

Abstract
Background
Intratumoral heterogeneity in glioblastoma multiforme (GBM) poses a significant barrier to therapy in certain subpopulation such as the tumor-initiating cell population, being shown to be refractory to conventional therapies. Oncolytic virotherapy has the potential to target multiple compartments within the tumor and thus circumvent some of the barriers facing conventional therapies. In this study, we investigate the oncolytic potential of myxoma virus (MYXV) alone and in combination with rapamycin in vitro and in vivo using human brain tumor–initiating cells (BTICs).
Methods
We cultured fresh GBM specimens as neurospheres and assayed their growth characteristics in vivo. We then tested the susceptibility of BTICs to MYXV infection with or without rapamycin in vitro and assessed viral biodistribution/survival in vivo in orthotopic xenografts.
Results
The cultured neurospheres were found to retain stem cell markers in vivo, and they closely resembled human infiltrative GBM. In this study we determined that (i) all patient-derived BTICs tested, including those resistant to temozolomide, were susceptible to MYXV replication and killing in vitro; (ii) MYXV replicated within BTICs in vivo, and intratumoral administration of MYXV significantly prolonged survival of BTIC-bearing mice; (iii) combination therapy with MYXV and rapamycin improved antitumor activity, even in mice bearing “advanced” BTIC tumors; (iv) MYXV treatment decreased expression of stem cell markers in vitro and in vivo.
Conclusions
Our study suggests that MYXV in combination with rapamycin infects and kills both the BTICs and the differentiated compartments of GBM and may be an effective treatment even in TMZ-resistant patients.
Keywords: brain tumor initiating cells, malignant glioma, myxoma virus, oncolytic virus, rapamycin
Although temozolomide (TMZ) improves the survival of patients with glioblastoma multiforme (GBM), TMZ resistance is a significant obstacle, and new treatments aimed at overcoming TMZ-resistant cell populations are needed. Recent research has focused on targeted molecular strategies using small-molecule inhibitors; however, despite promising preclinical successes, the results in the clinic are disappointing. Resistance is mediated by various mechanisms, including pathway cross-talk, low tissue concentrations, and selection for resistant mutations and/or resistant subpopulations.1,2
Oncolytic viruses (OVs), alone or in combination with small-molecule inhibitors, offer a promising alternative, as they circumvent several resistance mechanisms to targeted therapies. Several OVs have shown potential in malignant gliomas (MGs) preclinically and in clinical trials.3,4 These trials ubiquitously found that OVs are safe and have limited toxicities, but only a handful of clinical responses were seen. There may be several reasons why OVs have not been more effective clinically, including robust antiviral immune responses, limited intratumoral distribution, and endogenous subpopulations with resistance to OVs.
One subpopulation of interest is the putative cancer stem cell population, which has been isolated in a variety of human cancers,5–10 including MGs.11,12 These populations are both chemo- and radioresistant13–15 and may act as treatment-resistant “disease reservoirs.”16–19 Cells derived from gliomas that are cultured under neurosphere conditions and retain stem cell–like properties are referred to here as brain tumor–initiating cells (BTICs). Patient-derived BTICs express stem cell markers, self-renew, and have multilineage differentiation.16–20 We consider these cultures to be more appropriate than conventional cell lines for preclinical testing, as they accurately recapitulate the original tumor expression patterns, mutational status, and glioma phenotypes in vivo.17,21
We have previously found that myxoma virus (MYXV) is highly efficacious in conventional preclinical brain tumor xenograft models22,23 as well as in syngeneic GBM rat models.24 However, given the potential importance of BTIC cultures in modeling human disease and treatment resistance, we considered it important to assess MYXV in BTIC models. Previously, we found that combination therapy with rapamycin increased MYXV replication and overall treatment efficacy.23,24 It was also an attractive combination therapy because rapamycin and other mammalian targets of rapamycin (mTOR) inhibitors have been tested in GBM clinical trials.25–27 We and others have previously shown that rapamycin enhances MYXV and other OV infections through Akt activation23,24,27,28 and/or inhibition of type I interferon (IFN) responses.24,29
We consider MYXV an excellent OV candidate for several reasons: (i) it is a lagomorph poxvirus that is nonpathogenic for all other vertebrate species tested (including mice and humans) and has a track record for safety in humans; (ii) it has a large double-stranded DNA genome that can be genetically manipulated to hold large (>25 kB) therapeutically relevant genes; (iii) there is a lack of any acquired immunity (ie, preexisting antibodies) to MYXV in human populations.
Here we investigated the oncolytic potential of MYXV against BTICs, alone and in combination with rapamycin, in vitro and in vivo. Although several groups have investigated OV therapy in BTIC models,30–33 MYXV has only been tested in conventional cell lines. We show for the first time that BTICs are susceptible to MYXV in vitro and in vivo, including TMZ-resistant lines. We also report that combining MYXV with the mTOR inhibitor rapamycin further improves the in vivo efficacy of MYXV in half of the BTIC lines tested. This study demonstrates that MYXV with rapamycin is a potentially useful combination therapy in MG patients.
Materials and Methods
Isolation and Culture of BTICs From Clinical Brain Tumor Samples
Tumors were obtained from the Tumor Tissue Bank at Foothills Hospital, Calgary, Alberta, Canada. GBM surgical samples were obtained and transported to the laboratory. BTICs were cultured under neural stem cell promoting conditions as described previously.20,34,35 This study had institutional review board approval; it was under the Brain Tumor and Related Tissue Bank Protocol V2 and was approved by Foothills Hospital and the Conjoint Health Research Ethics Board. All brain tumor samples were obtained by informed written consent from patients. Briefly, tumor tissue was washed with phosphate buffered saline (PBS), then minced (0.5–1 mm3) and incubated for 5–10 min at 37°C in an enzyme cocktail of trypsin (0.25%) and DNase I (10 μg/mL) in PBS. Red blood cells were removed, then the tissue was strained through a 100-μm mesh and a 40-μm mesh and collected by centrifugation at 1500 rpm for 5 min. Cells were resuspended in stem cell media (NS-A Basal Medium plus NeuroCult NS-A Proliferation Supplements [Stem Cell Technologies] plus epidermal growth factor [20 ng/mL; Sigma] and basic fibroblast growth factor [20 ng/mL; Chemicon]). Brain tumor cell cultures were fed weekly and spheres were evident as early as 4–5 days following plating.
