

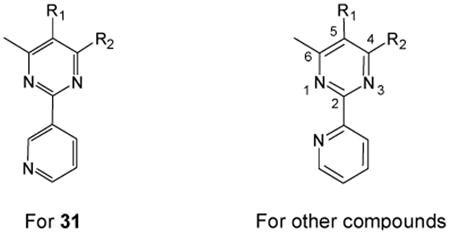
Abstract
Cellular protein synthesis is initiated with methionine in eukaryotes with few exceptions. Methionine aminopeptidases (MetAPs) which catalyze the process of N-terminal methionine excision are essential for all organisms. In mammals, type 2 MetAP (MetAP2) is known to be important for angiogenesis, while type 1 MetAP (MetAP1) has been shown to play a pivotal role in cell proliferation. Our previous high-throughput screening of a commercial compound library uncovered a novel class of inhibitors for both human MetAP1 (HsMetAP1) and human MetAP2 (HsMetAP2). This class of inhibitors contains a pyridinylpyrimidine core. To understand the structure-activity relationship (SAR) and to search for analogues of 2 with greater potency and higher HsMetAP1-selectivity, a total of fifty-eight analogues were acquired through either commercial source or by in-house synthesis and their inhibitory activities against HsMetAP1 and HsMetAP2 were determined. Through this systematic medicinal chemistry analysis, we have identified (1) 5-chloro-6-methyl-2-pyridin-2-ylpyrimidine as the minimum element for the inhibition of HsMetAP1; (2) 5′-chloro as the favored substituent on the pyridine ring for the enhanced potency against HsMetAP1; and (3) long C4 side chains as the essentials for higher HsMet AP1-selectivity. At the end of our SAR campaign, 25b, 25c, 26d and 30a–30c are among the most selective and potent inhibitors of purified HsMetAP1 reported to date. In addition, we also performed crystallographic analysis of one representative inhibitor (26d) in complex with N-terminally truncated HsMetAP1.
Keywords: Methionine aminopeptidase, Pyridinylpyrimidine, Anti-cancer
1. Introduction
With few exceptions, cellular protein synthesis is initiated with either an N-formylmethionine in eubacteria, mitochondria and plastids or a methionine in archaea and eukaryotes1. In eubacteria, the initiator methionine at the N-terminus of a significant portion of proteins is irreversibly removed,2 following the cotranslational deformylation catalyzed by peptide deformylase.3,4 In eukaryotes, the process of N-terminal methionine excision (NME) no longer requires peptide deformylase and became cotranslational.5 In all cells, NME is catalyzed by a family of ubiquitous enzymes named methionine aminopeptidases (MetAPs).6 As metalloproteases, MetAPs utilize one or two divalent metals to proteolytically cleave the initiator methionine from nascent peptides,7 with strict substrate specificity. Cleavage will only occur when the P1 residue is methionine and the side chain of P1′ residue is small and uncharged.8,9 Residues at the positions of P2′ and beyond also contribute to the efficiency of cleavage.8c,9
MetAPs are classified into two types. Type 1 enzymes originated from eubacteria, and type 2 originated from archaea. Only type 2 MetAP contains a characteristic insertion in its catalytic domain.6 Although archaea only have type 2 MetAPs and eubacteria only have type 1 MetAPs,7b,10,11 at least two genes encoding one cytosolic type 1 MetAP (MetAP1) and one cytosolic type 2 MetAP (MetAP2), respectively, can be found in all eukaryotic genomes.12,13 There is limited sequence identity between type 1 and type 2 MetAPs,12,13 but crystallographic analysis demonstrated that the catalytic domains of all MetAPs adopt the same pita-bread fold, in which the N- and C-terminal halves display pseudosymmetry about the active site.6 Moreover, the five metal binding residues and the shape of methionine-binding pocket are also highly conserved.6,14
The ubiquitous distribution of MetAP, their highly conserved structures and substrate specificity, suggest that NME may play an important role in cellular life. Mycoplasma genitalium has the smallest genome to support cellular growth in pure medium. One gene (map) encoding a MetAP has been found among its 382 essential genes.15 In eubacteria, the importance of MetAP is underscored by the lethality when the single map gene in Escherichia coli or Salmonella typhimurium was disrupted.10,11 In Saccharomyces cerevisiae, deletion of either map1 or map2 gene caused a slow growth phenotype, and double deletion of both map genes was lethal.12 In Arabidopsis thaliana, cytosolic MetAP1 and MetAP2s are functionally redundant. However, the complete inactivation of NME in the cytosol blocked the plant's development after germination.16 In animals, abrogation of cytosolic MetAP2 leads to organism-specific and tissue-specific developmental defects. Most of the defects caused either lethality or infertility.17 Small-interfering RNA (siRNA) mediated downregulation of either MetAP1 or MetAP2 significantly inhibited the proliferation of human umbilical vein endothelial cells, suggesting that both human MetAPs (HsMetAPs) are essential for cell proliferation.18 More recently, Hu et al. demonstrated that MetAP1 siRNA duplexes could delay the cell cycle progression of synchronized HeLa cells.19 Bengamides, a family of natural products isolated from marine sponges,20,21 were identified as nonselective inhibitors of both MetAP1 and MetAP2.22 The antitumor activities of bengamides in vitro21 and in vivo23 evince a pivotal role of MetAPs in human cells.
Over two decades ago, fumagillin, a natural metabolite of Aspergillus fumigatus, and its synthetic analogue TNP-470 (also known as AGM-1470) were discovered as potent inhibitors of angiogenesis.24,25 Subsequently, we and others identified MetAP2 as the primary target of fumagillin and TNP-470,26 and found that both inhibitors specifically inhibit MetAP2 without affecting MetAP1.27,28 Because the ablation of MetAP2's enzymatic activity preferentially inhibited endothelial cell proliferation, hence angiogenesis,29,30 TNP-470 displayed broad-spectrum activities against the growth of both primary and metastatic tumors in various animal models (for reviews, see31,32). TNP-470 was also effective against nutritionally induced obesity33, pulmonary hypertension34 and arthritis35. Inspired by fumagillin and its derivatives, there has been a renewed interest in developing MetAP2-selective inhibitors to treat cancer, rheumatoid arthritis and collagen-induced arthritis. Besides serving as a valuable prototype of angiogenesis inhibitors, fumagillin and its derivatives also greatly facilitated the investigation of MetAP2's functions in signal transduction, cell cycle progression and development of higher eukaryotes.
The shortage of potent and isoform-selective inhibitors of MetAP1 not only hampered the advancement of MetAP1 biology, but also restrained the evaluation of MetAP1 as a novel target for anti-antineoplastic therapy. Peptidyl hydroxamic acids were shown to inhibit human MetAP1 (HsMetAP1) in vitro. However, their inhibitory potency was low and lacked isoform selectivity with similar Ki values for the inhibition of both purified HsMetAP1 and human MetAP2 (HsMetAP2).36 Later, several barbiturate-based compounds were identified in an in vitro assay as low micromolar inhibitors of HsMetAP1. But their isoform selectivity is still unclear.37 We and others have previously reported that the derivatives of pyridine-2-carboxamide (such as compound 1, Figure 1) inhibited the purified type 1 MetAP from either prokaryotes (Escherichia coli) or eukaryotes (Saccharomyces cerevisiae and human) in vitro.19,38,39 Intriguingly, this class of inhibitors not only have low micromolar IC50 values against HsMetAP1 but also showed great selectivity for HsMetAP1 over HsMetAP2.19 However, we subsequently found that at low micromolar concentrations, 1 also had unexpected off-target celllular effects (a corrigendum will be published later). In addition to pyridine-2-carboxamides, a novel class of inhibitors containing 2-pyridinylpyrimidine (such as compound 2, Figure 1) was discovered in our group via a high-throughput screening of a commercial library.40 These pyridinylpyrimidines potently inhibited both purified HsMetAP1 and purified HsMetAP2 with moderate selectivity for HsMetAP1. Subsequently, crystallographic analysis of the catalytic domain of HsMetAP1 in complex with inhibitors revealed that both 1 and 2 mainly depended on the same auxiliary Co(II) (M3) in the active site to inhibit HsMetAP1 (Figure 2A and Figure 2B).19,40 However, Hu et al. showed that 1 and 2 could inhibit HsMetAPs inside HeLa cells.19,40 In addition, Chai and Ye demonstrated that an auxiliary metal-mediated quinolinyl sulfonamide (compound 3, Figure 1) inhibited cellular MetAP in bacteria as well, albeit at a high concentration.41
Figure 1.

