverse viral variants in the group of children who were chronically infected (>5 years of age) before HAART was initiated (Fig. 3A). The median viral diversity in plasma in this group was 0.008 (range, 0.002 to 0.042 substitutions per nucleotide) when the viral load was <50 copies/ml. In the early-treated group, in contrast, the median genetic diversity was significantly lower at 0.000 (range, 0 to 0.004; P < 0.008) (Wilcoxon rank test). Although the frequency of low-level viremia in the early-treated group was similar to that observed in the late-treated group, the viremia in the early-treated group was extremely homogeneous in character (Fig. 3B). There was minimal sequence variation detected despite a mean infection time of 3.3 years (range, 0.7 to 5.1 years) and the continuous production of low levels of virus for a mean treatment time with PIs of 3 years. These results suggest that early effective HAART preserves the state of HIV-1 evolution, arresting sequence variation to that which had occurred before HAART was initiated.
Another striking feature of HIV-1 in plasma during suppressive HAART was the lack of a clear relationship between the degree of divergence (genetic distance from the root of the tree) and the time of sampling. In the patients who were sampled at multiple time points, no temporal structure was identified in the phylogenetic trees. Representative trees showing the relationship between divergence and sampling time for patients C2, C8, C11, and C22 are shown in Fig. 4A. When the distance from the most recent common ancestor (MRCA) was calculated, no strong trend during suppressive HAART suggesting divergence was seen (Fig. 4B). Even when divergence in protease did occur due to treatment failure and the development of resistance (C11; Fig. 4), the plasma sequences obtained when viremia was suppressed by a new regimen did not show a clear temporal pattern. Viremia at <50 copies per milliliter involved a mixture of HIV-1 variants. Recently generated, drug-resistant variants commingled with more ancestral, drug-sensitive variants (Fig. 4). Thus, we found that in children in whom a successful HAART regimen was begun during chronic HIV-1 infection, viremia at <50 copies/ml lacked temporal structure and showed degrees of heterogeneity and divergence that reflected prior nonsuppressive therapy.
FIG. 4.

Maximum-likelihood phylogenetic analysis of HIV-1 protease gene sequences from individual study subjects. (A) Phylogenetic trees showing sequences obtained from plasma virus (diamonds) and from latently infected resting CD4+ T cells (circles). Time of sampling is indicated by the color scale. The red bracket used with the patient C11 data indicates sequences bearing drug resistance mutations. (B) Plots of genetic distance versus time on HAART for subjects who provided multiple specimens during HAART. An MRCA sequence was reconstructed for each subject, and distances from the MRCA to each sequence were calculated using the HKY85+G model. Sequences obtained from plasma (diamonds) and latently infected CD4+ T cells (circles) are indicated.
Plasma viremia at <50 copies/ml mainly involves HIV-1 variants that are wild type for protease.
To determine whether continuous production of low levels of plasma virus during effective HAART with PI-based regimens required or generated some level of PI resistance, we analyzed the nucleotide sequences for amino acid substitutions known to be associated with drug resistance to PIs. Sequence analysis revealed that amino acid substitutions at sites of polymorphisms were unique to each patient, confirming the patient-specific nature of the PCR products obtained at low template concentrations (Table 2). More importantly, in patients who were PI naïve when HAART was started, no drug resistance mutations were seen in 92% (107/116) of the independent HIV-1 variants cloned from plasma at a time when plasma virus levels were <50 copies/ml or during intermittent blips. In 11 of the 13 patients studied who had no prior history of treatment failure with PIs, no protease mutations were seen despite a mean of 3.6 years (range, 0.3 to 6.5 years) of continuous exposure to PIs (Table 2). In patient C10, for example, none of the 18 independent sequences obtained during a blip to 69 copies/ml after 4.7 years of a PI-based regimen showed PI resistance mutations, and none of the 13 independent sequences obtained when the viral load was below 50 copies/ml after 5 years of the PI-based regimen had PI resistance. Thus, in at least some children receiving potent antiretroviral therapy there can be continued release into the plasma of viruses sensitive to the most potent drug in the regimen for prolonged periods without the development of resistance mutations.