Generation of BTICs Stably Expressing Firefly Luciferase and Enhanced Green Fluorescent Protein
Stable BTIC cell lines expressing enhanced firefly luciferase (effLuc)36 and enhanced green fluorescent protein (eGFP) were generated using a self-inactivating lentiviral vector encoding the internal U3 region from murine stem cell virus, effLuc, the internal ribosomal entry site element from encephalomyocarditis virus, and eGFP.36 Virus was packaged in 293FT cells (provided by Dr Rabinovich, The University of Texas MD Anderson Cancer Center) using pMD2.G (VSV.G env) and pCMV–deltaR8.91 and concentrated 50× using Amicon Ultra-15 100 000 nominal molecular weight limit centrifugal concentration units (Millipore). Concentrated viral supernatants were used to transduce BTIC cell lines via spinfection for 2 h at 2200 rpm/30°C (approximate multiplicity of infection [MOI] = 10). After 72 h, eGFP expression was observed via fluorescent microscopy (Zeiss inverted microscope, Axiovert 200M) and used to calculate transduction efficiency by flow cytometry in fluorescence activated cell sorting (FACSCalibur; BD Biosciences). Bioluminescent activity based on effLuc was calculated using an IVIS 200 (in vivo imaging system; Caliper Life Sciences).
Viruses and the Oncolytic Assays In vitro
Viruses (MYXV, vMyx-Lac, and vMyx-MT5KO) were propagated and titrated by focus formation on baby green monkey kidney cells.37 UV-inactivated MYXV (dead virus [DV]) was prepared by irradiating virus with UV light for 2 h. Recombinant MYXV expressing firefly luciferase (vMyx-Fluc) was generated in the following manner. A recombination plasmid having firefly luciferase protein (Fluc) driven by a synthetic early/late (sE/L) poxvirus promoter and tandem-dimer tomato red fluorescent protein (TrFP) driven by poxvirus p11 late promoter was constructed using Gateway technology (Invitrogen). The plasmid cassette was inserted between the M135 and M136 gene locus of MYXV by homologous recombination. Three independent plasmids were used to make the final recombination plasmid. The sequence encoding Fluc was PCR amplified using gene-specific primers: 5′-GGGGGCACCATGGAAGACGCCAAAAACATAAAG-3′ (NcoI site italicized) and 5′-CCGCGCGCCCGGGTTACAATTTGGACTTTCCGCCCTTC-3′ (SmaI site italicized) and cloned into the plasmid pDONR221-P3P2-sE/L-GFP-M136 to replace the GFP cassette under sE/L promoter. The construction of the plasmid having TrFP and another plasmid having MYXV sequences corresponding to M135 and the partial sequence of M134 was described before.38 All 3 plasmids, along with the pcDNA3.2/capTEV-CT/V5-DEST_verA destination plasmid (Invitrogen), were then subjected to an LR recombination reaction using LR clonase II (Invitrogen) to generate the final plasmid construct. The MYXV-Fluc virus was created using the method described before for making other recombinant MYXV.38 The resultant virus produced red-fluorescent foci and was purified following 4 rounds of plaque purification.
To detect viability of BTICs following MYXV infection in vitro, BT012, BT025, BT042, and BT048 were plated in 96-well plates, then infected with different doses of MYXV (MOI = 0, 1, and 10). Cell viability was measured at 144 h post-infection by Alamar Blue (Invitrogen) assay as described by the manufacturer. The permissive cell line U87 and nonpermissive NIH 3T3 were used as positive and negative controls, respectively.
Viral replication was assessed by early (GFP) and late gene expression (β-galactosidase activity) after infection.22–24,37 Additionally, viral titers were performed by seeding BTICs at 5 × 104 cells/well in 6-well plates, then the cells were treated with MYXV (MOI = 1) for 2 h at 37°C. Cells were collected and washed and then recultured in fresh media until the designated time-points. Titers were calculated on Buffalo green monkey kidney cells as previously described.22–24,37
BTIC self-renewal (secondary sphere-forming) assays were performed by infecting BTICs with MYXV (MOI = 1, 3) or controls. Five days after treatment, spheres were dissociated into single-cell suspensions and viable cells were counted. Ten or 100 cells were then placed into 96-well plates and cultured for another 10 days, and secondary sphere formation was counted.
Combination Therapy: MYXV With Rapamycin In vitro
BTICs were pretreated with 100 nM rapamycin (LC Laboratories) for 2 h, then infected with MYXV (MOI = 1 for BT012, -25, and -48; MOI = 3 for BT042). Cell viability, viral replication, and self-renewal were performed as described.
Western Blotting and Immunofluorescence
Early and late gene expression was detected by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using early (MT7) and late (Serp-1) MYXV proteins as described previously.22–24 For stem cell markers, cells were harvested 24 h following treatment with rapamyin (100 nM), MYXV (5 MOI), or a combination of both (pretreatment with rapamycin for 2 h). Western blots were performed using 40–50 μg of total protein run on SDS-PAGE. Immunoflourescence was performed by spinning spheres onto slides with Cytofunnels (Thermoscientific A78710005). Western blot (WB) and immunofluorescence (IF) antibodies were Sox2 (1:1000 WB, 1:200 IF; Millipore AB5603), NANOG (1:1000 WB; Cell Signaling 3580), TuJ1 (1:1000 WB, 1:200 IF; Sigma T2200), and phospho-P70 (1:1000 WB; Cell Signaling 9204), detected using a goat anti-rabbit horseradish peroxidase (1:10 000; Pierce 31460) or goat anti-rabbit red fluorescent protein (1:200; Jackson 115.012.003). Musashi-1 (1 μg/mL WB; R&D MAB2628), nestin (1:1000 WB, 1:200 IF; Millipore MAB5326), and glial fibrillary acidic protein (1:1000 WB, 1:200 IF; Millipore MAB360) were detected using goat anti-mouse horseradish peroxidase (1:5000; Pierce 31430) or goat anti-mouse red fluorescent protein (1:200; Jackson 115.012.003).
Interferon Studies
Semiquantitative reverse transcriptase PCR was carried out on BTIC lines and the human foreskin fibroblast line HS68 (provided by Dr Karl Riabowol, University of Calgary, Canada). Cells were treated with 100 U/mL IFN-β (PBL Interferon Source), 25 μg/mL polyinosinic:polycytidylic acid (polyI:C) (high molecular weight; Invitrogen), DV (UV-inactivated MYXV), or 1.0 MOI of either MYXV or delta51–vesicular stomatitis virus (VSV). Cells and supernatants were harvested 24 h after treatment. Collected cells had RNA purified (Qiagen RNeasy Plus) and cDNA made (SuperScriptII, Invitrogen), and PCR amplification was done of specific IFN and IFN-responsive genes. Supernatants were collected, then 100 μL was added to HEK-Blue cell cultures (Invitrogen), and IFN production was measured per manufacturer's protocol.
Animal Experiments and Characterization of BTICs In vivo
Six to eight week-old female Severe Combined Immunodeficient (SCID) mice (CB17) from Charles River Laboratory were used in this study.