Structures of MetAP inhibitors 1-3.
Figure 2.

The X-ray crystal structures of the N-terminally truncated human MetAP1 (tHs MetAP1) in complex with 1 (2NQ6 [19]) (A) or 2 (2G6P [40]) (B) and (C). For all non-carbon atoms, nitrogen is blue, oxygen is red, sulfur is yellow and chlorine is green. In the active site of tHsMetAP1, Co(II) ions are shown as hot pink spheres, water molecules are shown as small red spheres and metal interactions are shown as dashed lines. In (A) and (B), 1 (carbon green), 2 (carbon purple) and the surrounding residues (carbon white) are shown as sticks. (C) In a surface-filling model of tHs MetAP1 (carbon white) in complex with Co(II) and 2, a HEPES molecule (carbon magenta) from the buffer is accommodated in a groove formed by Y196, G352 and W353. The HEPES molecule resides on the exterior of the phenyl group of 2, and generally parallels to the phenyl group.
Herein we describe a systematic medicinal chemistry approach to probing the structural requirements of pyridinylpyrimidine scaffold for selective inhibition of HsMetAP1. The X-ray crystal structure of compound 2 bound to the Co(II) form of the N-terminally truncated HsMetAP140 was used to guide our SAR studies. We set out to address: (1) the minimum elements of the pyridinylpyrimidine skeleton needed for inhibition of HsMetAP1; (2) the SAR of substitution at the C3′–C6′ positions of the pyridine ring; and (3) the optimal side chain at the C4 position of the pyrimidine ring. In addition to eight compounds purchased from commercial sources, we designed and synthesized fifty analogues based on the pyridinylpyrimidine scaffold and characterized their inhibitory activity with purified HsMetAP1 and HsMetAP2. We also performed crystallographic analysis of one representative inhibitor (26d) in complex with N-terminally truncated HsMetAP1 (PDB access code 4HXX).
2. Chemistry
Analogous to the procedure described by Medwid et al.,42 we started our synthesis of 2-pyridinylpyrimidine derivatives by transforming substituted picolinonitriles 4 into the corresponding picolinimidamides 5 (Scheme 1). Suitably substituted ethyl 3-oxobutanoates were condensed with 5 under basic condition using sodium methoxide and the resulting pyrimidinones 7 were refluxed with phosphoryl chloride to obtain 4-chloropyrimidines 8. The displacement of the 4-chloro group on pyrimidine 8 by phenethylamine was accomplished by refluxing the reaction mixture in methanol containing potassium carbonate. Thus, by using ethyl 2-fluoroacetoacetate, ethyl 2-methylacetoacetate, or ethyl acetoacetate in the condensation step to generate pyrimidinone 7, 2-pyridin-2ylpyrimidines 9a, 9b or 9c, respectively, were obtained as the final products. Pyrimidine derivatives carrying an ethylene diamine side chain 11, were synthesized by treating chloropyrimidine 8 with Boc-protected ethylyene diamine 10, and by treating the resulting product with trifluoroacetic acid. The pyrimidine derivatives 11 served as the intermediate in the synthesis of other side chain analogues of 2-pyridin-2-ylpyrimidine. Compound 13 was prepared by alkylating 5-chloro-6-methyl-2-(pyridin-2-yl)-3,4-dihydropyrimidine-4-thione 12 with phenethyl bromide as shown in Scheme 2.
Scheme 1.

Synthesis of 2-pyridinyl-6-methylpyrimidines. Reagents & Conditions: (i) MeONa/MeOH, rt; (ii) NH4Cl/MeOH, 65-74% yields for two steps; (iii) H3C-COCH(X)CO2Et (6), MeONa/MeOH, 45-53% yields; (iv) POCl3, reflux, 65-72% yields; (v) PhCH2CH2NH2, K2CO3/MeOH, 65 °C, 85-92% yield; (vi) (R)-H2NCH2CH(Ph)NHBoc (10), Et3N, THF, 82-95% yields; (vii) CF3CO2H/CH2Cl2, then aqueous NaHCO3, 81-86% yields. Yield for 9a, 9b, and 9c were 56%, 62%, and 49% over three steps, respectively.
Scheme 2.

Synthesis of 13. Reagents & Conditions: (i) K2CO3, PhCH2CH2Br, toluene, 60 °C, 92% yield.
Acylation of amines 11 provided chlorides 14 (Scheme 3) which were reacted with piperazine derivative 15 to afford amino alcohols 16a and 16b after desilylation. Additionally, replacement of chloride in 14 with Boc-protected piperazine 16 and subsequent deprotection produced a series of biaryl compounds (18a–18o) with different substituents on the pyridine ring. In a parallel scheme (Scheme 4), amine 11a was reacted with two piperazine-containing alkyl bromides (19a and 19b) and the resulting adducts were dprotected with TFA and TBAF respectively to give compounds 20a and 20b. Reductive amination of 11a with two piperazine-containing aldehydes (21a and 21b) furnished compounds 22a and 22b, after removal of the protecting groups. The sulfonamides 24a and 24b were assembled from condensation of the amine 11a with ethenesulfonyl chloride (Scheme 5). The resultant sulfonamide 23 was reacted with piperazines 15 and 17 to afford 24a and 24b upon deprotection with TFA or TBAF.
Scheme 3.

Synthesis of 2-pyridinylpyrimidine derivatives 16a, 16b and 18a–18p. Reagents & Conditions: (i) Cl(CH2)nCH2COCl, Et3N, THF, ∼80% yield; (ii) NaI, Na2CO3, DMF then TBAF, 67-74% yields; (iii) NaI, Na2CO3, DMF, then CF3CO2H, 62-79% yields.
Scheme 4.

Synthesis of additional 2-pyridinylpyrimidine derivatives 20a, 20b, 22a and 22b. Reagents & Conditions: (i) NaI, DMF then TFA or TB AF, 84-88% yields; (ii) NaBH3CN, HO Ac, MeOH, then TFA or TBAF, 55-60% yields.
Scheme 5.

Synthesis of sulfonamide derivatives 24a and 24b. Reagents & Conditions: (i) CH2=CHSO2Cl, Et3N, CH2Cl2, 91% yield; (ii) 15 or 17, EtOH 50 °C, 80-90% yields; (iii) TFA or TBAF, 80-84% yields.
Amides 25a–25e were prepared by simple condensation of pyridinylpyrimidine 11b with carboxylic acids (Scheme 6). A series of secondary amines (26a–26f and 27a–27h) were obtained via either reductive amination or a simple displacement reaction (Scheme 6). To synthesize compounds 30a–30d, we carried out alkylation reaction of 2-phenylacetonitrile with alkyl bromides (Scheme 7). The resultant alkylation products 28 were reduced with LAH to afford primary amines 29, which were coupled with the aryl chloride 11b to yield 30a–30d in good yields.
