

Summary
In normal brain, the side population (SP) phenotype is generated by ABC transporter activity and identifies stem cell and endothelial cell sub-populations by dye exclusion. By drug efflux, the ABCG2 transporter provides chemoresistance in stem cells and contributes to the blood brain barrier (BBB) when active in endothelial cells. We investigated the SP phenotype of mouse and human gliomas. In glioma endothelial cells, ABC transporter function is impaired, corresponding to disruption of the BBB in these tumors. By contrast, the SP phenotype is increased in non-endothelial cells that form neurospheres and are highly tumorigenic. In this cell population, Akt, but not its downstream target mTOR, regulates ABCG2 activity, and loss of PTEN increases the SP. This Akt-induced ABCG2 activation results from its transport to the plasma membrane. Temozolomide, the standard treatment of gliomas, although not an ABCG2 substrate, increases the SP in glioma cells, especially in cells missing PTEN.
Keywords: Side Population, PTEN/Akt, Glioma, ABCG2
Introduction
Glioblastoma multiforme (GBM) is the most prevalent and aggressive form of primary brain tumor. The infiltrative growth of glioma cells and their resistance to standard therapy have limited the development of successful treatment. In recent years, numerous studies have reported the presence of stem cells in solid tumors referred to as cancer stem-like cells (Regenbrecht et al., 2008; Yuan et al., 2004). Cancer stem-like cells represent a population of drug-resistant cells that can survive treatment and repopulate the tumor. These data led to new paradigms of tumorogenesis and chemotherapy focusing on tumor initiation and resistance of cancer stem-like cells to chemotherapeutic agents and radiation (Hambardzumyan et al., 2008a). The underlying mechanisms for such resistance have been attributed to their greater capacity for DNA damage repair, activation of survival pathways (Bao et al., 2006; Hambardzumyan et al., 2008b) and over-expression of ATP-binding cassette transporters (ABC-transporter) (Dean et al., 2005).
ABC transporters belong to a superfamily of membrane pumps that use ATP hydrolysis to efflux various endogenous compounds and xenobiotics from the cell (Szakacs et al., 2006). These proteins participate in tumor resistance by actively transporting drugs across the cell membrane, serving to protect cells from chemotherapeutic agents (Donnenberg and Donnenberg, 2005). ABCB1, ABCC1 and ABCG2 are the three main multidrug-resistance genes overexpressed in multiple cancers. In addition to the efflux of drugs, these pumps also possess the ability to exclude fluorescent dyes, such as the Hoechst 33342. Stem cells are frequently identified as the “side-population” (SP) by flow cytometry based on ABCG2-mediated efflux of Hoechst dye (Goodell et al., 1996). Stem cells can be isolated by FACS sorting to select cells that exhibit low level of Hoechst fluorescence intensity. SP cells were first isolated from bone marrow and more recently from various solid tumors and cell lines (Wu and Alman, 2008). These cells are pluripotent and have greater growth capacity and resistance to cytotoxic drugs as compared to non-SP cells (Hirschmann-Jax et al., 2004). Moreover, when isolated from solid tumors, SP cells are more tumorigenic after injection into nude mice (Ho et al., 2007; Patrawala et al., 2005).
In addition to their presence in normal cells and neoplastic stem cells, ABC transporters are even more active in brain endothelial cells, contributing to the blood–brain barrier (BBB) and playing a pivotal role in detoxification (Dean et al., 2005). This biological activity is critical with regard to treatment of brain tumor patients where drug penetration into the brain is limited by ABC transporter function in endothelial cells. Chemotherapy efficacy is further limited by ABCG2 expression in the cancer stem-like cells population that presumably repopulates the tumor after therapy (Robey et al., 2007).
Although much is known about the SP phenotype and ABC function in stem cells, several key questions remain unanswered regarding the role of ABCG2 in glioma biology. For example, what role does ABCG2 function have in the loss of BBB integrity in malignant gliomas? How do genomic alterations associated with glioma progression, such as loss of the tumor suppressor PTEN, and therapeutic events in glioma treatment with drugs such as temozolomide regulate ABCG2 function in cancer stem-like cells? Further, can the SP phenotype and ABCG2 activity be used as a marker for stem-like cells in gliomas, and how do genomic and therapeutic events modify its function? Finally, can these issues be modeled in mice?
In order to investigate the role of ABCG2 function in the glioma BBB and glioma cancer stem-like cells, we chose a well characterized mouse model for glioma. We used PDGF-induced gliomas generated by the RCAS-tv-a system (Shih et al., 2004) where concomitant deletion of PTEN can be achieved by delivery of the RCAS-Cre virus (Hu et al., 2005). In this study, we extend previous findings linking the stem cell properties of the side population and ABCG2 function to glioblastoma mouse models. We confirm the high tumorigenicity of SP cells, their self-renewal ability and resistance to chemotherapy. We also verify that the SP phenotype, which results from ABCG2 activity, is a stem cell marker for glioma cells. We show that regulation of ABCG2 function and localization to the plasma membrane are achieved by Akt. In the present study of gliomas in vivo, we found loss of ABC transporter function in glioma endothelial cells, correlating with the loss of the BBB integrity seen in glioma patients. Further, we provide the first evidence that loss of PTEN increases the SP phenotype of glioma cancer stem-like cells. In addition, temozolomide increases the SP fraction, especially in cells lacking PTEN. We show that temozolomide, while not a substrate for ABCG2, enriches the fraction of glioma cells with stem-like characteristics and elevated O6-methylguanine DNA methyltransferase (MGMT) expression. Finally, we report a more detailed mechanism of ABCG2 regulation: the PI3K/Akt pathway, but not the downstream target of Akt mTOR, specifically modulates the transporter's activity and also extend these observations to human cancer stem-like cells.
Results
Immunolocalization of ABCG2 in normal brain and PDGF-induced gliomas
The expression of ABC transporters at the BBB has been proposed as the major factor limiting drug penetration inside the brain with the consequent multidrug resistance of tumors. We initially used immunofluorescence staining of tissue samples to determine the location of ABCG2 expression in normal mouse brain and in brains harboring PDGF-induced gliomas. Consistent with previous reports, immunohistochemical analyses of normal brain sections revealed strong ABCG2 expression at the luminal side of capillary endothelial cells. Such immunoreactive cells were distributed throughout the brain, including the cortex, white matter tract and cerebellum (Figure S1A). In PDGF-induced glioma, ABCG2 localized to the large vessels of the tumor where it co-expressed with the endothelial cell marker Von Willebrand factor (vWF) (Figure 1A). Flow cytometry analysis for ABCG2 function usually identifies the side population as a very small fraction of cells in tumors; consistent with this, we found a small percentage of tumor cells expressing ABCG2. However, tumor samples exhibited some regions of cells positive for both ABCG2 and nestin, which were not endothelial cells (negative for vWF) (Figure 1B). These observations suggested that both endothelial cells and progenitor tumor cells express ABCG2.
Figure 1. ABCG2 localizes in vasculature and progenitor cells of PDGF-induced gliomas and identifies side population cells.