The stereotactic techniques used to implant tumor cells in the right putamen have been described in our previous work.25–27,40 BT012, BT025, BT042, and BT048 were implanted into the mouse brain to test BTIC tumor formation in vivo.22–24,41 Mice were monitored weekly, and tumor growth was monitored by MRI at the designated time-points (3 or 4 mo). Mice were anesthetized prior to MR imaging using ketamine (80 mg/kg) and xylazine (10 mg/kg) in saline, administered by i.p. injection. All protocols were reviewed and approved by the Animal Care Committee of the independent laboratory and the University of Calgary (protocol approved number M10044/NRC 10003). All animal work procedures were in accordance with the Guide to the Care and Use of Experimental Animals published by the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals issued by the National Institutes of Health. A standard imaging protocol consisting of multi-echo T2 sequences (repetition time = 5 s with 16 echos of 10 ms each) was performed. Imaging was performed at the University of Calgary Experimental Imaging Centre using a 9.4T Bruker horizontal bore MR system. Images were reviewed using Marevisi image software.20 Mice were sacrificed for confirmation of tumor growth, and brain sections were stained with hematoxylin and eosin. Immunohistochemistry of paraffin sections was performed using monoclonal antibodies against nestin, Sox2, Musashi (5 μg/mL; all purchased from R&D), anti-CD133 (1:50; Miltenyi), and anti-human nuclei (1:100; R&D). Primaries were kept at 4°C overnight and then detected using goat anti-mouse secondary antibody (Cedarlane). Slides were mounted, counterstained, and viewed with a Zeiss inverted microscope (Axiovert 200M) and a Carl Zeiss camera (AxioCam MRc).
CD133 expression by flow cytometry
Tumor cells from BTIC-bearing mice were recultured under neural stem cell–promoting conditions as described above. Recultured BTIC cells were dissociated into single cells and washed, resuspended in PBS 0.5% bovine serum albumin (PBS-BSA), then incubated with CD133/PE2-conjugated antibody or a mouse immunoglobulin G1 isotype control antibody (both 1:10, according to the manufacturer's instructions; Miltenyi Biotec) for 60 min at 4°C. Cells were then washed with 2 mL PBS-BSA, centrifuged at 800 rpm for 5 min, resuspended in 0.5 mL PBS-BSA, and analyzed by flow cytometry.20
Real-Time Monitoring of Virus Replication in Mice Bearing Intracranial BTIC Tumors
The experiments we have described here established BTIC models to assess real-time virus replication in vivo using vMyx-Fluc alone (intratumoral 2.4×106 focus forming units [FFUs]/animal at 3 wk after tumor administered) or vMyx-Fluc plus rapamycin. Rapamycin was 2.5 mg/kg/mouse i.p. daily for 2 wk started 1 day before viral administration. Animals were imaged using the Xenogen IVIS 200 system on days 0 (4 h), 3, 7, 14, 21, and 42 after viral administration. Total photon flux emission (photons/s) in the region of interest over the intracranial space was measured.24 Two mouse brains per time-point were collected for viral titers.
Efficacy Studies of MYXV With or Without Rapamycin in Mice Bearing Intracranial BTIC Tumors
Mice with intracranial, BTIC-derived tumors were injected with 5 × 106 FFUs of MYXV (or DV) intratumorally 21 days after tumor implantation. Animals were followed for survival until they lost ≥20% of body weight or had trouble ambulating, feeding, or grooming or until we terminated the experiment.
To evaluate the in vivo effects of MYXV + rapamycin combination therapy, BTIC-bearing mice 21 days after tumor implantation were divided into 4 groups (n = 5–7 animals): (i) DV control, (ii) rapamycin (2.5 mg/kg/daily i.p. for 6 wk), (iii) MYXV (5 × 106 FFUs/mouse) 2 times administered at 3 wk and 7 wk after implantation, and (iv) MYXV + rapamycin (starting at 1 day before MYXV infection).
Combination treatments were also performed on an “advanced” or “late-stage” tumor that grew for 60 or 80 days prior to treatment. Tumor size was monitored with MRI on days 80, 130, and 160 after tumor implantation. Two mice per group were sacrificed for histological analysis.
For monitoring tumor growth in vivo in real time, we labeled BT025 with eGFP and effLuc and imaged with the Xenogen system (IVIS 200). Mice were treated with DV, rapamycin (2.5 mg/kg/daily i.p. for 4 wk), MYXV (5 × 106 FFUs/mouse on days 21 and 42 after tumor implantation), or MYXV + rapamycin (4–7 mice per group).
Statistical Analysis
Statistical Analysis Software and GraphPad Prism version 4 were used for statistical analyses. Survival curves were generated by the Kaplan–Meier method. The average latency data were analyzed with the ANOVA 1- or 2-way test. All reported P-values were 2-sided and were considered to be statistically significant at P < .05.
Results
BTICs Are Susceptible to MYXV Infection and Killing In vitro Independently of TMZ Sensitivity
We first confirmed sensitivity of a panel of genetically distinct BTICs to TMZ.34 We found that BT012, BT025, and BT042 were resistant, while BT048 was sensitive to TMZ at the lowest doses (<10 μM), as we have shown previously, but all except BT042 became susceptible at higher doses of 100 and 200 μM (Supplementary material, Fig. S1A), doses used in the literature for conventional glioma cell lines42–44 (Supplementary material, Fig. S1B). We then evaluated the susceptibility of BTICs to infection with MYXV using several methods. All 4 BTICs were permissive to infection independently of TMZ sensitivity (Fig. 1A), as demonstrated by expression of both early (GFP from vMyx-GFP) and late (β-galactosidase from vMyx-Lac) viral genes. We found that progeny viral titers varied among BTICs and mirrored their susceptibility to initial infection (Fig. 1B). The titers from BT012 and BT025 were similar to U87 (positive control), and those from BT042 and BT048 were less than U87 (the NIH 3T3 cell line is a poorly permissive negative control22,40; Fig. 1B). To confirm a specific MYXV infection, expression of early (MT7) and late (Serp-1) viral proteins was detected by Western blotting (Fig. 1C).
Fig. 1.


Effects of MYXV on human BTICs in vitro. (A) All BTICs were permissive to infection, as shown by expression of both early (GFP) and late (β-galactosidase, X-gal staining) viral genes 96 h after infection with vMyxGFP or vMyx-Lac (MOI = 1). (B) The viral replication assay (72 h post-infection [p.i.]) showed that viral titers varied among different BTICs and mirrored their susceptibility to viral infection. (C) Western blots 72 h after MYXV infection (MOI = 5). Both early (MT7, 35 kDa) and late (Serp-1, 55 kDa) viral gene expression was seen in all BTICs; the least susceptible NIH 3T3 had the lowest levels. (D) Alamar Blue assay comparing the effects of MYXV on viability (MOI = 0, 1, and 10, 144 h p.i.). NIH 3T3 is shown as a negative control. (E) Self-renewal assay shows that MYXV inhibited secondary neurosphere formation in all BTICs 120 h p.i. *P < .05, **P < .001 for treatment compared with control by Student's t-test.