Scheme 6.

Synthesis of 2-(5-chloro-pyridin-2-yl)-pyrimidine derivatives 25a–25e, 26a–26f and 27a–27h. Reagents & Conditions: (i) Ph(CH2)nCO2H, EDCI, HOBt, DIPEA, 70-85% yields; (ii) Ar(CH2)nCHO, NaBH3CN, HO Ac, MeOH, 25-35% yields; (iii) NaI, Et3N, CH3CN, 37% yields.
Scheme 7.

Synthesis of 2-(5-chloro-pyridin-2-yl)-pyrimidine derivatives 30a–30d. Reagents & Conditions: (i) Ph(CH2)nBr, NaH, DMF, 0 °C, 50-65% yields; (ii) LAH, ether, 80-90% yields; (iii) 8, Et3N, THF, reflux, 70-80% yields.
3. Results and discussion
3.1 Improvement in potency and isoform selectivity of pyridinylpyrimidine derivatives against human MetAP1
The starting point for this work was compound 2, one of the best inhibitors of both Plasmodium falciparum MetAP1b and human cytosolic MetAPs (HsMetAP1 and HsMetAP2) identified through a high-throughput screening against approximately 175,000 compounds.40,43 According to the crystal structure of N-terminally truncated HsMetAP1 in complex with Co(II) and 2, compound 2 mainly relied on an auxiliary Co(II)(M3) in the active site of HsMetAP1 to inhibit the enzyme (Figure 2B).40 This auxiliary Co(II) adopted a distorted octahedral geometry, and was hexacoordinated by a pyrimidine nitrogen and a pyridine nitrogen from the inhibitor, an imidazole nitrogen from His212 of HsMetAP1 and three water molecules (W1, W2 and W3 in Figure 2B). In agreement with this vital role of pyridinylpyrimidine in coordinating the auxiliary Co(II), replacement of 2-pyridinyl group in 2 with phenyl, 3-pyridinyl or 4-pyridinyl group gave compounds exhibiting very poor potency against Plasmodium falciparum MetAP1b.43 Similarly, compound 31, a 2-(3-pyridinyl)-pyrimidine analogue, had no effect on human MetAP at concentrations up to 100 μM (Table 1). Together, these lines of evidence suggested that the 2-(2-pyridinyl)-pyrimidine capable of chelating a metal ion is a key pharmacophore for the inhibition of MetAP enzymes.
Table 1. Inhibition of purified recombinant human MetAPs by pyridinylpyrimidine derivatives.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Cmpd | R1 | R2 | IC50a (μM) | IC50 ratio (type 2/1) | |
|
| |||||
| HsMetAP1 | HsMetAP2 | ||||
| 2 | Cl | NHCH2CH2Ph | 0.86 ± 0.04 | 9.0 ± 0.9 | 10 |
| 9a | F | NHCH2CH2Ph | 4.6 ± 0.1 | 62 ± 9 | 13 |
| 9b | Me | NHCH2CH2Ph | >100 | >100 | NAb |
| 9c | H | NHCH2CH2Ph | >100 | >100 | NAb |
| 13 | Cl | SCH2CH2Ph | 10 ± 1 | >100 | >10 |
| 31 | Cl | NHCH2Ph | >100 | >100 | NAb |
| 32 | Cl | SCH2Ph | 11 ± 1 | >100 | >9.1 |
| 33 | Cl | N(Me)CH2Ph | 1.5 ± 0.1 | 13 ± 0.1 | 8.7 |
| 34 | Cl | NHMe | 8.9 ± 0.3 | 14 ± 1 | 1.6 |
| 35 | Cl |
|
1.0 ± 0.1 | >100 | >100 |
| 36 | Cl |
|
2.0 ± 0.1 | >100 | >50 |
| 37 | Cl | 4-(2-hydroxyl-ethyl)-piperazinyl | 1.1 ± 0.2 | >100 | >91 |
| 38 | Cl |
|
0.36 ± 0.03 | >100 | >278 |
IC50 values of compounds to inhibit human MetAPs were shown as mean ± SD of three determinations. The bottom values of all inhibitory dose-response curves were smaller than 10%.
NA: not available.
To determine the role of chlorine substitution at C5 of the pyrimidine ring (Table 1), three analogues were synthesized. Replacement of chlorine (2) with fluorine (9a) decreased the potency against HsMetAP1 and HsMetAP2 by 5.3-fold and 6.9-fold, respectively. Moreover, replacement of the chlorine with a methyl group (9b) or hydrogen (9c) led to inactive analogues, suggesting that chlorine is the optimal substituent at C5 of the pyrimidine ring.
Next, we assembled eight analogues with different substituents at C4 of the pyrimidine ring (Table 1). Swapping the secondary amine for thioether linkages (13 and 32) resulted in a significant decrease in potency against both HsMetAP1 and HsMetAP2 without any appreciable change in the isoform selectivity in favor of HsMetAP1. High similarity between the structures of the catalytic domains of HsMetAP1 and HsMetAP2 presents a formidable challenge in the design of type 1-selective inhibitors. In our previous report of Plasmodium falciparum MetAP1b inhibitors, a wide range of substituents at C4 were well tolerated by PfMetAP1 enzyme. However, when the X-ray crystal structure of HsMetAP1 in complex with 2 was superimposed on the structure of HsMetAP2, several potential steric clashes between the inhibitor and HsMetAP2 were revealed.40 The most severe steric clash was between the C4 side chain of 2 and the Tyr444 residue of HsMetAP2. Presumably, an unfavorable interaction like this could be exploited to improve the inhibitor's selectivity for HsMetAP1 over HsMetAP2. Indeed, decreases in size of C4 substituents diminished HsMetAP1 selectivity (33 and 34). On the contrary, incorporating longer and more polar C4 side chains generally yielded substantial increases in the selectivity for HsMetAP1 over HsMetAP2, and at the same time the potency against HsMetAP1 was retained (35–38).
The crystal structure of compound 2 in complex with N-terminally truncated HsMetAP1 also revealed an unexpected 2-[4-(2-hydroxylethyl)-1-piperazinyl]ethanesulfonic acid (HEPES) molecule from the buffer bound in a groove formed by Tyr196, Gly352 and Trp353. This HEPES molecule resided on the exterior of the phenyl group of 2 (Figure 2C). We postulated that the binding of pyridinylpyrimidine analogues to HsMetAP1 might benefit from extending C4 substituent of 2 all the way to the piperazine ring of HEPES. One such compound (24b in Table 2) was synthesized by fusing HEPES moiety to compound 2 via the C4 side chain. Although 24b did not gain appreciable potency against HsMetAP1, there was a considerable decrease in its potency against HsMetAP2. As a result, 24b achieved 158-fold selectivity for HsMetAP1 over HsMetAP2. In light of this increased selectivity for HsMetAP1, nine more pyridinylpyrimidine analogues were synthesized (Table 2). When the sulfonamide in the linker in 24b was replaced by either a secondary amine (20b) or an amide (16a), there was little change in potency and HsMetAP1-selectivity. Extending the amine linker and the amide linker by one carbon further improved the HsMetAP1-selectivity (22b and 16b). On the contrary, compared to 24b and 16a, shortening the side chain by removal of the 2-hydroxylethyl group led to 13-fold and 4-fold decreases in the HsMetAP1-selectivity of 24a and 18a, respectively. However, removing the same 2-hydroxylethyl group did not have any appreciable effect on two analogues with secondary amine linkers (20a vs. 20b, and 22a vs. 22b) and one analogue with a longer amide linker (18b vs. 16b). Thus, installation of a long side chain containing a piperazinyl group to the C4 position of the pyrimidine ring resulted in the largest improvement in HsMetAP1-selectivity.