Early drug-resistant variants were detected in two of the children whose primary PI regimens were suppressive. Patient C7 had detectable viremia (148 copies/ml) at the time of initiation of this study and a history of recent nonadherence. All of the six clones detected had a V82I substitution associated with low-level resistance to nelfinavir. On repeat analysis 3 months later, plasma virus levels were below 50 copies/ml, and we were unable to amplify plasma virus using our five-copy-sensitivity RT-PCR assay. In the second child, C40, one of the seven clones detected at study entry had the N88S substitution, and, at the second study visit 3 months later, two of the nine clones had amino acid changes at position 82 (V82I) associated with nelfinavir therapy. At 3 months later, none of these substitutions had become predominant, and the patient's viral load has remained undetectable at <50 copies/ml. Thus, plasma viremia below 50 copies/ml in patients on prolonged uninterrupted treatment with PI-containing regimens was comprised largely of HIV-1 that was sensitive to the relevant PI.
As expected, archival HIV-1 drug-resistant variants were detected in the two children (C45 and C11) who had prior treatment failure with a PI regimen and were known to have developed PI-resistant HIV-1 variants (Table 1). In patient C45, all 10 variants detected at a single time point had mutations at amino acid positions 54 (I54V) and 82 (V82A) associated with high-level drug resistance to ritonavir. This genotypic profile is consistent with the patient's treatment history of prior failure on a ritonavir regimen. Despite this finding, the patient's viral load remained undetectable (at <50 copies/ml) at a subsequent clinic visit. In this case, detection of archival PI-resistant variants did not represent impending treatment failure of the current suppressive regimen; rather, it likely reflected production of archival resistant viruses selected by a prior regimen. Similarly, for patient C11, drug-resistant variants harboring the D30N and N88D mutations that arose during prior treatment failure with a nelfinavir-containing regimen remained detectable in the plasma up to 6 months following the discontinuation of nelfinavir and suppression on a lopinavir-containing regimen. These highly resistant, archival protease HIV-1 variants were found to coexist in plasma with the more ancestral wild-type HIV-1 protease variants at plasma virus levels below 50 copies/ml (clones 2.1 and 7.1, from the second time point and clones 2, 5, and 7 from the third analysis) (Table 2 and Fig. 4A). The coexistence of wild-type, drug-susceptible HIV-1 and more divergent drug-resistant variants in plasma during suppressive therapy provides strong evidence for active production of archival variants from long-lived viral reservoirs.
Phylogenetic relatedness of plasma virus to replication-competent HIV-1 in the latent reservoir in resting CD4+ T-cells.
Because six of the children (C2, C7, C8, C10, C11, and C22) included in this study were previous participants in a 5-year longitudinal study of HIV-1 latency in resting CD4+ T cells, we compared the protease sequences of the persistent HIV-1 in plasma during effective HAART to those of the replication-competent HIV-1 recovered from the resting CD4+ T-cell reservoir. As previously reported (20), in all patients there was extensive comingling of the viral variants amplified from plasma at <50 copies/ml and HIV-1 recovered from the resting CD4+ T-cell compartment (Fig. 4A). Overall, there was no distinct clustering of plasma-derived sequences compared with that of the latent reservoir sequences.
Impact of early HAART on low-level viremia, HIV-1 diversity in plasma, and the HIV-1 antibody responses.
As discussed above, there was no apparent difference in the frequency of ongoing low-level viremia in children initiating HAART during acute, perinatally acquired HIV-1 infection compared to that seen with those treated during chronic infection (Table 2). Five of the six children in the early-treated group and all of the children in the late-treated group had viremia that was detectable using our sensitive RT-PCR assay. The one child for whom ongoing viremia was not detected was from the early-treated group. We were unable to amplify plasma virus obtained on two separate occasions 3 months apart.