To determine whether MYXV infection caused BTIC cytotoxicity, we utilized the Alamar Blue viability assay at 144 h after infection with MYXV (Fig. 1D). We found that 3 of 4 BTICs underwent extensive cell death, while BT042 underwent the least amount of cytotoxicity in vitro. We also found that BTIC self-renewal was significantly inhibited following infection with MYXV (Fig. 1E). Both cytotoxicity and self-renewal were independent of temozolomide sensitivity. Interestingly, despite similar titers, at shorter times post-infection (72 h), all BTICs underwent less cell death than the conventional cell line U87 (Supplementary material, Fig. S1C).
The mTOR Inhibitor Rapamycin Enhances MYXV Replication and Killing of BTICs In vitro
We found that pretreatment with rapamycin significantly enhanced cell killing in the BT025 and BT042 lines (P < .05) but not in BT012 and BT048 (Fig. 2A). We also found significant inhibition of self-renewal in the BT012, BT025, and BT042 lines (P < .05) but not in BT048 (Fig. 2B). We next assessed whether combination therapy increased viral replication; we found that MYXV titers were significantly increased with rapamycin pretreatment in BT025 and BT042 (P < .01) and trended upward in the BT012 and BT048 lines (Fig. 2C). Further, to assess the TMZ-sensitizing effect of our monotherapy or combination treatment, we added 100 μM TMZ to the combination treatment in vitro. Interestingly, at the higher dose of 100 μM, we did see an additive effect of TMZ with MYXV (Supplementary material, Fig. S1D). Further, only 2 of the cell lines, BT042 and BT048, achieved a benefit from the triple therapy of MYXV, rapamycin, and TMZ. This is especially interesting because BT042 was the most resistant to TMZ at this dose. This combination benefit may be of significant interest given the use of TMZ in the clinic and problems with TMZ resistance.
Fig. 2.

The mTOR inhibitor rapamycin promotes infection and killing by MYXV in half of the BTICs in vitro. (A) Cell viability assay (Alamar Blue) treated with MYXV (MOI = 1) + rapamycin (100 nM) in BTICs (5 days p.i). (B) Self-renewal of BTIC lines after pretreatment with rapamycin (100 nM) followed by MYXV infection. BTICs were infected with MYXV (MOI = 1); 5 days after infection they were replated and then after 10 days the spheres were counted. *P < .05 compared by Student's t-test with the MYXV alone. (C) Viral titers were obtained in BTIC lines after MYXV (MOI = 1) + rapamycin (100 nM) at 72 h p.i. Values represent mean FFUs ± SD from triplicate wells. **P < .01 compared by Student's t-test with the MYXV alone.
MYXV Replicated in the Tumors of BTIC-Bearing Mice Was Enhanced With Rapamycin
We found that cultured BTICs form infiltrative intracranial tumors by MRI and histology in representative lines BT025 and BT042 (Supplementary material, Fig. S2)20 and that these tumors express neural stem cell markers (CD133, nestin, Musashi-1, Sox2) in vivo (Table 1). We treated BT025- or BT048-bearing mice with a luciferase tagged MYXV (MYXV-Fluc) and monitored viral replication in vivo in real time using the Xenogen system. We chose these lines because of their differential responses to rapamycin in vitro (Fig. 2A–C). We found that the MYXV-Fluc virus replicated in both BT025 and BT048 in vivo and reached very high levels at 3 days and 3 weeks after viral administration (Fig. 3A–D). Rapamycin increased viral replication in BT025 (1-way ANOVA, P = .0081) but not in BT048 (1-way ANOVA, P = .0625; Fig. 3B and D). These results were confirmed by viral titers from BTIC tumors resected from mice (P < .05, P < .01; Fig. 3E and F).
Table 1.
Characteristics of BTICs in vivo
| #ID | Path | Spheres | CD133+ | Nestin | Musashi-1 | Sox2 | ANA |
|---|---|---|---|---|---|---|---|
| BT012 | GBM-r (IV) | Yes | 1.25% | + | + | + | + |
| BT025 | GBM-r (IV) | Yes | 4.26% | + | + | + | + |
| BT042 | GBM (IV) | Small | 2.13% | + | + | + | + |
| BT048 | GBM (IV) | Yes | 1.46% | + | + | + | + |
Abbreviations: Path, pathology; ANA, anti-human nuclei; r, recurrent tumor; IV, World Health Organization grade for tumor.
Fig. 3.



MYXV replicated in SCID mice bearing intracranial human BTICs and rapamycin promotes virus replication in some lines in vivo. (A and B) Representative viral bioluminescence images and quantification of the virus (photons/s) in BT025 line after treatment with MYXV + rapamycin at the different times. (C and D) Representative viral bioluminescence images and quantification of the virus (photons/s) in BT048 line after treatment with MYXV + rapamycin at the different times. (E and F) Viral titers (FFUs/g tumor tissue) in BT025- and BT048-bearing mice treated with MYXV + rapamycin (*P < .05, **P < .01 compared by Student's t-test with MYXV alone).
Intratumoral MYXV Significantly Prolongs Survival in BTIC-Bearing Mice, and Combination Therapy With Rapamycin Further Improves Survival in Half of the BTIC Lines
We next sought to determine whether intratumoral MYXV would prolong survival in these orthotopic xenografts. A single intratumoral administration of MYXV at 21 days post-implantation prolonged survival in all lines tested (log-rank test, P= .0001, Fig. 4A; P = .0016, Fig. 4B; P = .0012, Fig. 4C). Median survivals for MYXV-treated animals were 127 days for BT012 (95% confidence interval [CI] = 109–185 d), 72.5 days for BT025 (95% CI = 55–102 d), and 121 days for BT048 (95% CI = 102–131 d). Control-treated animals died with shorter median survivals (BT012, 96 d; BT025, 48.5 d; and BT048, 93 d).
Fig. 4.

Intratumoral (i.t.) administration of MYXV prolongs survival of BTICs in vivo. Kaplan–Meier survivals of BTIC-bearing mice after treatment with DV or MYXV (5 × 106 FFUs/mouse, single injection) showed MV-treated mice survived longer than DV-treated controls with (A) BT012 (log-rank test, P = .0001), (B) BT025 (log-rank test, P = .0016), and (C) BT048 (log-rank test, P = .0012). Arrows indicate time of viral inoculation.