Table 2. Inhibition of purified recombinant human MetAPs by pyridinylpyrimidine derivatives.
| ||||
|---|---|---|---|---|
|
| ||||
| Cmpd | R | IC50a (μM) | IC50 ratio (type 2/1) | |
|
| ||||
| HsMetAP1 | HsMetAP2 | |||
| 16a |
|
0.8 ± 0.1 | 114 ± 12 | 143 |
| 16b |
|
1.1 ± 0.1 | >300 | >272 |
| 18a |
|
0.6 ± 0.1 | 22 ± 4 | 37 |
| 18b |
|
1.1 ± 0.2 | >300 | >272 |
| 20a |
|
1.4 ± 0.2 | 108 ± 11 | 77 |
| 20b |
|
1.7 ± 0.5 | 101 ± 12 | 59 |
| 22a |
|
1.3 ± 0.5 | 188 ± 17 | 145 |
| 22b |
|
0.8 ± 0.2 | 197 ± 22 | 246 |
| 24a |
|
0.8 ± 0.1 | 9.4 ± 1.0 | 12 |
| 24b |
|
0.6 ± 0.1 | 95 ± 14 | 158 |
IC50 values of compounds to inhibit human MetAPs were shown as mean ± SD of three determinations. The bottom values of all inhibitory dose-response curves were smaller than 10%.
After a comparative analysis of twenty analogues with various side chains, we selected 18b as a lead to explore the SAR of substituents on the pyridine ring of pyridinylpyrimidine (Table 3). Compared to 18b, installation of either a methoxyl group or a chloro group at C6′ or C3′ reduced the potency against HsMetAP1 to different degrees (18c, 18d, 18e and 18f). On the contrary, the potency was enhanced by 4.1-fold via the introduction of a methoxyl substituent at the adjacent C5′ position (18g). Similarly, introduction of fluoro (18h), chloro (18i), bromo (18j), nitro (18k) or dimethylamino (18l) group at C5′ led to 3.3-fold to 31-fold increases in the anti-HsMetAP1 activities relative to 18b. Unfortunately, the potency against HsMetAP2 was simultaneously restored for 18h, 18i, 18k and 18l, significantly compromising HsMetAP1-selectivity. A pyrrolidinyl substituent, which is bigger than a dimethylamino group, was not well tolerated at C5′, as indicated by the reduced inhibitory potency and the loss of HsMetAP1-selectivity of 18m. In contrast to the substituents at C6′ and C3′, analogues with methoxy (18n) and chloro (18o) substituents at C4′ maintained their potency against HsMetAP1. However, the HsMetAP1-selectivity of 18n was attenuated by the relatively bigger C4′ substituent.
Table 3. Inhibition of purified recombinant human MetAPs by pyridinylpyrimidine derivatives.
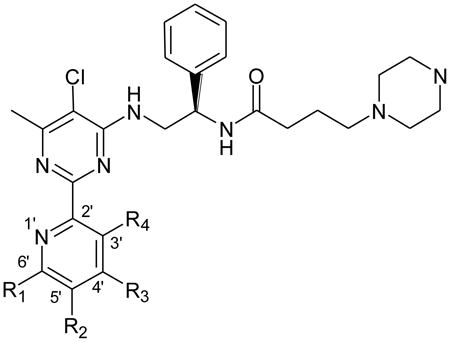
| ||||
|---|---|---|---|---|
|
| ||||
| Cmpd | Substituents (when not specified, R# = H) | EC50a (μM) | EC50 ratio (type 2/1) | |
|
| ||||
| HsMetAP1 | HsMetAP2 | |||
| 18c | R1 = OMe | 20 ± 6 | >30b | >1.5 |
| 18d | R1 = Cl | 7.2 ± 2.4 | >30b | >4.2 |
| 18e | R4 = OMe | 2.5 ± 1.3 (Bc = 0.15)* | >300 | >120 |
| 18f | R4 = Cl | 7.6 ± 0.4 | >300 | >39 |
| 18g | R2 = OMe | 0.27 ± 0.04 | >300 | >1111 |
| 18h | R2 = F | 0.049 ± 0.07 | 1.2 ± 0.3 | 24 |
| 18i | R2 = Cl | 0.036 ± 0.006 | 1.2 ± 0.3 | 33 |
| 18j | R2 = Br | 0.33 ± 0.01 | 53 ± 5 | 161 |
| 18k | R2 = NO2 | 0.30 ± 0.06 | 14 ± 1 | 47 |
| 18l | R2 = N(CH3)2 | 0.14 ± 0.05 | 1.7 ± 0.7 | 12 |
| 18m | R2 = pyrrolidinyl | 1.7 ± 0.3 (B = 0.36) | 1.1 ± 0.1 (B = 0.22) | 1.1 |
| 18n | R3 = OMe | 0.53 ± 0.07 | 14 ± 9 | 26 |
| 18o | R3 = Cl | 0.73 ± 0.03 | >300 | >411 |
EC50 values of compounds to inhibit human MetAPs were shown as mean ± SD of two to three determinations.
Higher concentrations have not been tested for these compounds.
B: bottom value of the inhibitory dose-response curve. It has been noted in parentheses only when B>10%.
As 18i exhibited impressive potency against HsMetAP1 and moderate selectivity over HsMetAP2, we decided to further explore the C4 side chain to improve HsMetAP1-selectivity (Table 4). First, swapping of the piperazinyl group in 18i to a phenyl group greatly improved HsMetAP1-selectivity (25a–25e), best exemplified by 25b and 25c. Except 25d, other compounds in this series retained submicromolar IC50 values for HsMetAP1. Secondly, changing the carbonyl group in side chain of 25a–25e to a methylene group afforded a new series of analogues (26a–26f) with submicromolar IC50 values and comparable selectivity for HsMetAP1 in comparison to 25a–25e. Thirdly, derivatizing 26d to 27a–27d or replacing the phenyl group at the end of the side chain with various heterocyclic aromatic rings (27e–27g) only led to modest (1.6-fold to 6.0-fold) reductions in the potency against HsMetAP1. Except 27c that potently inhibited the coupling enzyme in BcProIP-coupled HsMetAP2 assay, the remaining eight analogues in this series were inactive against HsMetAP2 at the highest concentrations tested. And lastly, 30a–30d were generated by using 3–6 carbon aliphatic linkers to replace the secondary amino linker that tied the two phenyl groups together in the C4 side chain of 26d. All analogues in this series bore excellent HsMetAP1-selectivity. And when the aliphatic linker was shortened, the compound appeared to be more potent (compare 30a with 30d), with analogue 30a being among the most selective and potent inhibitor of HsMetAP1 reported to date.
Table 4. Inhibition of purified recombinant human MetAPs by pyridinylpyrimidine derivatives.