Despite the prevalence of viremia in both the early- and late-treated groups, phylogenetic analysis revealed differences in the pace and character of sequence variation between the children with early suppressive therapy and those with late suppressive therapy. In the children initiating HAART during the first 4 months of life, there was little sequence variation (median genetic diversity, 0.00) between plasma virus clones amplified at below 50 copies/ml. In addition, there were no sequence variations in the protease gene between plasma viral variants and replication-competent HIV-1 in resting CD4+ T cells (Fig. 3B).
Importantly, and consistent with previous reports (26), all of the children tested in the early-treated group had limited or absent HIV-1-specific antibody responses (Fig. 5). While the absence of HIV-1-specific antibody and cellular immune responses has been used as an indicator of complete control of virus replication in children treated from early infancy, our results show that as determined by Western blot analysis, ongoing low-level viremia continues even in children who have negative or highly restricted HIV-1-specific antibody responses. Together, these results show that in early-treated children there is a continuous release into the plasma of low levels of HIV-1 virions that do not show evidence of genetic diversification in pol and that do not trigger a normal HIV-1-specific antibody response.
FIG. 5.

Representative HIV-1 Western blots of samples from children who were treated with HAART from early infancy (C101, C103, and C108) or who initiated HAART at later times (C10). Patient C102 had no viremia detectable using our RT-PCR assay. Ages at the time of HIV-1 antibody testing are indicated in parentheses. y, year.
DISCUSSION
Our study of 15 children with durable suppression of HIV-1 replication treated with HAART for up to 6 years demonstrates that HIV-1 viremia persists at plasma virus levels below 50 copies/ml in most if not all infected children on HAART. Viremia was even detected in a unique subset of children who started HAART in early infancy, some of whom reverted to HIV-1 seronegativity. We were able not only to detect viremia but also to clone and characterize the protease gene from the rare plasma virions that constitute viremia at this level. Amplification from low numbers of template molecules raises concerns about contamination, but phylogenetic analysis confirmed the patient-specific nature of the plasma viral sequences obtained. Most importantly, despite the finding of continued virus production in the setting of PI-based HAART regimens, HIV-1 viremia in children achieving durable suppression with their first PI-HAART regimen consisted primarily of HIV-1 variants that were wild type in protease. For the six subjects monitored longitudinally (C2, C7, C8, C10, C22, and C40), the median duration of continuous exposure to PIs was 3.2 years (range, 0.59 to 5.12). The median duration of follow-up was 21 months (range, 5 to 37 months). Despite the length of the observation period, accumulation of mutations in protease was not observed. These findings suggest that persistent low-level viremia is characteristic of HIV-1 infection during effective HAART and is comprised largely of PI-susceptible HIV-1 variants rather than protease variants with some degree of PI resistance.
Two potential processes might be involved in the stable persistence of HIV-1 viremia in the setting of potent antiretroviral therapy. One possibility is that viremia at plasma RNA levels of <50 copies/ml represents a new steady state, reflecting ongoing cycles of HIV-1 replication. Under these conditions, HIV-1 replication might be continuous but might occur only at low levels because of efficacy of the HAART regimen. Indeed, studies in HIV-1-infected adults have shown that the decay kinetics of plasma HIV-1 RNA during initial HAART can be accelerated when four- or five-drug HAART regimens are used (14, 29). This finding suggests that during standard HAART with three antiretroviral agents, newly infected cells are generated, albeit at a low level. If the stable persistence of HIV-1 viremia observed during HAART is in fact due to continuous cycles of HIV-1 replication, one might expect the genetic composition to show progressive divergence and the eventual emergence of drug-resistant variants (13). However, neither drug-resistant HIV-1 nor viral divergence was observed in the children who were fully compliant, as evidenced by durable suppression with their first PI regimen. It remains possible that the pace of evolution of drug resistance in protease is too slow for detection over the treatment period (36, 37). The pol gene is more stable than other regions (e.g., env) of the HIV-1 genome; therefore, it is possible that analysis of changes in env sequences over time might provide some evidence for evolution (16, 47). Nevertheless, the evolutionary changes that are likely to be of clinical significance in these patients are the changes affecting drug susceptibility, and these were not apparent in our study.