We then found that combination therapy with rapamycin improved upon the survival of MYXV treatment alone in BT025 and BT042 but not in BT012 and BT048 (Fig. 5). BT025-bearing mice treated with MYXV + rapamycin survived longer than mice treated with MYXV alone (log-rank test, P = .00895), rapamycin alone (log-rank test, P = .0183), or DV (log-rank test, P = .0002; Fig. 5A). Similarly, BT042-bearing mice (started treatment 60 days after tumor implantation to mimic more “advanced” clinical tumors) treated with combination therapy survived significantly longer compared with animals treated with rapamycin alone (log-rank test, P = .0343), MYXV alone (log-rank test, P = .0011), or DV alone (log-rank test, P = .0001; Fig. 5B).
Fig. 5.

Combination therapy with rapamycin improves survival in half of the BTIC lines in vivo. Kaplan–Meier plots of various BTICs treated with DV, rapamycin (2.5 mg/kg/daily), MYXV (2 injections of 5 × 106 FFUs/mouse), or both. (A) Survival of BT025-bearing mice after treatment. (B) Survival of BT042-bearing mice after treatment. (C) Survival of BT012-bearing mice after treatment. (D) Survival of BT048-bearing mice after treatment. Arrows indicate time of viral inoculation.
In contrast, animals bearing BT012 or BT048 did not clearly achieve any additional benefit from combination therapy with rapamycin. We found in BT012 and BT048 grafts that MYXV + rapamycin had no greater effect than rapamycin alone (log-rank test, P = .0778 and P = .6534, respectively) or MYXV alone (log-rank test, P = .0543 and P = .1608, respectively; Fig. 5C and D).
We repeated these combination therapy experiments using the Xenogen system and MRI to measure tumor growth in real time. We found that combination therapy was superior to monotherapy or the control group in BT025 GFP Fluc-bearing mice using bioluminescence imaging (Supplementary material, Fig. S3A and B). Combination therapy was also found superior in BT042-bearing mice as measured by MRI (Supplementary material, Fig. S3C, left 2 rows), histological examination (Supplementary material, Fig. S3C, right 2 rows), and tumor size quantification (Supplementary material, Fig. S3D).
MYXV Treatment Decreases Stem Cell Marker Expression in BTICs In vitro and In vivo
The current paradigm suggests that even within a BTIC culture, only a fraction of these cells are the putative cancer stem cells, while the remaining cells in the neurosphere represent partially committed/partially differentiated cells.16,17 In order to show that our therapy is effective against the potential disease reservoir, we examined the lines BT025, BT012, and BT048 for stem cell markers Sox2 and nestin and differentiation markers TuJ1 by immunofluorescence. Double staining lines BT025 and BT048 for Sox2 and nestin found that only a portion of the cells simultaneously stained Sox2 and nestin (Supplementary material, Fig S4A). TuJ1, a neuronal differentiation marker, was also seen in only a proportion of the cells, likely representing partially committed cells down the neuronal path. Importantly, MYXV was able to infect Sox2-, nestin-, and TuJ1-positive and -negative cells in all the lines tested (Fig. 6A). The ability of MYXV to infect Sox2- and nestin-positive cells suggests that the stem cell compartment is indeed susceptible to this therapy. To further examine the loss of stem cell markers with an expanded repertoire of markers, with or without combination therapy, we examined all 4 lines by Western blot. We found that loss of Sox2 and/or Musashi-1 occurred shortly after MYXV infection (Fig. 6B). The extent of this loss was directly associated with the ability of the BTIC line to produce a productive MYXV infection. The more sensitive lines BT025 and BT012 showed the greatest marker loss, while the more resistant lines BT048 and, especially, BT042 showed little loss of expression with treatment. Further, nestin was downregulated in the more sensitive lines BT025 and BT012, while NANOG did not change in response to MYXV infection alone. Interestingly, rapamycin alone resulted in little or no loss of expression, and combination therapy appeared to be similar to MYXV treatment alone in all cases. This loss of stem cell marker in response to infection could be a result of immediate destruction of the stem cell population within the sphere or due to the engagement of a differentiation program. To examine this further, we looked at MYXV infection compared with low serum treatment. Interestingly, MYXV seemed to be able to downregulate stem cell markers better than 1% serum in the line BT012, while this ability was comparable in BT025. BT048 was resistant to marker loss in response to serum and MYXV (Supplementary material, Fig. S4B). Interestingly, these results closely correlated to the ability of these to retain sphere-forming abilities following infection (Fig. 2B). However, comparing these cells visually, there was an obvious differentiation phenotype in the serum-treated cells (Supplementary material, Fig. S4C) that was lacking in the MYXV-treated cells (Fig. 1A; Supplementary material, Fig. S4C). This suggests to us that the loss of these stem cell markers represents the loss of this compartment rather than MYXV-induced differentiation.
Fig. 6.