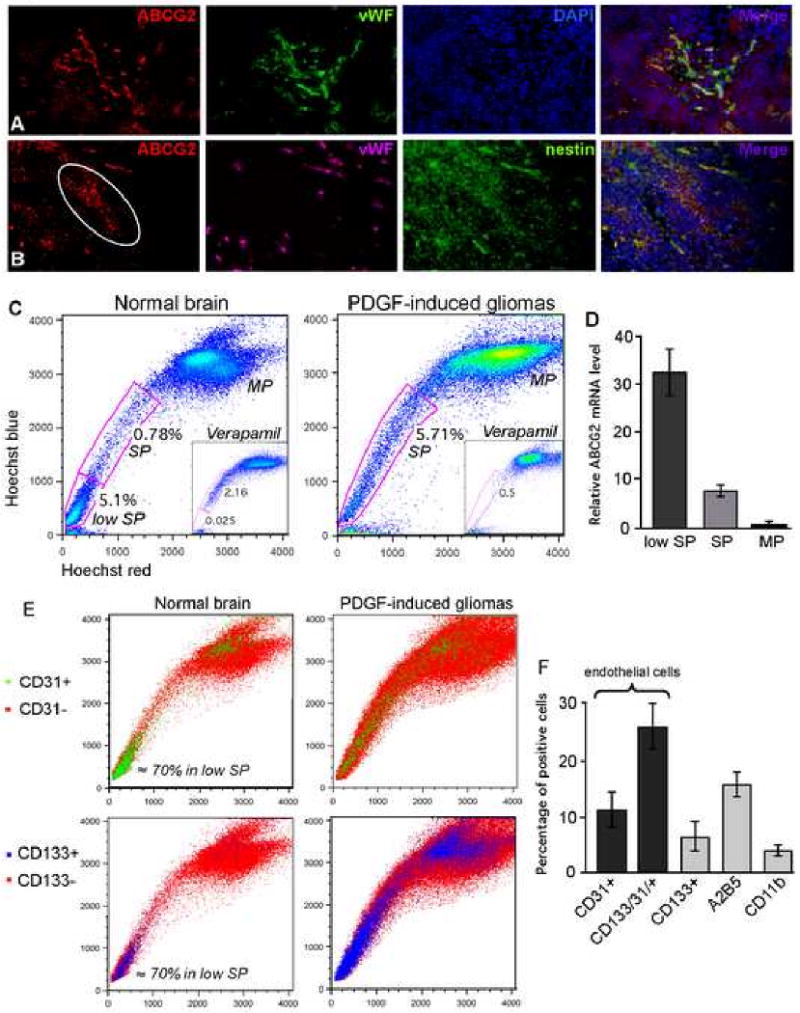
(A) ABCG2 (red) localizes in blood vessels of gliomas, where it is co-expressed with Von Willebrand factor (vWF) (green); DAPI counterstained in blue.
(B) ABCG2 (red), vWF (purple) and nestin (green) shows that some nestin immunoreactive cells are positive for ABCG2 in both endothelial cells and tumor progenitor cells.
(C) Hoechst 33342 dye exclusion assay on cells isolated from normal brain cortex identifies a SP and low SP fraction, while a uniform SP fraction is detected in PDGF-induced gliomas. Incubation with verapamil abolished the SP and low SP; MP corresponds to main population.
(D) Q-RT-PCR confirms high expression levels of ABCG2 in the low SP and SP as compared to the MP in PDGF-induced tumors.
(E) Cell surface phenotype of SP cells shows that in normal brain, the majority of low SP cells are positive for CD31 and/or CD133. In tumor samples, the CD31+ and CD133+ cells are spread over the three populations, indicating a lower ability of endothelial cells to exclude the dye.
(F) Quantification of double staining for Hoechst and cell surface markers (CD31, CD133, A2B5 and CD11b) in SP cells isolated from mouse gliomas.
Identification of a side population in normal brain and PDGF-induced gliomas
To characterize the cells that actively contribute to the SP phenotype, we performed a Hoechst dye exclusion assay on cells isolated from the normal brain cortex of 5 different mice, as well as cells isolated from PDGF-induced tumors. As most cells accumulate Hoechst 33342, SP cells can be isolated by dual-wavelength flow cytometry based of their ability to efflux this dye, a process that requires the action of the ABC transporters. We identified a side population among cells isolated from normal mouse brain cortex (Figure 1C). This SP fraction was generally small, ranging from 0.5 to 0.8% of total cells, and was accompanied by a distinct population of cells termed the “low side population” (low SP). Low SP cells were present in large numbers (c.a. 6 to 8%) and very actively excluded Hoechst 33342. Incubation in the presence of 100μM verapamil, known to block ABC transporter activity, abolished the low SP and part of the SP. In contrast to what was seen with normal brain tissue, Cells isolated from solid tumor exhibited a different pattern of Hoechst dye exclusion, with a uniform SP representing 4 to 8% of the total population (Figure 1C). While a low SP has already been described in normal brain, its relative absence in the tumor samples was unexpected (Mouthon et al., 2006). Q-RT-PCR for ABCG2 confirmed the high level of expression of ABCG2 transporter in the low SP and SP, i.e. 30 and 10 fold-increases respectively as compared to the main population (MP), confirming that ABCG2 contributes to the SP phenotype (Figure 1D). The ABCB1a (MDR1a) transporter was also found to be highly expressed in the low SP and SP fractions, while the level of ABCB1b (MDR1b) and ABCC1 (MRP1) transporters was not elevated in these fractions (Figure S1B).
Cell surface phenotype of SP cells from normal brain and PDGF-induced gliomas
To further characterize the cells present within the SP and low SP, cells isolated from normal mouse brain or PDGF-induced gliomas were subjected to double staining for Hoechst and various cell surface markers. Staining of cells isolated from normal brain cortex for CD31, a marker of endothelial cells, showed that up to 70% of the positive cells are concentrated into the low SP area (Figure 1E). This observation indicates that the low SP is highly enriched in endothelial cells, and is consistent with the immunolocalization of ABCG2 at the vasculature described above. As CD133 has been proposed to be a marker of endothelial progenitor cells, haematopoietic and neuronal stem cells in normal and malignant tissues, we looked at the expression profile of CD133 within the Hoechst populations. In normal brain, CD133+ cells were located mainly in the low SP, where a high proportion of these cells were also positive for CD31, indicating an endothelial cell origin (Figure 1E).
In contrast to the normal brain, where CD31 cells were predominantly found in the low SP, the CD31+ cells in gliomas were spread over the low SP, SP and MP, implying a reduction of ABC transporter function in the endothelial cells in these tumors. (Figure 1E). The CD133+ cells were more abundant in PDGF-induced gliomas as compare to normal brain (4% ± 1.5 versus 12% ± 3.3), and were also distributed across the three Hoechst populations. Results for quantification of the staining in SP fraction isolated from tumor-bearing mice are presented in Figure 1F. Up to 35% of the cells were endothelial cells (CD31+ and CD31+/CD133+) and 5% were CD133 single positive cells. Finally, 15% of the cells labeled for the oligodendrocyte progenitor marker A2B5, and 4% for the microglia marker CD11b. Based on immunostaining, a majority of tumor cells expressing ABCG2 were also nestin positive. These data further indicate that the SP population of cells is composed of both endothelial cells and cells expressing stem and progenitor markers. Previous reports have documented that the BBB is disrupted in human and animal models of brain tumors resulting in leakage of IV contrast material into tumors (McConville et al., 2007; Provenzale et al., 2005). In line with this observation, the absence of low SP in cells isolated from solid tumors suggests that their endothelial cells possess lower levels of ABC transporter expression or activity. This is supported by the wide distribution of CD31+ cells amongst the SP and MP, demonstrating that endothelial cells indeed possess a lower capacity to eflux the Hoechst dye.