The persistence of low-level viremia in children on HAART can also be understood in the context of the latent reservoir for HIV-1. Although resting CD4+ T cells harboring replication-competent HIV-1 are present at a low frequency (1 per million resting CD4+ T cells), they can be readily detected using cellular activation methods in all infected individuals (6, 12, 33, 40, 45). It is therefore plausible that resting CD4+ T cells harboring latent replication-competent HIV-1 are continuously activated for virus production during HAART. In our phylogenetic analysis, we found extensive commingling of protease sequences from plasma virus and replication-competent HIV-1 recovered from resting CD4+ T cells during HAART. Several genetic studies in HIV-1-infected adults have implicated viral reservoirs, including the resting CD4+ T-cell reservoir, as sources of rebound viremia following treatment discontinuation (4, 9, 22, 46). In patients failing antiretroviral therapy with multidrug-resistant HIV-1, wild-type HIV-1 becomes predominant when the drugs are discontinued (7). The only documented site where archival, wild-type, replication-competent HIV-1 can coexist for long periods of time with drug-resistant variants generated during treatment failure is the latent reservoir in resting CD4+ T cells (38). Therefore, the reemergence of wild-type HIV-1 in the plasma of patients who clearly have had viral divergence with progressive high-level drug-resistant virus provides further evidence for the active contribution of latent viral reservoirs to plasma virus. The continued production of drug-sensitive HIV-1 that we observed in children on suppressive HAART can be explained by the release of virus from stable reservoirs.
The failure to detect PI-resistant variants in most patients was not due to any inherent bias in the assay. Possible early PI-resistant variants were detected in two children (C7 and C40) who had no history of failure on a PI regimen, one with suboptimal adherence and the other in the setting of fully suppressive HAART. In patient C7, a V82I substitution was present in all clones amplified during a blip to 148 copies/ml following recent nonadherence. This substitution, while not typically observed with nelfinavir failure, could represent a preexisting polymorphism or an uncommon early resistance mutation (23). At 3 months later, the viral load was <50 copies/ml and we were unable to detect persistent low-level viremia with our assay. In patient C40, changes at two sites, a N88S mutation at the first analysis and a V82I substitution at the second analysis, were seen in individual clones. Neither was detected at a subsequent time point. While these amino acid substitutions may represent polymorphisms, they have also been observed early in the course of nelfinavir treatment failure. Again, the detection of early drug resistance mutations did not represent impending treatment failure. Therefore, while PI-resistant variants may arise at plasma virus levels below 50 copies/ml in the presence of drug-selective pressure, drug-resistant variants do not become predominant or accumulate additional mutations when plasma virus levels are maintained below 50 copies/ml.
On the basis of the results of this study, we propose that every day a small subset of the cells within the latent reservoir become activated through an encounter with an antigen or another activating stimuli and that the virus released by these cells contributes to the low-level viremia. These viruses may undergo some additional rounds of replication. However, our results suggest that this additional replication is limited sufficiently that resistance mutations do not accumulate. This model explains why the reservoir for HIV-1 in resting CD4+ T cells does not decay with prolonged HAART even though there is continuous activation of cells in the reservoir. When additional rounds of replication are occurring, the number of latently infected cells that must be activated to fuel this replication may be such a small fraction of the reservoir that decay will not be significant even over a time scale of years (40). Using an exponential decay model (31), we calculate that the number of productively infected