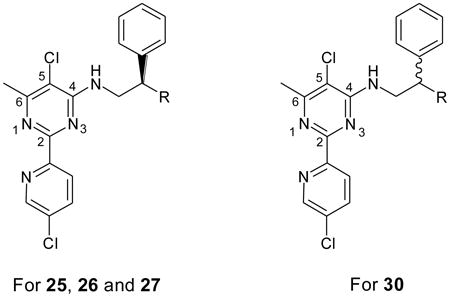
| ||||
|---|---|---|---|---|
|
| ||||
| Cmpd | R | EC50a (μM) | IC50 ratio (type 2/1) | |
|
| ||||
| HsMetAP1 | HsMetAP2 | |||
| 25a | NHCOCH2Ph | 0.70 ± 0.52 | 28 ± 6 | 40 |
| 25b | NHCO(CH2)2Ph | 0.093 ± 0.042 | >30b | >323 |
| 25c | NHCO(CH2)3Ph | 0.16 ± 0.12 | >30b | >188 |
| 25d | NHCO(CH2)4Ph | 1.3 ± 0.2 (Bc = 0.12) | >30b | >23 |
| 25e | NHCO(CH2)5Ph | 0.67 ± 0.30 | >30b | >45 |
| 26a | NHCH2Ph | 0.22 ± 0.7 | 31 ± 5 | 141 |
| 26b | NH(CH2)2Ph | 0.26 ± 0.05 | >100 | >384 |
| 26c | NH(CH2)3Ph | 0.26 ± 0.06 | >300 | >1154 |
| 26d | NH(CH2)4Ph | 0.20 ± 0.03 | >100 | >500 |
| 26e | NH(CH2)5Ph | 0.78 ± 0.19 | >100 | >138 |
| 26f | NH(CH2)6Ph | 0.71 ± 0.21 | >30b | >42 |
| 27a | NH(CH2)4-(4-phenolyl) | 0.42 ± 0.06 | >300 | >714 |
| 27b | NH(CH2)4-(4-chlorophenyl) | 0.57 ± 0.10 | >300 | >526 |
| 27c | NH(CH2)4-(4-cyanophenyl) | 0.52 ± 0.07 | 16 ± 7d | 31 |
| 27d | NH(CH2)4-(4-propoxylphenyl) | 1.2 ± 0.2 | >300 | >250 |
| 27e | NH(CH2)4-(2-furyl) | 0.33 ± 0.08 | >300 | >909 |
| 27f | NH(CH2)4-(2-thienyl) | 0.38 ± 0.18 | >300 | >789 |
| 27g | NH(CH2)4-(2-quinolyl) | 0.69 ± 0.08 | >300 | >435 |
| 27h | NH(CH2)4-(1-pyrrol) | 0.50 ± 0.09 | >100 | >200 |
| 30a | (CH2)2Ph | 0.063 ± 0.024 (Bc = 0.14) | >200 | >3175 |
| 30b | (CH2)3Ph | 0.16 ± 0.01 (Bc = 0.13) | >200 | >1250 |
| 30c | (CH2)4Ph | 0.20 ± 0.11 | >200 | >1000 |
| 30d | (CH2)5Ph | 2.3 ± 1.8 | >200 | >87 |
EC50 values of compounds to inhibit human MetAPs were shown as mean ± SD of two to three determinations.
Higher concentrations have not been tested for these compounds.
B: bottom value of the inhibitory dose-response curve. It has been noted in parentheses only when B>10%.
In the counter screening, compound 27c was identified as a potent inhibitor of the coupling enzyme, BcProIP. 10 μM and 100 μM of 27c inhibited 27% and 97% of the activity of BcProIP in MetAP assay buffer, in the presence of 600 μM Pro-pNA and 10 μM CoCl2.
3.2 The X-ray crystal structure of N-terminally truncated human MetAP1 in complex with compound 26d
The crystal structure of N-terminally truncated (Δ1–80) human MetAP1 (tHsMetAP1) in complex with Co(II) and 26d was determined to 2.09 Å resolution (Table 5). The locations of cobalt ions were identified by a difference electron density map computed with the anomalous diffraction signal (Figure 3A). Two Co(II) are presented at the bottom of the active site (Figure 2C), the ubiquitous positions for metal ions (M1 and M2) in the MetAP family. In addition, a third Co(II) (Co3) is located at a place (M3) close to the active site, which has previously been identified in the complexes of tHsMetAP1 with two specific classes of inhibitors.19,40 Among them, compound 2 has a pyridinylpyrimidine core similar to that of compound 26d. In the crystal structures of tHsMetAP1 in complex with 2 (PDB access code 2G6P, reported in [40]) and 26d, both pyridine nitrogen (N1′) and pyrimidine nitrogen (N1) are bound to Co3 (Figure 3B), displaying a near-perfect octahedral coordination which is completed by the nitrogen atom from the side chain of His212 and three water molecules.
Table 5. X-ray data collection and refinement statistics of the N-terminal truncated (Δl–80) human MetAP1 (tHsMetAP1) in complex with 26d.
| Crystal | tHsMetAP1 w. 26d |
|---|---|
| Space group | P21 |
| Cell dimensions | |
| a (Å) | 47.5 |
| b (Å) | 77.4 |
| c (Å) | 48.0 |
| β (deg) | 90.9 |
| X-ray data collection statistics | |
| X-ray Source | FR-E+/Raxis IV |
| Wavelength (Å) | 1.54178 |
| Resolution range (Å) (HighRes shell) | 50.00-2.09 (2.16-2.09) |
| Collected Reflections | 71,126 |
| Unique Reflections | 20,226 |
| I/σ | 29.6 (2.8) |
| Completeness (%) | 97.9 (86.6) |
| Rmerge (%) | 11.7 (49.0) |
| Refinement statistics | |
| Rcryst (%) | 0.18 (0.27) |
| Rfree (%) | 0.25 (0.33) |
| R.m.s deviations | |
| Bond length (Å) | 0.013 |
| Angle (deg) | 1.1533 |
| Monomer in ASU | 1 |
| Total Atoms | 2,586 |
| Protein atoms | 2,400 |
| Water molecules | 152 |
| Ligand | 34 |
| B-factor (ΔHsMetAP1)(Å2) | 41.8 |
| B-factor (26d)(Å2) | 62.0 |
| B-factor (H2O)(Å2) | 55.0 |
Figure 3.

The X-ray crystal structure (4HXX) of the N-terminally truncated human MetAP1 (tHs MetAP1) in complex with 26d (cyan) (A) and (C), and a superimposition of 4HXX with the crystal structure (2G6P [40]) of tHsMetAP1 with 2 (purple) (B) and (D). A HEPES molecule (carbon purple) in 2G6P was from the buffer. In (A) and (B), the stereo views of the active site of tHsMetAP1 show that both 26d and 2 (sticks) mainly depend on an auxiliary Co(II) (Co3, green sphere) to bind the H212 residue (sticks) of HsMetAP1. Water molecules are shown as red spheres and metal interactions are shown as dashed lines. In (C) and (D), the surface-filling views of tHsMetAP1 (surface colored gray) show the cavity in the active site which accommodates 26d and 2. The C4 side chains of 26d and 2 interact with different parts of the enzyme. For all non-carbon atoms, nitrogen is blue, oxygen is red, sulfur is yellow and chlorine is green.