SP cells are enriched for neurosphere-forming ability
We then determined the extent that SP cells behave as stem cells. After Hoechst staining, the three populations from four different brains were separated by FACS sorting and equal numbers of cells were cultured in neurosphere medium (supplemented with EGF and bFGF). In all cases, the SP cells from gliomas formed more neurospheres than the MP and low SP cells (Figure 2A). By contrast, none of the three cell populations isolated from adult normal brain cortex contained cells able to form neurospheres. These results indicate the specific presence of cancer stem-like cells within the SP fraction of solid tumors.
Figure 2. SP cells possess stem cell properties and are enriched in neurospheres.
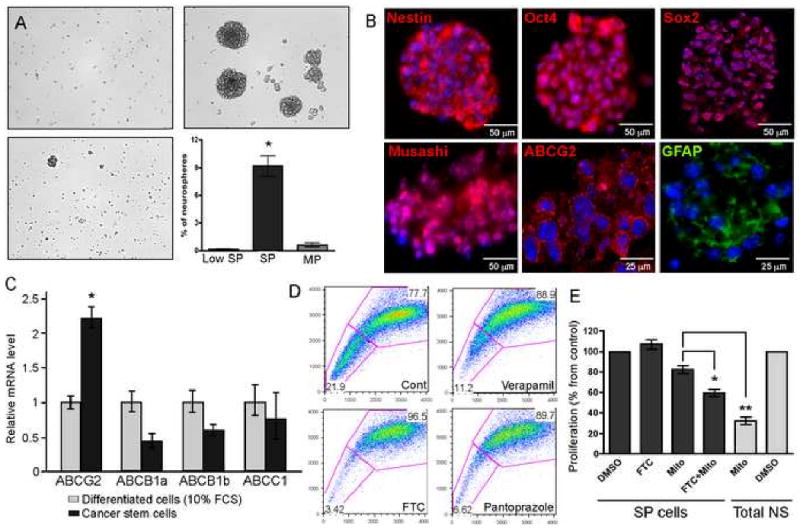
(A) Culture of sorted low SP, SP and MP cells in neurosphere medium shows that only SP cells are able to form neurospheres.
(B) Total neurosphere cultures prepared from PDGF-induced tumors stain positive for various stem cell markers such as nestin, Oct4, Sox2, Musashi, ABCG2 and GFAP. Bars = 25 and 50 μm.
(C) Q-RT-PCR showing relative ABC transporter mRNA levels in neurospheres as compared to the same cells cultured in DMEM supplemented with 10% FCS. Note that only ABCG2 expression increases under stem cell conditions.
(D) Hoechst dye exclusion assay on neurospheres reveals a high percentage of SP cells. Incubation with Verapamil partially abolished the SP fraction while Pantoprazole and Fumitremorgin C (FTC) almost totally abolished the SP phenotype.
(E) Sorted SP cells isolated from neurospheres are highly resistant to mitoxantrone as compared to the total population, but were sensitized by the co-incubation with FTC.
Neurospheres are enriched in the side population, which mainly results from the activity of ABCG2
Since the SP in vivo accounts for a small fraction of the cells and contains a high number of endothelial cells, we used neural stem cell culture to enrich for cancer stem-like cells from mouse gliomas. Cancer stem-like cells cultures were obtained by transferring cells isolated from different PDGF-driven tumors into neurosphere medium. These spheres contained cells that predominantly stained with antibodies to stem cell markers such as nestin, Oct-4, Sox-2, musashi and also to some level to ABCG2 (Figure 2B). ABCB1, ABCC1 and ABCG2 are the three main transporters known to posses the ability to exclude drugs and to be present in stem cells of different tumor types. Analysis of ABC transporter expression in tumor cells cultured in neurosphere medium, as opposed to DMEM supplemented with 10% FBS, showed that ABCG2 is the only transporter upregulated in glioma cancer stem-like cells; the expression of ABCB1a/b and ABCC1 was even reduced (Figure 2C). This observation is consistent with the concept that the ABCG2 transporter is the most specific stem cell marker of this family (Islam et al., 2005). As the SP represents a high concentration of stem cells, Hoechst dye exclusion of neurospheres was expected to yield a large SP fraction. As expected, 10 to 20% of cells from neurospheres were located in the SP fraction (Figure 2D). There were no CD31 positive cells in neurosphere culture, indicating that the SP phenotype is not due to the presence of endothelial cells (data not shown). Q-RT-PCR confirmed the higher level of ABCG2 expression in the SP fraction (Figure S1C). The relative expression of ABCB1a and ABCC1 was also found to be increased in the SP fraction, which is not surprising given that Hoechst dye is a substrate for various ABC transporters (Figure S1C). We then pre-incubated neurospheres with fumitremorgin C (FTC), a potent and specific inhibitor of ABCG2 (Rabindran et al., 2000), to determine the contribution of ABCG2 to the SP phenotype. Addition of FTC reduced the SP phenotype from 22% to 4%, confirming the dominant role that ABCG2 plays in the SP phenotype (Figure 2D). Pantoprazole, known to block ABCG2 activity (and to a lesser extent ABCB1) also diminished the amount of SP cells to 7% of total cells. Verapamil, a non-specific inhibitor of ABC transporters (and more potent inhibitor of ABCB1 and ABCC1) only reduced the SP fraction to 11%.
SP cells isolated from neurospheres propagate the neurosphere phenotype and are resistant to mitoxantrone
We next analyzed the self-renewal capacity of SP cells using the neurosphere-forming assay. Neurospheres cultured from four different PDGF-induced glioma-bearing mice were dissociated and stained with Hoechst. SP and MP cells were then sorted and cultured in neurosphere medium. SP cells from neurospheres are endowed with a higher ability to generate secondary neurospheres: 20.4 ± 1.3% of SP cells as compared to 4.6 ± 0.4% of MP cells (p<0.001) (Figure S1D). The rare spheres formed from the MP fraction may results in part from the low contamination with SP cells, as shown by the re-analysis of the sample directly after sorting (Figure S1D, right panel); it is also possible that rare stem cells exist that do not efflux Hoechst dye. Overall, this data demonstrates that SP cells are enriched in cancer stem-like cells characteristics and self-renewal ability.
We then evaluated the drug resistance property of the side population; SP and MP neurosphere cultures were exposed to 50 nM mitoxantone and analyzed for their proliferation rate using an MTT assay. Mitoxantrone has been reported to be a substrate of ABCG2 and its efflux by the transporter is a determinant mechanism of drug resistance. As stated above, MP cells possess a lower ability to form neurospheres, consequently the initial proliferative rate of these cells was below the detection limit of the assay. We found that SP cells are more resistant to mitoxantrone, and were sensitized by concurrent blockade of ABCG2 using FTC (Figure 2E). In SP cells, the quantitative decrease in proliferation induced by incubation with both mitoxantrone and FTC did not reach that produced by mitoxantrone alone in total neurosphere culture. This suggests that additional mechanisms, other than efflux by ABC transporters, might also be involved in drug resistance. One possibility proposed recently (Bao et al., 2006) involves an increased capacity of stem cells for DNA damage repair. Taken together, these data indicate that ABCG2 is expressed in neurospheres, allows for the selection of SP cells, and participates in resistance to chemotherapy.