cells required to give rise to plasma virus levels of between 5 and 50 copies per ml in a child weighing 30 kg (estimated total extracellular fluid volume, 6 liters) ranges between 1,000 and 10,000 cells, respectively. This calculation assumes that N, the number of virions produced per productively infected cell, is 1,000 virions (21), and that the clearance rate constants c, for plasma virions, and δ, for productively infected CD4+ T cells, are 23 day−1 and 0.7 day−1, respectively (29). If all of these productively infected cells were to be generated by the activation of cells in the latent reservoir and if all of the activated cells were to die, then decay of the reservoir would be expected. Because the reservoir is stable, it is necessary to postulate that not all of the productively infected cells come from the reservoir. Our model suggests that virus released by cells reactivated from latency may infect some other cells secondarily. The stability of the reservoir could also reflect the process of proliferative renewal that maintains the memory T-cell compartment at roughly constant levels throughout life (42). In this case, proviral DNA is preserved by cell division and should show genetic stability consistent with the previous observations reported by Ruff et al. on the stability of this reservoir in children (38). It is also possible that the stability of the reservoir reflects survival of some latently infected cells after a cytokine-induced state of partial activation in which the cells become permissive for some level of virus production but do not die as quickly as antigen-stimulated cells. Indeed, in vitro studies have shown resting CD4+ T cells can be partially activated by cytokine signals to result in HIV-1 production (44). Infected cells may be more likely to persist during antiretroviral treatment due to waning cytotoxic T lymphocyte responses (2). Finally, if the burst size is significantly greater (for example, 50,000), then the activation of fewer than 200 latently infected cells per day would give rise to a plasma virus level of 50 copies/ml. At this rate of activation, only limited decay of the latent reservoir would be expected. Thus, there are several possible mechanisms that would allow the pool of latently infected cells to contribute to ongoing viremia without showing substantial decay.
In summary, the data presented above provide evidence that during effective HAART, HIV-1 infection is dynamic, with the persistent production of virus that is sensitive to the PIs and that is related to latent HIV-1 in resting CD4+ T cells. This dynamic model of HIV-1 infection during suppressive HAART is consistent with recent studies of the suppressed state in HIV-1-infected adults (8, 28). Understanding the contribution of viral reservoirs to the fueling of virus replication during effective HAART in children is important for targeting future therapeutic strategies for HIV-1 infection. Furthermore, knowledge of the clinical implications of low levels of plasma virus during HAART is of paramount importance as more sensitive plasma HIV-1 RNA assays are incorporated into care and are used to guide therapeutic decisions.
Acknowledgments
Funding and support: this work was supported by an Elizabeth Glaser Pediatric AIDS Foundation Award (D. Persaud), Doris Duke Charitable Foundation Awards (D. Persaud and R. F. Siliciano), and NIH grants AI43222 and AI51178 (R. F. Siliciano).
REFERENCES
- 1.Blankson, J. N., D. Persaud, and R. F. Siliciano. 2002. The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 53:557-593. [DOI] [PubMed] [Google Scholar]
- 2.Bucy, R. P. 1999. Immune clearance of HIV type 1 replication-active cells: a model of two patterns of steady state HIV infection. AIDS Res. Hum. Retrovir. 15:223-227. [DOI] [PubMed] [Google Scholar]
- 3.Cavert, W., D. W. Notermans, K. Staskus, S. W. Wietgrefe, M. Zupancic, K. Gebhard, K. Henry, Z. Q. Zhang, R. Mills, H. McDade, C. M. Schuwirth, J. Goudsmit, S. A. Danner, and A. T. Haase. 1997. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science 276:960-964. [DOI] [PubMed] [Google Scholar]