26d buries about 468 Å2 upon binding tHsMetAP1 from a total accessible area of 753 Å2. Similar to 2, the pyridine ring of 26d is fitted in a hydrophobic pocket which is normally occupied by the side chain of the initiator methionine (Figure 3C). But when compared with 2, an extra chloro group is present at the C5′ position of the pyridine ring in 26d. This chloro group points towards the phenyl group of Phe198 which constitutes the bottom of the hydrophobic pocket for methionine side chain. In comparison to the pyridinylpyrimidine core of 2 in the structure 2G6P, 26d is displaced up to 1 Å out of the hydrophobic pocket for methionine side chain to accommodate the C5′ chloro substituent (Figure 3B). Therefore for 26d, the distances from C5′ of the pyridine ring of the inhibitor to C1 and C4 of the phenyl group in Phe198 increase from 3.87 Å and 4.64 Å in 2G6P to 4.77 Å and 5.70 Å, respectively. However, the pyridinylpyrimidine core of 26d still maintains the octahedral interaction with Co3 and Co3 is in a slightly changed position. Just like 2 in the structure 2G6P,40 the pyridine ring and the pyrimidine ring of 26d are still coplanar. In the complex of tHsMetAP1 with 26d, the distances from Co3 to the imidazole nitrogen of His212 and from Co3 to the pyridine nitrogen (N1′) of 26d remain the same as the two distances in 2G6P (2.20 Å, 2.13 Å). However, the distance from Co3 to the pyrimidine nitrogen (N1) of the inhibitor decreases from 2.32 Å in 2G6P to 2.27 Å. The inhibitor binding site does not change significantly, as the main chain remains within 0.25 rmsd of the previously described structure of tHsMetAP1 with 2. The fact that the pyridinylpyrimidine core of 26d is forced out of the hydrophobic pocket for methionine side chain up to 1 Å does not necessarily mean that the chloro substitute at C5′ sabotages the inhibitory potency of 26d. To the contrary, since 18i was 22 times more potent against purified HsMetAP1, the interaction between this chloro group and the hydrophobic pocket may even contribute to the binding affinity of 26d to HsMetAP1. But to satisfy the geometry of the metal coordination, there would be a limit for the displacement of the pyridinylpyrimidine core. This limit may help to explain why the inhibitors with larger substituents at C5′ (18g, 18j, 18k, 18l and 18m) were less potent against purified HsMetAP1 than the inhibitors with smaller substituents such as a fluoro group (18h) or a chloro group (18i).
In the crystal structure of tHsMetAP1 with 2 (PDB access code 2G6P), an unexpected HEPES molecule from the buffer is accommodated in a hydrophobic groove formed by Tyr196, Gly352 and Trp353 on the exterior of the phenyl group of 2.40 Because extra interactions with the enzyme may lead to higher potency, we extended the C4 side chain of the inhibitors to exploit potential hydrophobic interactions with this groove. In the structure 2G6P, the phenyl group in the C4 side chain of 2 curls back and stacks over the pyrimidine ring of the same compound. Although the C4 side chain in compound 26d is exactly an extended version of the equivalent in 2, it does not curls back and interact with Tyr196, Gly352 and Trp353. Instead, the C4 side chain of 26d adopts a wild-open conformation in which the additional aliphatic segment of the side chain and the second phenyl group at its end extend out towards the external surface of the enzyme and binds to a small hydrophobic pocket created by the side chain of Lys132 (Figure 3C). To accommodate the end of the C4 side chain of 26d, the side chain of Lys132 moves up to 4.74 Å towards the external surface of the enzyme. This interaction most likely keeps the C4 side chain of 26d in the open conformation and prevents it from curling onto the pyridinylpyrimidine core of the inhibitor.
A superimposition of the crystal structure of tHsMetAP1 in complex with compound 26d and the crystal structure of HsMetAP2 in complex with L-methionine (PDB access code 1KQ9, reported in [44]) revealed a possible structural basis for the selectivity of 26d for HsMetAP1 over HsMetAP2 (Figure 4). Compared with HsMetAP1, the 63-amino acid insertion (from residue 382 to 444) in the catalytic domain of HsMetAP2 results in a smaller entrance to its active site. In addition, the smaller distance between His231 and His339 of HsMetAP2 (5.38 Å vs. 5.89 Å between the equivalent His212 and His310 in HsMetAP1) also narrows the entrance. In the present conformation, the long C4 side chain of 26d is likely to clash with Tyr444 of HsMetAP2, and the C5′ chloro substituent on the pyridine ring of 26d might clash with Pro220 which defines the bottom of the binding pocket for methionine side chain in HsMetAP2.
Figure 4.

A superimposition of the crystal structure of N-terminally truncated HsMetAP1 (surface colored grey) in complex with 26d (sticks, carbon cyan) and the crystal structure of HsMetAP2 (wires and ribbons, colored teal) in complex with L-methionine (1KQ9 [44]). The residues of HsMetAP2 are shown as teal sticks. The numbering throughout is based on HsMetAP2. Compared with the catalytic domain of HsMetAP1, the insertion (382–444) in the catalytic domain of HsMetAP2 results in a smaller entrance to the active site. In particular, Y444 clashed with 26d. D442 and H339 on the other side of the inhibitor also narrow the binding site for 26d. The L-methionine and the surface filling model of HsMetAP2 are not shown for the sake of clarity. For all non-carbon atoms, nitrogen is blue and chlorine is green.
4. Conclusion
Starting from 5-chloro-6-methyl-N-(2-phenylethyl)-2-pyridin-2-ylpyrimidine-4-amine (2) as a hit in a high-throughput screening and based on the X-ray crystal structure of N-terminally truncated HsMetAP1 (tHsMetAP1) in complex with Co(II) and 2, we set out to identified the key pharmacophore for the inhibition of MetAP enzymes. The deleterious effects on inhibitor potency by the replacement of the 2-pyridin-2-yl group with a 2-pyridin-3-yl group, as well as the crystal structure of 26d bound in the active site,of tHsMetAP1, confirm the key interactions between the auxiliary cobalt ion and the N1 and N1′ nitrogens. We also established chlorine as the optimal substituent at C5 of the pyrimidine ring. Therefore, 5-chloro-6-methyl-2-pyridin-2-ylpyrimidine has been revealed as the key pharmacophore for the potent inhibition of HsMetAP1. A potential steric clash between the C4 substituent of 2 and the Tyr444 residue of HsMetAP2 has been exploited to improve the inhibitor's selectivity for HsMetAP1 over HsMetAP2. Assays with a variety of commercially available pyridinylpyrimidine analogues demonstrated that decreases in size of C4 substituents diminished the selectivity for HsMetAP1, while the installation of longer side chains at C4 increased the selectivity in general. In the effort to extend the C4 side chain to exploit hydrophobic interactions with the enzyme, we discovered that a long C4 side chain containing a piperazinyl group greatly enhanced selectivity for HsMetAP1 (18b). Based on 18b, a variety of substituents on the pyridine ring have been explored and we found that small substituents at C4′ could be tolerated, substituents at C6′ or C3′ reduced the potency against HsMetAP1, and small substituents at C5′ greatly improved the potency but compromised the HsMetAP1 selectivity (18h and 18i). In the further exploration of the C4 side chain on the basis of 18i, we successfully restored the HsMetAP1 selectivity of inhibitors by swapping the piperazinyl group to various aromatic groups. In addition, we uncovered that the replacement of the carbonyl group with a methylene group and the exchange of the secondary amino linker for aliphatic linkers in the C4 side chain were both well tolerated. In the end, we obtained a group of compounds containing both high nanomolar potency against 0.15 μM of purified HsMetAP1 and hundreds/thousands fold selectivity over HsMetAP2. Thus, these compounds may serve as leads for the development of therapeutical agents against cancer.
5. Experimental
5.1. Chemistry
5.1.1. General methods
Unless stated otherwise, all non-aqueous reactions were carried out under ambient atmosphere in oven-dried glassware. Indicated reaction temperatures refer to those of the reaction bath while room temperature (rt) is noted as 25 °C. All solvents were of reagent grade purchased from Fisher Scientific or VWR and used as received. Commercially available starting materials and reagents were purchased from Acros, Aldrich, or TCI America and were used as received. Eight commercially available 2-pyridinylpyrimidines (31–38) were purchased from Maybridge (Cambridge, United Kingdom), and their masses were confirmed by API-MS.