Loss of PTEN increases the SP fraction and highly favors the MDR phenotype
Akt has been shown to regulate the SP phenotype in bone marrow cells, and similar effects were reported in breast cancer cells after PTEN loss (Zhou et al., 2007). To test whether loss of PTEN could modulate the SP in gliomas, we compared the amount of SP cells in neurospheres isolated from PDGF-induced gliomas in both PTEN intact and PTEN-deficient mice (by Cre recombinase). Five different tumors were analyzed in each group; loss of PTEN and increased in pAKT level in neurospheres were confirmed by immunoblot (Figure S1E). The loss of PTEN produced a pronounced increase of the SP fraction; the PTEN deleted cells contained 33.1 ± 4.0% SP cells versus 15.5 ± 1.5% in PTEN intact (Figure 3A). While mitoxantrone induced an arrest of PDGF-neurospheres in the G0/G1-phase, the higher level of SP cells in PTEN-deleted cells resulted in increased resistant to mitoxantrone, an effect that was reversed by incubation with FTC (Figure S1F). Incubation of neurospheres with the PI3 kinase inhibitor LY294002 (20 μM) strongly reduced the proportion of SP cells in both PDGF (3.6 ± 0.4% fold) and PDGF/PTEN deleted (3.0 ± 0.7% fold) tumors, confirming that PI3K regulates the SP phenotype (Figure 3A). We confirmed this observation using the PI3K inhibitor GDC-0941 and found that it reduced the amount of SP cells in a dose-dependent manner (Figure S2A). No substantial difference in ABCG2 protein and mRNA levels were detected upon treatment with LY294002 (Figure 3E and Figure S2B), suggesting that PI3K activity regulates ABCG2 function rather than its expression. None of the other ABC transporters tested showed a difference in mRNA levels after treatment with LY294002 (Figure S2B). LY294002 has been reported to induce a translocation of ABCG2 from the plasma membrane to the endoplasmic reticulum. To determine if such a mechanism also occurs in glioma cancer stem-like cells, we stained PTEN-deleted cells for ABCG2 after 24h treatment with LY294002. We observed a change in the localization of the protein, presenting a diffuse cytoplasmic pattern as compared to the membrane staining in the control (Figure 3B).
Figure 3. Loss of PTEN and temozolomide treatment increase the SP fraction; SP cells with PTEN loss are highly tumorigenic.
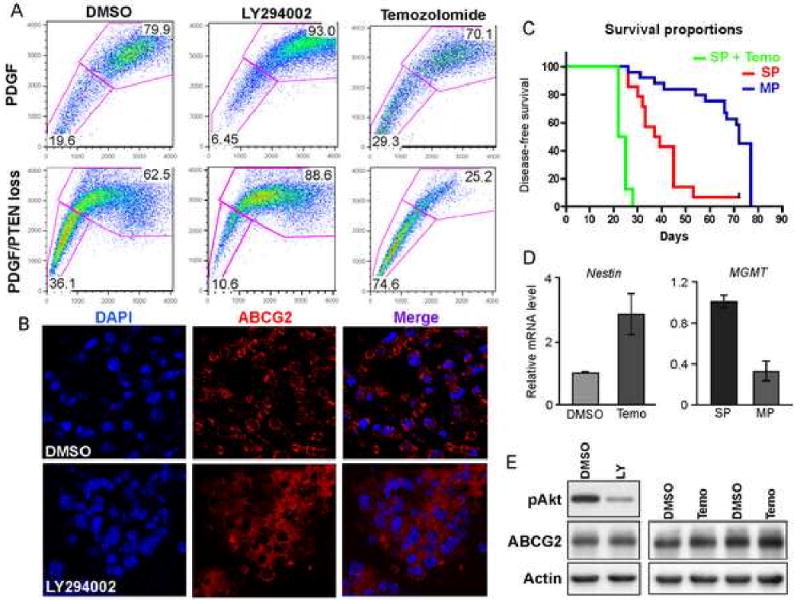
(A) Loss of PTEN in neurospheres strongly increases the SP phenotype, which is partially block by the incubation with the PI3K inhibitor LY294002. Long-term treatment of neurospheres with 100 μM temozolomide induces an increase in the amount of SP cells, which is greatly favored by PTEN deletion.
(B) ABCG2 (red) localizes at the cell membrane in untreated cells as compared to the cytoplasmic staining upon treatment with LY294002.
(C) Kaplan-Meier survival curve shows that SP cells isolated from neurospheres with PTEN loss possess a higher tumorigenic potential than the MP cells; temozolomide treatment increases the tumorigenicity of SP cells.
(D) Q-RT-PCR in neurospheres showing increased nestin expression after treatment with temozolomide; MGMT expression is enriched in SP cells.
(E) Western Blot analysis for ABCG2 indicates that LY294002 (LY) and temozolomide (T) don't modulate ABCG2 expression as compared to DMSO treated cells.
The SP cells are highly tumorigenic
We then determined the relative tumorigenic potential of side population cells isolated from neurospheres derived from PDGF driven PTEN deleted tumors. We have already demonstrated in our mouse model that loss of PTEN correlates with higher-grade and tumor latency. Following Hoechst staining, we sorted SP and MP cells: 1×104 cells were injected intracranially in newborn pups (20 mice per group). When mice presented symptoms of intracranial pathology such as lethargy, weight loss and macrocephaly the animals were scarified and evaluated for tumors by histology. In general, these secondary tumors exhibited features of high-grade gliomas such as microvascular proliferation and pseudopalisading necrosis (Figure S2C). In two cases, we noticed subcutaneous proliferation on the head of mice injected with MP cells, but not in the cranial lesions; these mice were not included in the analysis. Kaplan-Meier survival curves indicated that in the absence of PTEN, SP cells are highly tumorigenic as compared to MP cells, showing higher tumor frequency and shorter latency (p < 0.0001) (Figure 3C). As observed for the low sphere-formation ability of MP cells, tumor formation induced by this fraction might also result from the contamination with SP cells, or the presence of stem-like cells in the non-SP compartment.
We verified that the lower frequency of tumors obtained after the injection of MP cells was not a result of Hoechst dye toxicity by measuring cell viability 10 days after sorting and culture of the SP and MP, as well as the total cell population stained or not with the dye. In all cases, viability was not affected (Figure S2D). In parallel, we incubated total neurosphere culture from PDGF-induced tumors for 90 min with Hoechst and analyzed the cell cycle distribution 10 days later. No apoptosis or differences in cell cycle distribution were observed (Figure S2D, right panel).
To determine whether ABCG2 is itself tumorigenic, we injected DF1 cells transfected with a RCAS-ABCG2 vector into Ntv-a/Ink4a−/−Arf−/− newborn pups and assessed the tumor incidence. ABCG2 activity was confirmed by the ability of transfected DF1 cells to exclude Hoechst dye (Figure S3A). Of the 25 mice tested, no tumors or lesions were detected up to 6 months after injection. By contrast, RCAS-PDGF induces high-grade gliomas in the vast majority of mice with this background in less than 12 weeks. Unlike the high-grade gliomas formed in the in the Ntv-a/Ink4a−/−Arf−/− mice, RCAS-PDGF induces low-grade gliomas in Ntv-a/wt mouse background (Ink4a/Arf intact) (Dai et al., 2001). Therefore, we determined if ABCG2 could increase tumor incidence and grade, or reduce latency time in these mice with lower grade tumors. Co-injection of DF1 cells expressing RCAS-PDGF and RCAS-ABCG2 produced the same number and grade of tumors as for RCAS-PDGF alone. This data suggest that ABCG2 is not tumorigenic by itself, nor does it enhance PDGF tumorigenicity. Rather the SP phenotype, which mainly results from ABCG2 activity, is a useful marker to isolate tumor-initiating cancer stem-like cells.