- 4.Chun, T. W., R. T. Davey, Jr., M. Ostrowski, J. J. Shawn, D. Engel, J. I. Mullins, and A. S. Fauci. 2000. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 6:757-761. [DOI] [PubMed] [Google Scholar]
- 5.Chun, T. W., D. Finzi, J. Margolick, K. Chadwick, D. Schwartz, and R. F. Siliciano. 1995. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1:1284-1290. [DOI] [PubMed] [Google Scholar]
- 6.Chun, T. W., L. Stuyver, S. B. Mizell, L. A. Ehler, J. A. Mican, M. Baseler, A. L. Lloyd, M. A. Nowak, and A. S. Fauci. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 94:13193-13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deeks, S. G., T. Wrin, T. Liegler, R. Hoh, M. Hayden, J. D. Barbour, N. S. Hellmann, C. J. Petropoulos, J. M. McCune, M. K. Hellerstein, and R. M. Grant. 2001. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N. Engl. J. Med. 344:472-480. [DOI] [PubMed] [Google Scholar]
- 8.Dornadula, G., H. Zhang, B. VanUitert, J. Stern, L. Livornese, Jr., M. J. Ingerman, J. Witek, R. J. Kedanis, J. Natkin, J. DeSimone, and R. J. Pomerantz. 1999. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 282:1627-1632. [DOI] [PubMed] [Google Scholar]
- 9.Dybul, M., M. Daucher, M. A. Jensen, C. W. Hallahan, T. W. Chun, M. Belson, B. Hidalgo, D. C. Nickle, C. Yoder, J. A. Metcalf, R. T. Davey, L. Ehler, D. Kress-Rock, E. Nies-Kraske, S. Liu, J. I. Mullins, and A. S. Fauci. 2003. Genetic characterization of rebounding human immunodeficiency virus type 1 in plasma during multiple interruptions of highly active antiretroviral therapy. J. Virol. 77:3229-3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783-791. [DOI] [PubMed] [Google Scholar]
- 11.Finzi, D., J. Blankson, J. D. Siliciano, J. B. Margolick, K. Chadwick, T. Pierson, K. Smith, J. Lisziewicz, F. Lori, C. Flexner, T. C. Quinn, R. E. Chaisson, E. Rosenberg, B. Walker, S. Gange, J. Gallant, and R. F. Siliciano. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5:512-517. [DOI] [PubMed] [Google Scholar]
- 12.Finzi, D., M. Hermankova, T. Pierson, L. M. Carruth, C. Buck, R. E. Chaisson, T. C. Quinn, K. Chadwick, J. Margolick, R. Brookmeyer, J. Gallant, M. Markowitz, D. D. Ho, D. D. Richman, and R. F. Siliciano. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295-1300. [DOI] [PubMed] [Google Scholar]
- 13.Frenkel, L. M., Y. Wang, G. H. Learn, J. L. McKernan, G. M. Ellis, K. M. Mohan, S. E. Holte, S. M. De Vange, D. M. Pawluk, A. J. Melvin, P. F. Lewis, L. M. Heath, I. A. Beck, M. Mahalanabis, W. E. Naugler, N. H. Tobin, and J. I. Mullins. 2003. Multiple viral genetic analyses detect low-level human immunodeficiency virus type 1 replication during effective highly active antiretroviral therapy. J. Virol. 77:5721-5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grossman, Z., M. Feinberg, V. Kuznetsov, D. Dimitrov, and W. Paul. 1998. HIV infection: how effective is drug combination treatment? Immunol. Today 19:528-532. [DOI] [PubMed] [Google Scholar]
- 15.Gulick, R. M., J. W. Mellors, D. Havlir, J. J. Eron, C. Gonzalez, D. McMahon, D. D. Richman, F. T. Valentine, L. Jonas, A. Meibohm, E. A. Emini, and J. A. Chodakewitz. 1997. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 337:734-739. [DOI] [PubMed] [Google Scholar]
- 16.Gunthard, H. F., S. D. Frost, A. J. Leigh-Brown, C. C. Ignacio, K. Kee, A. S. Perelson, C. A. Spina, D. V. Havlir, M. Hezareh, D. J. Looney, D. D. Richman, and J. K. Wong. 1999. Evolution of envelope sequences of human immunodeficiency virus type 1 in cellular reservoirs in the setting of potent antiviral therapy. J. Virol. 73:9404-9412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammer, S. M., K. E. Squires, M. D. Hughes, J. M. Grimes, L. M. Demeter, J. S. Currier, J. J. Eron, Jr., J. E. Feinberg, H. H. Balfour, Jr., L. R. Deyton, J. A. Chodakewitz, M. A. Fischl, and the AIDS Clinical Trials Group 320 Study Team. 1997. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. N. Engl. J. Med. 337:725-733. [DOI] [PubMed] [Google Scholar]
- 18.Hammond, J., C. Calef, B. Larder, R. F. Schinazi, and J. W. Mellors. 1999. Mutations in retroviral genes associated with drug resistance, p. 542-591. In C. Kuken, B. Foley, B. Hahn, and M. Preston (ed.), Human retroviruses and AIDS. Theoretical biology and biophysics. Los Alamos National Laboratory, Los Alamos, N.M.