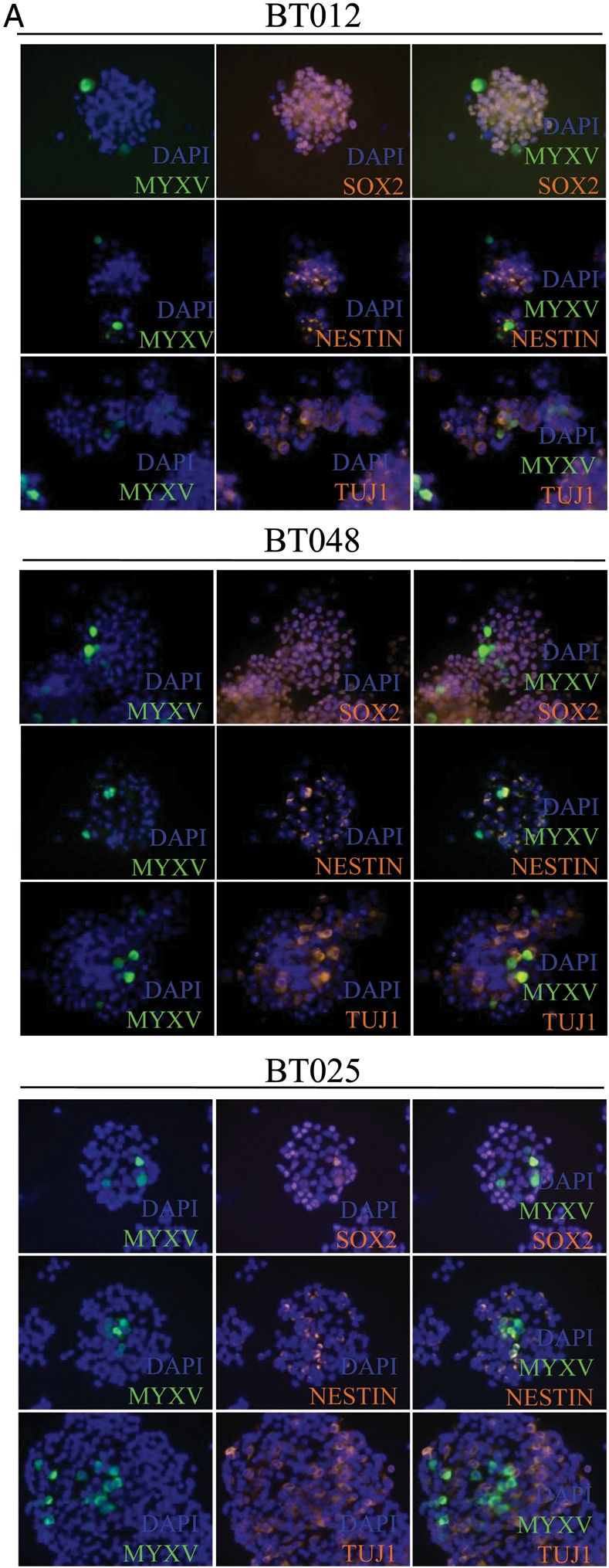

MYXV infection of BTICs resulted in loss of stem cell markers in vitro and in vivo. (A) BTIC lines BT012, -25, and -48 spheres were infected with 1 MOI of MYXV GFP and stained for nestin, Sox2, and TuJ1 24 h after infection. MYXV is able to infect both nestin- and Sox2-positive cells, demonstrating its ability to target the stem cell fraction within the sphere. (B) All BTICs lost protein expression of stem cell markers Sox2 and/or Musashi-1 (MSI) following MYXV infection with 5 MOIs. Protein collected 24 h post-infection with MYXV with (M + R) or without (M) a 2-h pretreatment with 100 nM rapamycin compared with control (CT) or rapamycin alone (R). Phospho-P70 (pP70) blots demonstrate that the concentration of rapamycin we used was sufficient to inhibit mTOR activity. (C) BT025 was implanted in SCID mice and treated with MYXV, rapamycin, or the combination therapy starting 5 wk post-implantation for a total of 2 wk. Tumors were resected and flow cytometery performed looking at human CD133 cell surface expression. Figures are a result of a pool of 5 animals per group, and percent positive results reflect a subtraction of the isotype control for each group.
Next we looked to see whether stem cell markers were lost in vivo following therapy. We chose to interrogate the BT025 line, as this showed the most significant survival improvement following combination therapy. We harvested 7 week BT025 tumors treated with rapamycin, MYXV, or the combination therapy for 2 weeks. Tumors were dissociated and stained with CD133 for measurement by flow cytometry. We found that the BT025 tumors expressed 5.0% CD133+ cells in vivo, with this number decreasing to 1.4% and 0.7% after rapamycin or MYXV therapy, and 0.3% after combination therapy (Fig. 6C). These data demonstrate that all of our treatments decreased the CD133+ fraction of these tumors in vivo and suggests that the MYXV and combination therapies are most efficacious.
The mTOR Inhibitor Rapamycin Promotes Infection of Half of the Tested Human BTICs
We next wanted to determine the mechanism of enhancement of MYXV infection by rapamycin in BTICs. We previously found in established rat MG cell lines that rapamycin inhibited type-I IFN expression.24 In contrast, here BTICs did not produce IFN in response to MYXV infection either at the level of message (Fig. 7A) or at that of secreted protein (Fig. 7B). Interestingly, BT012, BT025, and BT048 produced IFN in response to VSV or polyI:C at the transcriptional level, but BT042 failed to produce any functional type-I IFN (Fig. 7A). Therefore, rapamycin did not enhance infection in vitro by inhibiting IFN transcription or translation in BTICs as has been previously described.24,29 Interestingly, we have yet to see MYXV induce an IFN response in human cancer lines, fibroblasts, or primary astrocytes (Supplementary material, Fig. S5).
Fig. 7.


Promotion of MYXV infection in human BTICs by rapamycin is independent of type-I IFN and dependent on the viral MT5 gene in half of the BTICs. (A) Real-time PCR of BTIC lines 24 h post-stimulation (CT, control; IFN, 10 U/mL IFN-β; PIC, 25 μg/mL polyI:C; MV, 1.0 MOI MYXV; VSV, 1.0 MOI of delta51–vesicular stomatitis virus) for type-I IFN and type-I IFN stimulated genes. (B) Absorbance readings of HEK-Blue Assay (Invitrogen) measuring secreted type-I IFN from BTICs 24 h posttreatment (n = 4). Error bars indicate standard error. (C) Quantified GFP expression after infection with 1.0 MOI wild-type MYXV or vMyxMT5KO at 72 h p.i. (D) Representative viral titers on BTICs and glioma cell lines after infection with MYXV and vMyxMT5KO at 72 h p.i.
We next wanted to determine whether the ability of MYXV to infect human BTICs was associated with different activated signaling pathways, as we found in non-BTIC glioma cell lines.23,40,45 Infection by MYXV in some tumor cells depends on the activation of cellular Akt by the viral MT5 protein, which directly interacts with Akt to favor infection by MYXV.28,45 To determine the requirement for MT5-mediated activation of Akt in BTICs, we assessed and quantified the early viral gene expression marker GFP in our BTIC panel using both wild-type MYXV and vMyxMT5KO (in which the MT5 gene is deleted) and compared the results with differentiated glioma cell lines that were previously characterized as type I (U87), type II (U373), and type III (GL261).23 We found that GFP expression with wild-type MYXV or vMyxMT5KO differed among the BTICs (Fig. 7C and Supplementary material, Fig. S6) and confirmed these differences using viral titers (Fig. 7D). The levels of infection between the 2 viruses were comparable in U87, BT012, and BT048, but we found significantly lower than titers with the MT5 mutant in U373, BT025, and BT042. These data suggest that BT012 and BT048 are type I cells and that BT025 and BT042 are type II cells.
Discussion
We report here for the first time that primary human gliomas grown in neurosphere culture as BTICs are infected and killed by oncolytic MYXV. Importantly, MYXV also killed TMZ-resistant BTICs, which suggests that MYXV might be effective in TMZ-resistant patients. All BTIC lines tested were susceptible to MYXV infection, viral replication, and cytotoxicity in vitro. Further, all mice bearing intracranial BTIC-derived tumors were found to have significantly enhanced survival with MYXV treatment alone. Notably, MYXV treatment was less effective in BTICs in vitro and in vivo than the conventional brain tumor cell lines. We have previously shown that MYXV treatment, with23 or without22 rapamycin, results in durable “cures” in conventional xenografts. In this study, we found that our animals eventually recurred, suggesting that MYXV does not fully infect and kill this disease reservoir. This discrepancy may highlight the importance of using patient-derived BTICs as a preclinical model and gives us the opportunity to investigate how to further optimize this therapy.