Temozolomide in vitro induces an increase of the SP fraction in neurospheres
Recent work has suggested that cancer stem-like cells are the source of chemotherapy resistance in cancer with the potential to repopulate a tumor after therapy. Temozolomide, in combination with radiation, is currently the standard treatment for patients with glioblastomas. To investigate whether temozolomide could induce the MDR phenotype in cancer stem-like cells, we evaluated the resistance to temozolomide of neurospheres derived from PDGF-induced gliomas. We performed a step-wise exposure with increasing doses of temozolomide over two weeks and assessed for the SP fraction. Four different tumors were analyzed: the treatment led to a 1.5 ± 0.04 fold increase in the SP fraction, reaching up to 30% of total cells (p<0.05) (Figure 3A). As our data above demonstrates that PTEN loss in gliomas increases SP cell number, we hypothesized that PTEN loss leads to greater temozolomide resistance. Indeed, long-term treatment of PDGF/PTEN-deleted neurospheres with temozolomide induced a 2.2 ± 0.1 fold increase in the SP fraction as compared to the untreated spheres; in some cases, the amount of SP cells even reached 75% of total cells (Figure 3A). The fold increase in SP induced by temozolomide was significantly higher in neurospheres isolated from PDGF/PTEN-deleted tumors than PDGF alone (p<0.05). These data suggests that loss of PTEN has an important role in the regulation of the MDR phenotype, especially under the stress of cytotoxic therapy. We then sorted the SP cells from the temozolomide-treated neurospheres and assessed for their tumorigenic potential (8 mice per group; same number of implanted cells). We found that temozolomide treatment significantly shortened the tumor latency time as compared to the SP cells isolated from untreated neurospheres (p<0.01) (Figure 3C). The observed increase in SP cells produced by temozolomide treatment is accompanied by a parallel increase in nestin-expressing cells, as shown by a 2.24 ± 0.62 fold elevation in nestin mRNA (p<0.05) (Figure 3D, left panel). These observations demonstrate that temozolomide enrich for highly tumorigenic cells and cells with stem-like cell characteristics.
To investigate whether the elevated SP phenotype could also result from increased ABCG2 expression, time-course assays with temozolomide were performed and the neurospheres analyzed for ABCG2 expression at the protein and mRNA levels. Short-time exposures (2 to 24h) did not produce any changes in ABCG2 protein or mRNA levels, nor was the activity of the transporter affected, as measured by mitoxantrone efflux (Figure 3E; Figure S2E and F). In summary, the above data are consistent with temozolomide either being a substrate for ABCG2, or alternatively with temozolomide treatment inducing an enrichment of naturally resistant stem cells through a mechanism other than direct selection.
Localization of ABCG2 at the cell membrane regulates its activity and depends on post-translational modifications
To investigate whether the increase in SP phenotype induced by temozolomide resulted from an active transport of the drug by ABCG2, we generated a human glioma cell line U87-MG that stably expressed hABCG2 (U87-ABCG2). While neither SP cells nor ABCG2 protein were found in cells infected with the empty vector (U87-Cont), those stably expressing ABCG2 displayed a band at 72 kDa and a clear SP fraction (i.e. 7%) that was blocked by FTC (Figure 4A and B; Figure S3B). Western blot of ABCG2 from lysates derived from these cells showed multiple bands, suggesting the acquisition of post-translational modifications. A sample highly enriched for SP cells derived from double sorting (SP2; i.e. 80% of SP cells) predominantly exhibited a high MW form of the ABCG2 protein. In contrast, this band was absent in non-SP cells, indicating that additional post-translational modifications are associated with ABCG2 activity in SP cells. ABCG2 is known to be N-glycosylated at Asn596, although this modification is not essential for its activity (Diop and Hrycyna, 2005). We tested whether the higher MW form observed in the sorted SP cells line could result in part from glycosylation. PGNase treatment of SP cell extracts produced a marked decrease in the apparent Mr, demonstrating that this ABCG2 glycoform is more extensively N-glycosylated (Figure S3D).
Figure 4. U87MG-ABCG2 stable cell line is resistant to mitoxantrone but not temozolomide.
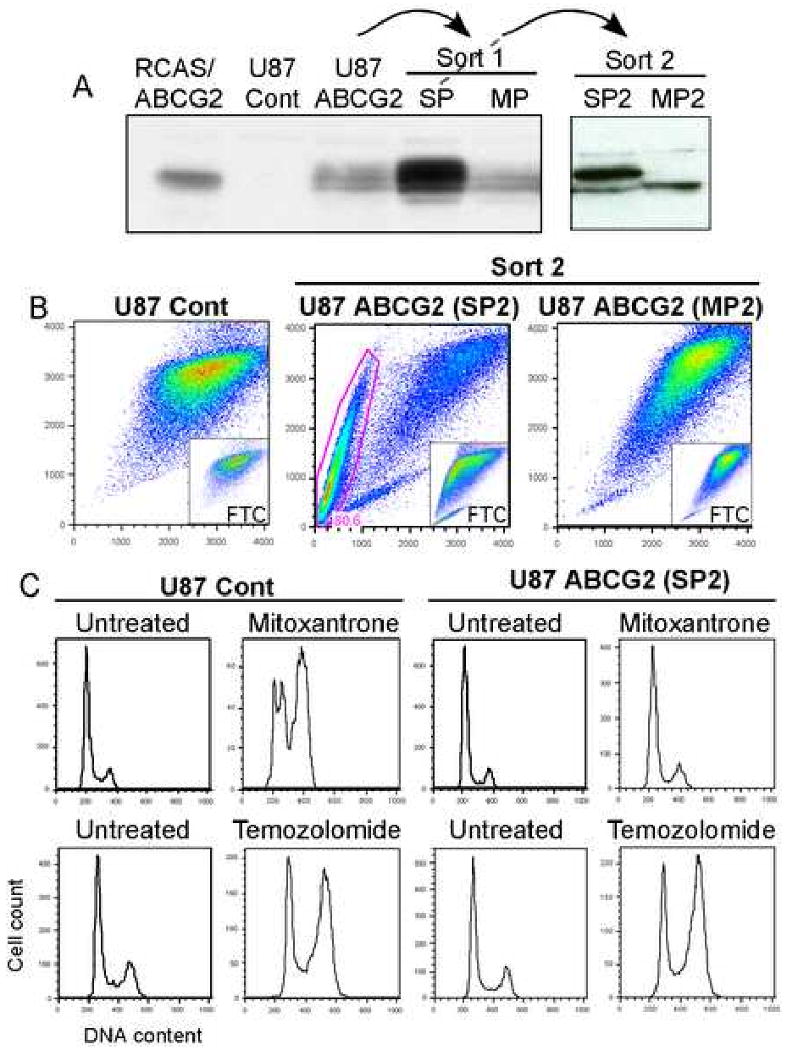
(A) U87-ABCG2 cell line presents a 72kDa band accompanied by post-translational modifications that correlates with high ABCG2 activity (B).
(C) The SP2 cells (SP cells enriched by two sorting) are highly resistant to mitoxantrone but not temozolomide.