- 19.Havlir, D. V., N. S. Hellmann, C. J. Petropoulos, J. M. Whitcomb, A. C. Collier, M. S. Hirsch, P. Tebas, J. P. Sommadossi, and D. D. Richman. 2000. Drug susceptibility in HIV infection after viral rebound in patients receiving indinavir-containing regimens. JAMA 283:229-234. [DOI] [PubMed] [Google Scholar]
- 20.Hermankova, M., S. C. Ray, C. Ruff, M. Powell-Davis, R. Ingersoll, R. T. D'Aquila, T. C. Quinn, J. D. Siliciano, R. F. Siliciano, and D. Persaud. 2001. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. JAMA 286:196-207. [DOI] [PubMed] [Google Scholar]
- 21.Hockett, R. D., J. M. Kilby, C. A. Derdeyn, M. S. Saag, M. Sillers, K. Squires, S. Chiz, M. A. Nowak, G. M. Shaw, and R. P. Bucy. 1999. Constant mean viral copy number per infected cell in tissues regardless of high, low, or undetectable plasma HIV RNA. J. Exp. Med. 189:1545-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imamichi, H., K. A. Crandall, V. Natarajan, M. K. Jiang, R. L. Dewar, S. Berg, A. Gaddam, M. Bosche, J. A. Metcalf, R. T. Davey, Jr., and H. C. Lane. 2001. Human immunodeficiency virus type 1 quasi species that rebound after discontinuation of highly active antiretroviral therapy are similar to the viral quasi species present before initiation of therapy. J. Infect. Dis. 183:36-50. [DOI] [PubMed] [Google Scholar]
- 23.King, R. W., D. L. Winslow, S. Garber, H. T. Scarnati, L. Bachelor, S. Stack, and M. J. Otto. 1995. Identification of a clinical isolate of HIV-1 with an isoleucine at position 82 of the protease which retains susceptibility to protease inhibitors. Antivir. Res. 28:13-24. [DOI] [PubMed] [Google Scholar]
- 24.Learn, G. H., Jr., B. T. Korber, B. Foley, B. H. Hahn, S. M. Wolinsky, and J. I. Mullins. 1996. Maintaining the integrity of human immunodeficiency virus sequence databases. J. Virol. 70:5720-5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, S. L., A. G. Rodrigo, R. Shankarappa, G. H. Learn, L. Hsu, O. Davidov, L. P. Zhao, and J. I. Mullins. 1996. HIV quasispecies and resampling. Science 273:415-416. [DOI] [PubMed] [Google Scholar]
- 26.Luzuriaga, K., M. McManus, M. Catalina, S. Mayack, M. Sharkey, M. Stevenson, and J. L. Sullivan. 2000. Early therapy of vertical human immunodeficiency virus type 1 (HIV-1) infection: control of viral replication and absence of persistent HIV-1-specific immune responses. J. Virol. 74:6984-6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luzuriaga, K., H. Wu, M. McManus, P. Britto, W. Borkowsky, S. Burchett, B. Smith, L. Mofenson, and J. L. Sullivan. 1999. Dynamics of human immunodeficiency virus type 1 replication in vertically infected infants. J. Virol. 73:362-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malderelli, F., A. Wiegand, S. Palmer, M. Kearney, V. Boltz, M. Polis, J. Falloon, J. Mican, R. Davey, D. Rock, S. Liu, A. Planta, Metcalf, J. Mellors, and J. Coffin. 2003. Persistence of stable quantifiable viremia in patients on antiretroviral therapy despite suppression of plasma HIV-1 RNA to less than 50 copies/ml. 10th Conf. Retrovir. Opport. Infect. 2003:466. [Google Scholar]
- 29.Markowitz, M., M. Louie, A. Hurley, E. Sun, M. Di Mascio, A. S. Perelson, and D. D. Ho. 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J. Virol. 77:5037-5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moyle, G. J., and D. Back. 2001. Principles and practice of HIV-protease inhibitor pharmacoenhancement. HIV Med. 2:105-113. [DOI] [PubMed] [Google Scholar]