Anticipating the use of combination therapy clinically, we found that the mTOR inhibitor rapamycin improved efficacy of MYXV in vitro and in vivo in half of the BTICs tested. Rapamycin is particularly appealing because it may also act as an immunosuppressant in vivo to further enhance viral replication.24,29,46 These types of effects may not be seen in these xenograft models due to species-specific stroma/tumor interactions and the necessity for immunocompromised mice. Others have reported that rapamycin enhances infection with MYXV and other OVs in other cancers,28,29,41,47,48 but this is the first manuscript to our knowledge to test this in tumor-initiating cell models. These findings suggest that combination therapy of MYXV plus an mTOR inhibitor may be a rational approach when advancing this therapy into the clinic.
Therapies that treat BTICs in addition to the more differentiated cells in the tumor are of considerable interest. Several OVs have also been studied in BTIC models in vitro and in vivo, including oncolytic adenovirus,30,31 herpes virus,32 and vaccinia virus,41 and all these studies demonstrated the capacity of OVs to be efficacious in this model system. Our study now adds MYXV to the list of OVs capable of infecting and killing these cells and further strengthens its candidacy for clinical application.
In addition to their ability to treat the putative cancer stem cell compartment, there are several reasons why OVs are excellent candidates for combination therapy with targeted monotherapies and conventional chemotherapeutics. Firstly, OVs have the potential to provoke a multipronged attack on a tumor, with the potential to kill cancer cells directly through viral infection, through the induction of antiviral/antitumor cytokines from the tumor stroma, and possibly through antagonizing the immune system to attack the tumor. This multipronged approach is believed to circumvent some of the classical resistance mechanisms that have plagued targeted therapies and conventional chemotherapeutics. Secondly, OVs are replication competent within the tumor, as we have demonstrated in vivo, which should obviate quantitative concerns about establishing intratumoral therapeutic concentrations. Based on our preclinical data, delivery of a small number of infectious virions to the target cells should be sufficient for replication and intratumoral spread of virus to occur. We are proponents of oncolytic poxviruses (ie, MYXV and vaccinia virus), specifically as they do not require defined cell surface receptors for tumor infection, which would otherwise limit the therapy to subsets of patients. Further, their sizable genomes allow for future genetic manipulations to specifically target genes and therapies to the tumor or to enhance immunological responses for further therapeutic benefit (ie, JX-59441,49). For example, to enhance the safety of MYXV, we may explore targeted deletion mutants (eg, vMyx063KO, vMyx135KO)40 that remain oncolytic but have been rendered nonpathogenic even in their natural hosts (European or domestic rabbits).
Ultimately, although finding significant therapeutic efficacy, we did not achieve a “cure” in any animals, as we have shown previously.22,23 The factors that limit MYXV infection of BTICs are unknown, but we made several novel observations. First, human BTIC lines, as with all human lines we have tested, do not produce type-I IFN after MYXV infection, suggesting that tumoral IFN signaling in vitro may not be an important limitation for MYXV virotherapy. Further, given the incompatibility between murine IFN and the human IFN receptor,50,51 rapamycin-enhanced replication in vivo must function independently of limiting stromal-produced IFN, as has been previously shown in syngeneic models.24,29 This is an interesting observation because the type-I IFN response has been shown to enhance the therapeutic efficacy of some OVs.39 Secondly, the large increase in efficacy of the combination therapy in half of the BTICs tested suggests that using a “pharmacoviral”52 approach, whether an mTOR inhibitor or another chemotherapeutic, will likely be the optimal strategy when translating these preclinical data into the clinic.
We are aware of several limitations of our study. First, our model does not adequately assess host immune responses to the virus because our mice were immunocompromised. This may have falsely biased toward a finding of greater effectiveness than would occur in patients with (relatively) intact immune systems. Second, the precise mechanism of enhancement of infection in BTICs by rapamycin is unknown, and the combination therapy does not result in durable cures in vivo. This gives us the opportunity to improve efficacy by using other rapalogues or to use chemical library screens to identify drugs that may even further improve MYXV infection/killing in BTICs, as has previously been performed.52 This is currently under investigation in our laboratory. Third, the importance of BTICs mediating treatment resistance and recurrence in patients has yet to be proven. As such, in spite of being widely used today, this model may ultimately be irrelevant in the evaluation of experimental therapeutics. Finally, although we expect that intracranial injections of MYXV will be safe on the basis of extensive preclinical work and the safety of other OVs in glioma clinical trials, our model's own clinical trial will be needed to ultimately determine its safety in patients.
Supplementary Material
Conflict of interest statement. None declared.
Funding
This work was supported by the National Cancer Institute of Canada with funds raised by the Canadian Cancer Society (P.A.F., D.S.), by a Program Project Grant from the Terry Fox Foundation (J.B., P.A.F., D.S.), and by the V Foundation (P.A.F.). F.J.Z. was (during this work) and is funded by Alberta Innovates Health Solutions, a Vanier Scholarship from the Canadian Institutes of Health Research (CIHR), and an Izaak Walton Killam scholarship. B.A.M. was funded by CIHR. G.M.'s lab is funded by NIH grants (R01 AI080607, R21 CA149869, and R01 CA138541) and by the Bankhead Coley Foundation (1BT02).