ABCG2 is known to be expressed at the cell membrane and at the endoplasmic reticulum. We initially hypothesized that the band at higher MW detected by WB corresponds to the active form of the protein, which localizes to the cell membrane. To address this question, we performed immunostaining for ABCG2: the protein was detected both in the cytoplasm and at the cell membrane in ABCG2-SP2 cells, while it localized only in the cytoplasm in non-SP cells (MP2) (Figure 5A). As a second approach, we used FACS analysis with an anti-hABCG2 antibody that recognizes only the cell surface protein (without permeabilization): we observed a shift in staining intensity in SP2 cells as compared to non-SP cells as well as those infected with the empty vector (median staining intensity was 9.5 for SP cells versus 3.3 for non-SP cells) (Figure 5A).
Figure 5. PI3K/Akt pathway regulates ABCG2 activity in U87MG stable cell line and the SP phenotype of human neurospheres.
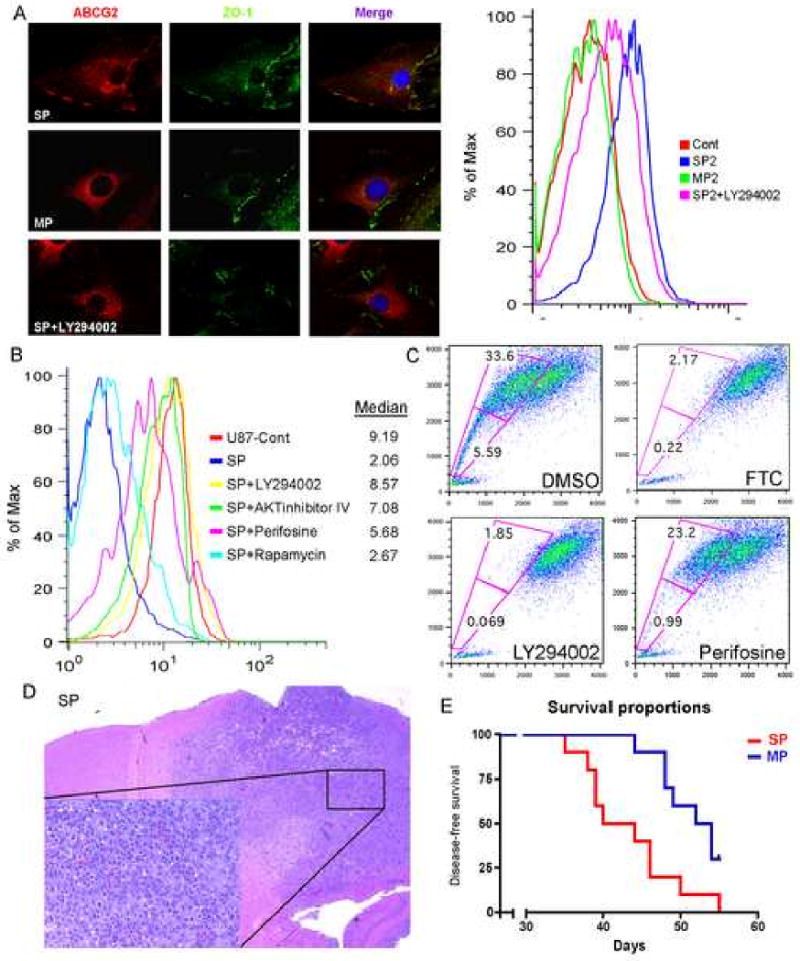
(A) Immunostaining for hABCG2 (red) shows a membrane staining in SP2 cells versus a cytoplasmic staining in MP2 cells; ZO-1 (green) delimitate the membrane. In the right panel, FACS analysis using cell surface hABCG2 antibody confirms high staining intensity in SP2 cells. Blocking of PI3K with LY294002 induced a change in ABCG2 localization, from the cell membrane to the cytoplasm.
(B) Mitoxantrone exclusion assay in SP2 cells after incubation with LY294002, Akt inhibitor IV, perifosine and rapamycin. Blocking of the PI3K and Akt pathways abolished the ability of ABCG2 to efflux mitoxantrone.
(C) Hoechst staining of human neurosphere culture shows the presence of a SP that was blocked by FTC. Treatment with LY29004 totally abolished the SP fraction while perifosine displayed a stronger effect in blocking the lower SP fraction.
(D) H&E of nude mice injected with SP cells isolated from human GBM neuropsheres.
(E). Kaplan-Meier survival curve shows that SP cells isolated from human neurospheres are more tumorigenic then MP cells.
Temozolomide is not a substrate for ABCG2 but induces its activity by a mechanism other than selection
Our observation that temozolomide selects for higher SP could simply be explained by the drug itself being a substrate for ABCG2. To test this hypothesis, we used the ABCG2-expressing U87-MG cell line to test for temozolomide resistance. Mitoxantrone served as control and we assessed the cell cycle distribution after 48h of incubation. In cells infected with the empty vector, 25nM mitoxantrone induced a clear G2M arrest while the U87-ABCG2 cell line was resistant to the drug (Figure 4C). We then analyzed cell cycle distribution after treatment with temozolomide; we were unable to detect any differences between the two cell lines, indicating that ABCG2 expression does not provide any resistance to temozolomide (Figure 4C). As a second approach, we performed a competition assay in which an excess of a first substrate competitively blocks the efflux of a dye. As a control, we measured the intracellular accumulation of mitoxantrone, with and without co-incubation with 10 μg/ml Hoechst. The dye induced a shift in the intracellular fluorescence of mitoxantrone (Figure S3C). By contrast, temozolomide was unable to produce any changes in mitoxantrone accumulation, even at very high concentration (1.2mM). Taken together, these results suggest that although temozolomide induces SP phenotype in glioma cells, it is not a substrate for ABCG2.
To determine which mechanism contributes to the increase in the SP phenotype, we looked at the level of O6-methylguanine DNA methyltransferase (MGMT): increased expression and elevated activity of MGMT has been shown to be a key mechanism involved in the resistance of gliomas to temozolomide (Kitange et al., 2008). We compared the MGMT mRNA level in SP and MP cells isolated from PDGF/PTEN-deleted neurospheres (prepared from four different tumors). We found that MGMT expression is increased by 3.1 fold in SP cells (p < 0.01) (Figure 3D, right panel), consistent with the resistance of SP cells to temozolomide. The elevation of MGMT expression in SP cells was accompanied by a 25% increase in temozolomide resistance as compared to MP cells (Figure S2E).
The PI3K and Akt pathways regulate ABCG2 activity
The data showing that PTEN loss increases the SP suggest that the PI3K pathway regulates ABCG2 activity. To shed light on this issue, we used the sorted SP cells from U87-ABCG2 cell line and assayed for their ability to efflux mitoxantrone by FACS analysis. Cells were treated for 6 h with Akt inhibitors (except 2 h for Akt inhibitor IV) and stained for 1h with mitoxantrone. As SP2 cells possess high ability to efflux mitoxantrone, the intracellular intensity was low (i.e. median fluorescence intensity of 2.06) as compared to the U87-Cont cells (median value of 9.19) (Figure 5B). We detected similar effects to the regulation of the SP phenotype in neurospheres: incubation with the PI3 kinase inhibitor LY294002 and Akt inhibitor IV totally abolished the ability of the transporter to exclude the drug (median value of 8.57 and 7.08 respectively). Perifosine (5 to 45 μM), an inhibitor of the Akt pathway, partially lowered the extent of mitoxantrone efflux (median value of 5.68), while the mTOR inhibitor Rapamycin at 0.1 or 1 nM produced a limited effect (2.67). We also observed that blocking of PI3K in these cells using LY294002 induced a translocation of ABCG2 from the cell membrane to the cytoplasm, as shown by intracellular immunofluorescence staining of the transporter and reduction in cell surface staining measured by FACS analysis (median fluorescence value of 5.6 as compared to 9.5 for untreated cells) (Figure 5A). These results suggest that the effect of PTEN loss on the elevation of ABCG2 activity shown above is due to the PI3K/Akt activity, but not mTOR, potentially by the modulation of ABCG2 localization.