Analytical thin layer chromatography (TLC) was performed using Analtech Uniplates (silica gel HLF, W/UV254, 250 μm). Visualization was achieved using 254 nm UV light, or additionally by staining with iodine, or ceric ammonium molybdate stain. Crude products were purified by air-flashed column chromatography over silica gel (0.06–0.2 mm, 60 Å, from Acros) using indicated eluents. Melting points were recorded on a Mel-Temp II apparatus and are uncorrected. NMR data were collected on a Varian Unity-400 (400 MHz 1H, 100.6 MHz 13C) or Bruker-Spectrospin 400 MHz spectrometers. Chemical shifts are reported in ppm (δ) with the solvent resonance or 0.1% tetramethylsilane contained in the deuterated solvent as the internal reference. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, m = multiplet, br = broad, app = apparent, exch = exchangeable), coupling constants (J, reported in Hertz, Hz), and number of protons. Low resolution mass spectra were acquired on a Thermo-Finnigan MAT, LCQ Classic ESI-mass spectrometer or Voyager DE-STR, MALDI-TOF (Applied Biosystems) instruments. The MALDI-samples were prepared by mixing the droplets of the sample solutions in chloroform or methanol and 2,5-dihydroxybenzoic acid solution in acetone, where the latter served as the matrix. Data are reported in the form m/z (molecular ion).
Samples were analyzed for purity on an Agilent LC-MS system consisting of 1200 Infinity Series LC equipped with an autoinjector, 1260 Diode-array detector (DAD), and 6120 Quadrupole MS detector. A Pursuit XRs C18 column (3 μm, 4.6 × 150 mm) was used while eluting with 60:40 acetonitrile/water (with 0.1 % TFA) as an isocratic mobile phase set at a flow rate of 0.8 mL/min. DAD output was processed for 254, 274, 294 nm while simultaneously screening for the desired mass in API-ES mode of the MS detector (mass range: 100 to 1000, positive mode, fragmenter set at 200). Purity of final compounds was determined to be ≥94%, using a 20 μL injection (approximately 1 mM in acetonitrile) with quantitation by area under the curve (AUC) at 254 and 274 nm. The retention time (tR) is reported in minutes with the AUC given in parentheses.
5.1.2. Synthesis of tetrasubstituted pyrimidines (9a–9c)
Tetrasubstituted pyrimidines 9a–9c were synthesized following a three-step procedure described by Medwid et al.45 Briefly, amidine hydrochloride 5 was prepared from 2-cyanopyridine by treating with 1.25 M HCl in methanol for 18 h and the resulting white solid was used without further purification to condense with different acetoacetates (ethyl 2-fluoroacetoacetate, ethyl 2-methylacetoacetate and ethyl acetoacetate in the cases of 9a, 9b and 9c, respectively) under basic conditions. The pyrimidinones 7 were reacted with phosphorus oxychloride at 65 °C for 24 h and after a basic work-up they were purified by flash column chromatography over silica gel. Subsequent displacement of the 4-chloro group on pyrimidines 8 with phenethylamine under refluxing conditions with potassium carbonate in methanol yielded the final products 9a-9c which were purified by column chromatography over silica gel (eluent: 5–10% EtOH—DCM). Pyridinyl pyrimidines 9a– 9c were analyzed for purity using an Agilent LC-MS system described in the general methods section. The tR values refer to the retention time when the samples were analyzed using Pursuit XRs C18 column (3 μm, 4.6 × 150 mm) with 60:40 acetonitrile/water (0.1% TFA) as an isocratic mobile phase.
5.1.2.1. 5-Fluoro-6-methyl-N-phenethyl-2-(pyridin-2-yl)pyrimidin-4-amine (9a)
Yield: 56% (over three steps). tR: 4.029 min (99.1%). 1H NMR (400 MHz, acetone-d6): δ 2.38 (d, J = 1.8 Hz, 3H), 2.96 (t, J = 6.5 Hz, 2H), 3.03 (br s, 1H), 3.55 (t, J = 6.5 Hz, 2H ), 7.21 (m, 5H), 7.61 (ddd, J = 8.2, 2.5, & 1.9 Hz, 1H), 8.05 (ddd, J = 8.2, 2.5, & 1.9 Hz, 1H), 8.42 (d, J = 8.2 Hz, 1H), 8.72 (d, J = 5.6 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 170.25, 161.72, 155.19, 149.83, 147.46, 139.03, 137.90, 136.94, 129.00, 128.88, 126.77, 124.35, 123.64, 40.31, 35.79, 17.19. MALDI-TOF: m/z 309 (M+H) +, 331 (M+Na) +.
5.1.2.2 5,6-Dimethyl-N-phenethyl-2-(pyridin-2-yl)pyrimidin-4-amine (9b)
Yield: 62% (over three steps). tR: 7.163 min (94.6%). 1H NMR (400 MHz, acetone-d6): δ 2.25 (s, 3H), 2.71 (s, 3H), 2.93 (t, J = 6.8 Hz, 2H), 3.42 (t, J = 6.8 Hz, 2H ), 7.13 (m, 5H), 7.41 (ddd, J = 8.1, 5.5, & 2.1 Hz, 1H), 7.85 (ddd, J = 8.1, 5.5, & 2.1 Hz, 1H), 8.1 (d, J = 8.1 Hz, 1H), 8.72 (d, J = 5.5 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 168.13, 164.17, 157.05, 154.93, 149.46, 139.76, 136.58, 128.91, 128.37, 126.33, 124.91, 123.69, 117.94, 41.29, 34.67, 25.11, 14.73. MALDI: m/z 308 (M+H) +, 330 (M+Na) +, 346 (M+K) +.
5.1.2.3. 6-Methyl-N-phenethyl-2-(pyridin-2-yl)pyrimidin-4-amine (9c).46
Yield: 49% (over three steps). tR: 6.328 min (95.8%). 1H NMR (400 MHz, acetone-d6): δ 2.32 (s, 3H), 2.93 (t, J = 7.6 Hz, 2H), 3.65 (m, 2H ), 6.92 (t, J = 7.8 Hz, 2H), 7.24 (t, J = 8.1 Hz, 3H), 7.58 (d, J = 7.8 Hz, 3H), 7.66 (ddd, J = 8.2, 5.6, & 1.9 Hz, 1H), 7.86 (dd, J = 8.2 & 1.9 Hz, 1H), 8.08 (dd, J = 5.6 & 1.9 Hz, 1H), 8.13 (br s, 1H), 8.76 (d, J = 5.6 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 168.02, 165.51, 162.41, 153.22, 149.13, 147.95, 137.98, 129.05, 126.85, 122.50, 113.48, 38.98, 30.62, 24.19. MALDI-TOF: m/z 291 (M+H) +.