- 31.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188-191. [DOI] [PubMed] [Google Scholar]
- 32.Persaud, D., T. Pierson, C. Ruff, D. Finzi, K. R. Chadwick, J. B. Margolick, A. Ruff, N. Hutton, S. Ray, and R. F. Siliciano. 2000. A stable latent reservoir for HIV-1 in resting CD4+ T lymphocytes in infected children. J. Clin. Investig. 105:995-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Persaud, D., Y. Zhou, J. M. Siliciano, and R. F. Siliciano. 2003. Latency in human immunodeficiency virus type 1 infection: no easy answers. J. Virol. 77:1659-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierson, T., J. McArthur, and R. F. Siliciano. 2000. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 18:665-708. [DOI] [PubMed] [Google Scholar]
- 35.Posada, D., and K. A. Crandall. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817-818. [DOI] [PubMed] [Google Scholar]
- 36.Rouzine, I. M., and J. M. Coffin. 1999. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc. Natl. Acad. Sci. USA 96:10758-10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rouzine, I. M., A. Rodrigo, and J. M. Coffin. 2001. Transition between stochastic evolution and deterministic evolution in the presence of selection: general theory and application to virology. Microbiol. Mol. Biol. Rev. 65:151-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruff, C. T., S. C. Ray, P. Kwon, R. Zinn, A. Pendleton, N. Hutton, R. Ashworth, S. Gange, T. C. Quinn, R. F. Siliciano, and D. Persaud. 2002. Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure. J. Virol. 76:9481-9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406-425. [DOI] [PubMed] [Google Scholar]
- 40.Siliciano, J. D., J. Kajdas, D. Finzi, T. C. Quinn, K. Chadwick, J. B. Margolick, C. Kovacs, S. J. Gange, and R. F. Siliciano. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9:727-728. [DOI] [PubMed] [Google Scholar]
- 41.Smith, D. B., J. McAllister, C. Casino, and P. Simmonds. 1997. Virus “quasispecies”: making a mountain out of a molehill? J. Gen. Virol. 78:1511-1519. [DOI] [PubMed] [Google Scholar]
- 42.Sprent, J., and C. D. Surh. 2002. T cell memory. Annu. Rev. Immunol. 20:551-579. [DOI] [PubMed] [Google Scholar]
- 43.Swofford, D. L. 2000. PAUP*, phylogenetic analysis using parsimony (*and other methods). Version 4.0b4a. Sinauer, Sunderland, Mass.
- 44.Unutmaz, D., V. N. KewalRamani, S. Marmon, and D. R. Littman. 1999. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J. Exp. Med. 189:1735-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong, J. K., M. Hezareh, H. F. Gunthard, D. V. Havlir, C. C. Ignacio, C. A. Spina, and D. D. Richman. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291-1295. [DOI] [PubMed] [Google Scholar]
- 46.Zhang, L., C. Chung, B. S. Hu, T. He, Y. Guo, A. J. Kim, E. Skulsky, X. Jin, A. Hurley, B. Ramratnam, M. Markowitz, and D. D. Ho. 2000. Genetic characterization of rebounding HIV-1 after cessation of highly active antiretroviral therapy. J. Clin. Investig. 106:839-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, L., B. Ramratnam, K. Tenner-Racz, Y. He, M. Vesanen, S. Lewin, A. Talal, P. Racz, A. S. Perelson, B. T. Korber, M. Markowitz, and D. D. Ho. 1999. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 340:1605-1613. [DOI] [PubMed] [Google Scholar]