The PI3K/Akt pathway regulates the SP phenotype in human neurospheres as well
We then evaluated the SP fraction of neurospheres prepared from 4 different human GBM specimens. Human tumor neurospheres exhibited a clear SP fraction, which was blocked by co-incubation with FTC, indicating that ABCG2 also participates in the SP phenotype of human cancer stem-like cells (Figure 5C and Figure S3E). Inhibition of PI3K with LY294002 substantially reduced the SP fraction, while perifosine was effective mainly against the SP cells located at the bottom of the fraction. These data suggest that the PI3K and Akt pathways regulate the SP phenotype in human neurospheres and underline a common mechanism of regulation between human and mouse cancer stem-like cells.
We finally assessed the tumorigenic potential of SP and MP cells isolated from human GBM neurosphere cultures. Two hundred thousands cells were stereotactically injected to the brain of nude mice (10 per group). Animals were sacrificed when presenting symptoms and evaluated for tumors by histology. All mice developed tumors that resemble features of human high grade gliomas (Figure 5D). Although tumors eventually formed in the majority of mice in both population, the latency for tumor formation was significantly shorter in the SP population (p<0.01) when compared to the MP cells, consistent with the mouse data (Figure 5E).
Discussion
Both the relative rarity of cancer stem-like cells in tumors and the absence of reproducible specific markers for glioma stem cell limit identification and isolation of this important cellular subpopulation. In this study we used the efflux of Hoechst dye, which allows identification of the side population phenotype, to isolate both tumor endothelial cells and tumor-initiating cells with cancer stem-like cells characteristic from mouse and human glioblastomas.
One of the main molecular changes accompanying progression of gliomas to high grade which increased stem cell character and enhances resistance to chemotherapy is the loss of PTEN and consequent elevation of Akt pathways activity (Hu et al., 2005). Increased activity of the Akt pathway is associated with many forms of resistance and is found in a population of radiation resistant cancer stem-like cells in medulloblastomas where Akt inhibition appears to sensitize the cell population for radiation-induced apoptosis (Hambardzumyan et al., 2008b). Our data provide further connections between the Akt pathway, stem-like character, and therapeutic resistance, suggesting that in addition to the induction of radiation resistance in medulloblastomas, activation of this pathway also enhances the ability of glioma cancer stem-like cells to expel drugs.
Although temozolomide is currently the first-line standard of care for the treatment of glioma, temozolomide increases the aggressiveness of surviving glioma cells by several mechanisms. We report in this study that temozolomide increases the number of SP cells, especially from PTEN-deleted gliomas. If temozolomide were a substrate for ABCG2, the data would suggest a selection for cells that efflux the drug. However, our data and other (Chua et al., 2008) indicate it is not an ABCG2 substrate, implying that the increase in the SP phenotype might result from the enrichment of cells with stem-like properties. Therefore, in the process of increasing the number of cells in tumors with stem-like properties, temozolomide may render surviving cells even more resistant to subsequent treatment with drugs that are substrates for ABCG2. Ubiquitous distribution of temozolomide was reported into all human tissues including the brain, suggesting that the BBB efflux pumps do not contribute to temozolomide resistance (Ostermann et al., 2004). Interestingly, we found that SP cells overexpress MGMT as compared to MP cells. The MGMT enzyme is responsible for the removal of alkyl groups induced by alkylating agents, suggesting that the resistance to temozolomide observed in glioma cancer stem-like cells might be related in part to its alkylating activity. A general trend towards high expression of several DNA repair genes, including MGMT, has been demonstrated in heamatopoeitic stem cells (Casorelli et al., 2007), contributing to the higher drug resistance of cancer stem-like cells.
The SP cells from PTEN-deleted neurospheres were substantially more tumorigenic than the non-SP cells, as shown by a significant decrease in tumor latency. Moreover, temozolomide further enriched for highly tumorigenic cells, suggesting that temozolomide treatment could favor tumor recurrence. Because the SP phenotype in glioma cancer stem-like cells is mainly mediated by ABCG2, as shown by the almost complete abolition of the SP phenotype when blocking of its function, we subsequently studied the oncogenic potential of ABCG2. Although ABCG2 expression appears not to be oncogenic itself, selecting for the Hoechst dye exclusion phenotype, conferred by this transporter, highly enriches for tumor-promoting cells. Cancer stem-like cells simultaneously present impaired oncogenes/tumor suppressors expression and drug resistance abnormalities, two distinct properties that can be mutually exclusive. ABCG2 main function is to efflux compounds to protect the cells from cytotoxic agents, a drug-resistance characteristic of cancer stem-like cells, which defines the SP phenotype.
One focus of the current study was to elucidate the mechanisms regulating ABCG2 function. We noticed that although PI3K activity and temozolomide treatment changed the activity of ABCG2 in neurospheres, the expression levels of the mRNA and protein were unaffected. In mouse and human, inhibiting the PI3K/Akt pathway, but not mTOR, strongly decreased the activity of the transporter by modulating its localization at the cell membrane. These data suggest that therapeutic blockade of either PI3K or Akt might reduce ABCG2 function in cancer stem-like cells in vivo and improve chemotherapy efficacy for drugs that are substrates of this transporter.
In summary, our studies support a role for ABCG2 as a molecular determinant of the side population phenotype in glioma stem-like cells, a phenotype characterized by both tumorigenic and chemoresistant properties. Furthermore, we report that loss of the tumor suppressor PTEN, an important genomic alteration associated with glioma progression, greatly increases the SP and further enhances the MDR phenotype upon the standard treatment with temozolomide.
Experimental Procedures
Hoechst 33342 staining and flow cytometry
Cells were dissociated from normal brain cortex or tumors and resuspended at 106 cells/ml in Hanks' balanced salt solution containing 2% fetal bovine serum (FBS), 10 mM Hepes, 25 U/ml penicillin G and 25 μg/ml streptomycin. Cells were preincubated at 37°C for 30 minutes with or without 100μM verapamil (Sigma-Aldrich, St. Louis), 25μM pantoprazole (MSKCC) or 5μM fumitremorgin C (Sigma-Aldrich) to inhibit ABC transporters and incubated for 90 minutes at 37°C with 5 μg/ml Hoechst 33342 (Sigma-Aldrich). Cells were incubated on ice for 10 min and washed with ice cold HBSS. For immunostaining, cells were blocked with normal goat serum and incubated 30 min on ice with FITC anti-mouse CD31 (1:100), PE anti-mouse CD133 (1:100), PE anti-mouse CD11b (1:100) (eBioscience, San Diego, CA, USA) anti-A2B5 (1:200) (R&D Systems, Minneapolis, MN USA) followed by Streptavidin-Phycoerythrin (1 :400) (R&D Systems). FITC or PE isotype controls were used as negative control (1:100). For staining of U87MG cell line, cells were trypsinized, blocked for 15 min at room temperature with 1ug mouse IgG/105 cells and incubated 30 min on ice with APC anti-hABCG2 or APC isotype control (1:10). Hoechst dye was excited at 407 nm by trigon violet laser and its dual wavelengths were detected using 450/40 (Hoechst 33342-Blue) and 695/40 (Hoechst 33342-Red) filters. FITC and PE were excited at 488 nm by octagon blue laser and fluorescence was detected using 530/30 and 675/20 filters, respectively. Fl4 excitation of APC was at 633 nm and emission at 660 nm. Dead cells were excluded by gating on forward and side scatter and eliminating PI-positive population. The data were analyzed by FlowJo (Ashland, OR, USA).