Supplementary Material
References
- 1.Huang TT, Sarkaria SM, Cloughesy TF, Mischel PS. Targeted therapy for malignant glioma patients: lessons learned and the road ahead. Neurotherapeutics. 2009;6(3):500–512. doi: 10.1016/j.nurt.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro-oncology. 2010;12(3):304–316. doi: 10.1093/neuonc/nop068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zemp FJ, Corredor JC, Lun X, Muruve DA, Forsyth PA. Oncolytic viruses as experimental treatments for malignant gliomas: using a scourge to treat a devil. Cytokine Growth Factor Rev. 2010;21(2–3):103–117. doi: 10.1016/j.cytogfr.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Mohyeldin A, Chiocca EA. Gene and viral therapy for glioblastoma: a review of clinical trials and future directions. Cancer J. 2012;18(1):82–88. doi: 10.1097/PPO.0b013e3182458b13. [DOI] [PubMed] [Google Scholar]
- 5.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):3. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 6.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 8.Fang D, Nguyen TK, Leishear K, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65(20):9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 10.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 11.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 12.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 13.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 14.Salmaggi A, Boiardi A, Gelati M, et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia. 2006;54(8):850–860. doi: 10.1002/glia.20414. [DOI] [PubMed] [Google Scholar]
- 15.Fu J, Liu ZG, Liu XM, et al. Glioblastoma stem cells resistant to temozolomide-induced autophagy. Chin Med J. 2009;122(11):1255–1259. [PubMed] [Google Scholar]
- 16.Chen R, Nishimura MC, Bumbaca SM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17(4):362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 17.Dirks PB. Brain tumor stem cells: the cancer stem cell hypothesis writ large. Mol Oncol. 2010;4(5):420–430. doi: 10.1016/j.molonc.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venere M, Fine HA, Dirks PB, Rich JN. Cancer stem cells in gliomas: identifying and understanding the apex cell in cancer's hierarchy. Glia. 2011;59(8):1148–1154. doi: 10.1002/glia.21185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012;149(1):36–47. doi: 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly JJ, Stechishin O, Chojnacki A, et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells. 2009;27(8):1722–1733. doi: 10.1002/stem.98. [DOI] [PubMed] [Google Scholar]
- 21.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):12. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 22.Lun X, Yang W, Alain T, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65(21):9982–9990. doi: 10.1158/0008-5472.CAN-05-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lun XQ, Zhou H, Alain T, et al. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67(18):8818–8827. doi: 10.1158/0008-5472.CAN-07-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lun X, Alain T, Zemp FJ, et al. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010;70(2):598–608. doi: 10.1158/0008-5472.CAN-09-1510. [DOI] [PubMed] [Google Scholar]
- 25.Chang SM, Wen P, Cloughesy T, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23(4):357–361. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 26.Galanis E, Buckner JC, Maurer MJ, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group study. J Clin Oncol. 2005;23(23):5294–5304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 27.Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5(1):e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanford MM, Barrett JW, Nazarian SH, Werden S, McFadden G. Oncolytic virotherapy synergism with signaling inhibitors: rapamycin increases myxoma virus tropism for human tumor cells. J Virol. 2007;81(3):1251–1260. doi: 10.1128/JVI.01408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alain T, Lun X, Martineau Y, et al. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc Natl Acad Sci U S A. 2010;107(4):1576–1581. doi: 10.1073/pnas.0912344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang H, Gomez-Manzano C, Aoki H, et al. Examination of the therapeutic potential of delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99(18):1410–1414. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- 31.Skog J, Edlund K, Bergenheim AT, Wadell G. Adenoviruses 16 and CV23 efficiently transduce human low-passage brain tumor and cancer stem cells. Mol Ther. 2007;15(12):2140–2145. doi: 10.1038/sj.mt.6300315. [DOI] [PubMed] [Google Scholar]
- 32.Wakimoto H, Kesari S, Farrell CJ, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69(8):3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanai R, Rabkin SD, Yip S, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104(1):42–55. doi: 10.1093/jnci/djr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blough MD, Westgate MR, Beauchamp D, et al. Sensitivity to temozolomide in brain tumor initiating cells. Neuro-oncology. 2010;12(7):756–760. doi: 10.1093/neuonc/noq032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blough MD, Beauchamp DC, Westgate MR, Kelly JJ, Cairncross JG. Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J Neurooncol. 2011;102(1):1–7. doi: 10.1007/s11060-010-0283-9. [DOI] [PubMed] [Google Scholar]
- 36.Rabinovich BA, Yang Y, Etto T, et al. Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proc Natl Acad Sci U S A. 2008;105(38):4. doi: 10.1073/pnas.0804105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smallwood SE, Rahman MM, Smith DW, McFadden G. Myxoma virus: propagation, purification, quantification, and storage. Curr Protoc Microbiol. 2010 doi: 10.1002/9780471729259.mc14a01s17. Chapter 14:Unit 14A 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bartee E, Mohamed MR, Lopez MC, Baker HV, McFadden G. The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J Virol. 2009;83(2):498–511. doi: 10.1128/JVI.01376-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9(9):1163–1176. doi: 10.1517/14712590903170653. [DOI] [PubMed] [Google Scholar]
- 40.Barrett JW, Alston LR, Wang F, et al. Identification of host range mutants of myxoma virus with altered oncolytic potential in human glioma cells. J Neurovirol. 2007;13(6):549–560. doi: 10.1080/13550280701591526. [DOI] [PubMed] [Google Scholar]
- 41.Lun X, Chan J, Zhou H, et al. Efficacy and safety/toxicity study of recombinant Vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther. 2010;18(11):1927–1936. doi: 10.1038/mt.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fisher T, Galanti G, Lavie G, et al. Mechanisms operative in the antitumor activity of temozolomide in glioblastoma multiforme. Cancer J. 2007;13(5):335–344. doi: 10.1097/PPO.0b013e318157053f. [DOI] [PubMed] [Google Scholar]
- 43.Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11(4):448–457. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- 44.Roos WP, Batista LF, Naumann SC, et al. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene. 2007;26(2):186–197. doi: 10.1038/sj.onc.1209785. [DOI] [PubMed] [Google Scholar]
- 45.Wang G, Barrett JW, Stanford M, et al. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc Natl Acad Sci U S A. 2006;103(12):4640–4645. doi: 10.1073/pnas.0509341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Araki K, Ellebedy AH, Ahmed R. TOR in the immune system. Curr Opin Cell Biol. 2011;23(6):707–715. doi: 10.1016/j.ceb.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu X, Tao L, Rivera A, Zhang X. Rapamycin enhances the activity of oncolytic herpes simplex virus against tumor cells that are resistant to virus replication. Int J Cancer. 2011;129(6):1503–1510. doi: 10.1002/ijc.25808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas DL, Doty R, Tosic V, et al. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol Immunother. 2011;60(10):1461–1472. doi: 10.1007/s00262-011-1045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JH, Oh JY, Park BH, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther. 2006;14(3):361–370. doi: 10.1016/j.ymthe.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 50.Uze G, Lutfalla G, Bandu MT, Proudhon D, Mogensen KE. Behavior of a cloned murine interferon alpha/beta receptor expressed in homospecific or heterospecific background. Proc Natl Acad Sci U S A. 1992;89(10):4774–4778. doi: 10.1073/pnas.89.10.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin XQ, Beckham C, Brown JL, Lukashev M, Barsoum J. Human and mouse IFN-beta gene therapy exhibits different anti-tumor mechanisms in mouse models. Mol Ther. 2001;4(4):356–364. doi: 10.1006/mthe.2001.0464. [DOI] [PubMed] [Google Scholar]
- 52.Diallo JS, Le Boeuf F, Lai F, et al. A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers. Mol Ther. 2010;18(6):1123–1129. doi: 10.1038/mt.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.