Intracranial injection of mouse and human glioma cells
For assessment of tumorigenicity, mouse or human neurospheres were stained with Hoechst 33342 for subsequent sorting of SP and non-SP cells. 1×104 mouse cells were injected into the cortex new-born pups cortex (Ntv-a/Ink4a−/−Arf−/− PTENflox mice).
For human cells, injection were performed using stereotactic fixation device device (Stoelting, Wood Dale, IL). Mice used for these experiments were nude at age range 5-6 week-old. Mice were anaesthetized by an i.p. injections of ketamine (0.1 mg/g) and xylazine (0.02 mg/g). 1μl of 2×105 SP and MP sorted cell suspensions were delivered using 30-gauge needle attached to Hamilton syringe. Locations were determined by using mouse atlas (Franklin and Paxinos, 2007). Coordinates for injections in right hemisphere are AP-2mm from bregma, Lat-0.5mm (left or right) depth-1mm from dural surface. Mice were monitored carefully for strong symptoms of tumor development and where sacrificed (lethargy, head tilt).
Flow cytometric detection of mitoxantrone accumulation and cell cycle analysis
The ability of neurospheres and U87MG cell lines to extrude mitoxantrone was measured with a FACS calibur flow cytometer (Becton Dickson Medical Systems, Sharon, MA, USA), equipped with an argon laser. Cells were preincubated with 5μM fumitremorgin C for 30 min and 3 μM mitoxantrone was added and the cells were incubated for 60 min at 37°C, 5% CO2. Cells were washed with ice cold PBS and mitoxantrone fluorescence of 10, 000-gated events was logarithmically measured at a laser excitation wavelength of 635 nm through a 670 nm band-pass filter. Measurements were performed on duplicate samples and experiments were performed in triplicate.
Statistical Analysis
Comparison between two groups was made using Student's t test. Data represent the mean ± SD. P values of < 0.05 were considered statistically significant. The differences in in vitro growth among treatments were tested using the analysis of covariance (ANCOVA). For Kaplan–Meier survival curves, the Tarone–Ware method was used.
Supplementary Material
Acknowledgments
We thank Jim Finney, Zhang Qunchao, Alicia Pedraza-Fernandez, Benjamin Lee and Muge Akpinar for technical assistance; Massimo Squatrito and Amanda Katz for critical reading of this manuscript and technical assistance; the flow cytometry and molecular core facility. This work was supported by NIH grant U54CA126518, American Brain tumor Association (in memory of George Kamberos) and Pediatric Brain Tumor Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Casorelli I, Pelosi E, Biffoni M, Cerio AM, Peschle C, Testa U, Bignami M. Methylation damage response in hematopoietic progenitor cells. DNA repair. 2007;6:1170–1178. doi: 10.1016/j.dnarep.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Chua C, Zaiden N, Chong KH, See SJ, Wong MC, Ang BT, Tang C. Characterization of a side population of astrocytoma cells in response to temozolomide. Journal of neurosurgery. 2008;109:856–866. doi: 10.3171/JNS/2008/109/11/0856. [DOI] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & development. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- Diop NK, Hrycyna CA. N-Linked glycosylation of the human ABC transporter ABCG2 on asparagine 596 is not essential for expression, transport activity, or trafficking to the plasma membrane. Biochemistry. 2005;44:5420–5429. doi: 10.1021/bi0479858. [DOI] [PubMed] [Google Scholar]
- Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; New York: 2007. [Google Scholar]
- Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183:1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Becher OJ, Holland EC. Cancer stem cells and survival pathways. Cell Cycle. 2008a;7:1371–1378. doi: 10.4161/cc.7.10.5954. [DOI] [PubMed] [Google Scholar]
- Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes & development. 2008b;22:436–448. doi: 10.1101/gad.1627008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner MK. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer research. 2007;67:4827–4833. doi: 10.1158/0008-5472.CAN-06-3557. [DOI] [PubMed] [Google Scholar]
- Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7:356–368. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MO, Kanemura Y, Tajria J, Mori H, Kobayashi S, Hara M, Yamasaki M, Okano H, Miyake J. Functional expression of ABCG2 transporter in human neural stem/progenitor cells. Neurosci Res. 2005;52:75–82. doi: 10.1016/j.neures.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, Wu W, James CD, Sarkaria JN. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-oncology. 2008 doi: 10.1215/15228517-2008-090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConville P, Hambardzumyan D, Moody JB, Leopold WR, Kreger AR, Woolliscroft MJ, Rehemtulla A, Ross BD, Holland EC. Magnetic resonance imaging determination of tumor grade and early response to temozolomide in a genetically engineered mouse model of glioma. Clin Cancer Res. 2007;13:2897–2904. doi: 10.1158/1078-0432.CCR-06-3058. [DOI] [PubMed] [Google Scholar]
- Mouthon MA, Fouchet P, Mathieu C, Sii-Felice K, Etienne O, Lages CS, Boussin FD. Neural stem cells from mouse forebrain are contained in a population distinct from the ‘side population’. J Neurochem. 2006;99:807–817. doi: 10.1111/j.1471-4159.2006.04118.x. [DOI] [PubMed] [Google Scholar]
- Ostermann S, Csajka C, Buclin T, Leyvraz S, Lejeune F, Decosterd LA, Stupp R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin Cancer Res. 2004;10:3728–3736. doi: 10.1158/1078-0432.CCR-03-0807. [DOI] [PubMed] [Google Scholar]
- Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer research. 2005;65:6207–6219. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- Provenzale JM, Mukundan S, Dewhirst M. The role of blood-brain barrier permeability in brain tumor imaging and therapeutics. AJR Am J Roentgenol. 2005;185:763–767. doi: 10.2214/ajr.185.3.01850763. [DOI] [PubMed] [Google Scholar]
- Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer research. 2000;60:47–50. [PubMed] [Google Scholar]
- Regenbrecht CR, Lehrach H, Adjaye J. Stemming Cancer: Functional Genomics of Cancer Stem Cells in Solid Tumors. Stem Cell Rev. 2008 doi: 10.1007/s12015-008-9034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Polgar O, Deeken J, To KW, Bates SE. ABCG2: determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007;26:39–57. doi: 10.1007/s10555-007-9042-6. [DOI] [PubMed] [Google Scholar]
- Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer research. 2004;64:4783–4789. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Wu C, Alman BA. Side population cells in human cancers. Cancer Lett. 2008 doi: 10.1016/j.canlet.2008.03.048. [DOI] [PubMed] [Google Scholar]
- Yuan X, Curtin J, Xiong Y, Liu G, Waschsmann-Hogiu S, Farkas DL, Black KL, Yu JS. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, Margolick JB, Liotta LA, Petricoin E, 3rd, Zhang Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16158–16163. doi: 10.1073/pnas.0702596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.