5.1.3. Synthesis of 5-chloro-4-methyl-6-(phenethylthio)-2-(pyridin-2-yl)pyrimidine (13)
Anhydrous potassium carbonate (415 mg, 3 mmol) was added to a solution of 5-Chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-thione (238 mg, 1 mmol) in toluene (15 mL) and the suspension was stirred at 60 °C for 20 min. Phenethyl bromide (222 mg, 1.2 mmol) was added to the reaction mixture and the stirring was continued for an additional 8 h. The reaction mixture was cooled to rt, quenched with with water (25 mL) and the mixture was extracted with EtOAc (2×20 mL). The combined organic layer was concentrated and the crude product was purified by flash column chromatography over silica gel (eluent: EtOH/CHCl3=5:95) to afford pyrimidine as an off-white solid. Yield: 314 mg (92%). tR: 6.103 min (96.1%). 1H NMR (400 MHz, acetone-d6): δ 2.71 (s, 3H), 2.91 (t, J = 7.6 Hz, 2H), 3.45 (t, J = 7.6 Hz, 2H ), 7.15 (m, 5H), 7.37 (app. dd, J = 8.2 & 5.6 Hz, 1H), 7.84 (app t, J = 8.2 Hz, 1H), 8.1 (d, J = 8.2 Hz, 1H), 8.75 (d, J = 5.6 Hz, 1H). 13C NMR (125 MHz, CDCl3): δ 171.50, 158.82, 155.79, 154.93, 140.36, 136.49, 129.07, 128.89, 126.95, 125.03, 111.45, 35.81, 32.22, 22.83. MALDI-TOF: m/z 343 (M+H) +, 365 (M+Na) +.
5.1.4. Typical procedure for synthesizing compounds 16 and 18
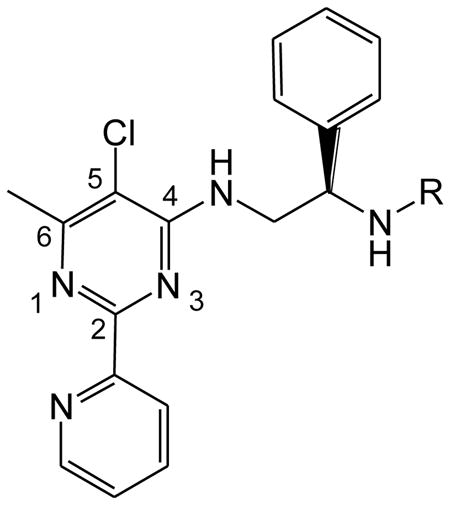
To the solution of 4,5-dichloro-6-methyl-2-(pyridin-2-yl)pyrimidine (8a, 612 mg, 2.5 mmol) and (R)-tert-butyl-2-amino-1-phenylethylcarbamate (783 mg, 3.3 mmol) in THF (10 mL) was added Et3N (516 mg, 5.1 mmol). The mixture was heated at 50 °C for 12 h, at which point TLC analysis indicated complete transformation. The reaction mixture was cooled to room temperature and diluted with ethyl acetate. After washed with water and brine, the organic phase was dried (Na2SO4), filtered and concentrated to give the corresponding condensation product as a light yellow solid, which was treated at room temperature with TFA (3 mL) and CH2Cl2 (10 mL) for 1 h before being concentrated in vacuo. The resulting TFA salt was dissolved in EA, and then washed with aq. NaHCO3 and brine. The organic phase was dried over Na2SO4. Evaporation and column chromatography on silica gel (eluent: 5–10% EtOH—DCM or similar conditions) afforded 11a (R1–R4 = H, X = Cl) as the foamy solid (722 mg, 85%): [α]D20 = -43.6 (c 0.1, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.73 (s, 1H), 8.31 (d, J = 7.2 Hz, 1H), 7.73 (t, J = 7.5 Hz, 1H), 7.27-7.36 (m, 6H), 6.02 (s, 1H), 4.28 (s, 1H), 3.88-3.92 (m, 1H), 3.70-3.72 (m, 1H), 3.34 (br s, 2H), 2.52 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 161.5, 159.5, 157.8, 154.9, 149.8, 142.8, 136.7, 128.8, 127.8, 126.4, 124.4, 123.5, 112.7, 55.1, 48.3, 22.2; ESIMS m/z 340.1 [M + H]+; HR-ESIMS (m/z) Calcd. for C18H19N5Cl [M + H]+ 340.1324, found 340.1323.
4-chlorobutanoyl chloride (345 mg, 2.5 mmol) was added drop wisely to the solution of 11a (426 mg, 1.3 mmol) and TEA (254 mg, 2.5 mmol) in anhydrous THF (6 mL) at 0 °C. After being stirred for 0.5 h, reaction mixture was diluted with ethyl acetate, washed with brine. The organic phase was dried over Na2SO4, filtered and concentrated to give the crude product, which was dissolved in DME and then added to a solution of NaI (195 mg, 1.3 mmol), anhydrous Na2CO3 (207 mg, 1.95 mmol) and 1-(2-(tert -butyldimethylsilyloxy)ethyl)piperazine (1.4 mmol) or tert-butyl piperazine-1-carboxylate (254 mg, 1.37 mmol) in DME. After being stirred at 90 °C for 18 h, the mixture was cooled to rt, and diluted with ethyl acetate, washed with water and brine. Treatment of crude product with a solution of TB AF in THF or a mixture of TFA and CH2Cl2 followed by ethyl acetate work up afforded 16a or 18a in about 30% overall yield.
N-((R)-2-(5-chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethyl)-3-(4-(2-hydroxyethyl)piperazin-1-yl)propanamide (16a) [α]D20 = -41.6 (c 0.16, CHCl3); 1H NMR (300 MHz, CDCl3): δ 9.36 (d, J = 8.2 Hz, 1H), 8.79 (d, J = 4.5 Hz, 1H), 8.40-8.46 (m, 1H), 7.83 (t, J = 7.8 Hz, 1H), 7.30-7.44 (m, 5H), 6.29 (t, J = 4.8 Hz, 1H), 5.29-5.37 (m, 1H), 3.91-4.13 (m, 2H), 3.56 (t, J = 5.4 Hz, 2H), 2.60 (s, 3H), 2.48-2.55 (m, 2H), 2.14-2.46 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 172.9, 161.6, 159.5, 158.2,154.9, 149.8, 139.4, 136.8, 129.0, 128.1, 126.8, 124.5, 123.6, 112.7, 59.2, 57.8, 53.7, 53.5, 52.6,52.3, 47.4, 31.8, 22.3; ESI-MS m/z 524.3 (M + H) +; HRMS (ESI) calcd for C27H35N7O2Cl (M + H) + 524.2535, found 524.2531.
N-((R)-2-(5-chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethyl)-4-(4-(2-hydroxyethyl)piperazin-1-yl)butanamide (16b) [α]D20 = -34.4 (c 0.25, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.83-8.79 (m, 1H), 8.44 (d, J = 7.8 Hz, 1H), 7.84 (dt, J = 1.5 Hz, 7.5 Hz, 1H), 7.76 (d, J = 6.6 Hz, 1H), 7.42-7.28 (m, 5H), 6.06 (t, J = 5.4 Hz, 1H), 5.32-5.23 (m, 1H), 4.15-4.03 (m, 1H), 3.95-3.86 (m, 1H), 3.57 (t, J = 5.4 Hz, 2H), 2.61 (s, 3H), 2.51-2.11 (m, 12H), 1.76-1.63 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.3, 161.8, 1595, 158.4, 154.8, 149.9, 139.8, 136.8, 128.9, 127.9, 126.7, 124.6, 123.5, 59.1, 57.7, 57.1, 54.9, 52.9, 52.7, 46.9, 34.2, 29.7, 22.5, 22.3. ESI-MS m/z 538.2 (M + H) +; HRMS (ESI) calcd for C28H37ClN7O2 (M + H) + 538.2692, found 538.2700.
N-((R)-2-(5-chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethyl)-3-(piperazin-1-yl)propanamide (18a) [α]D20 = -44.6 (c 0.3, CHCl3); 1H NMR (300 MHz, CDCl3): δ 9.37 (d, J = 7.8 Hz, 1H), 8.77-8.81 (m, 1H), 8.43 (d, J = 7.8 Hz, 1H), 7.82 (t, J = 7.8 Hz, 1H), 7.39-7.42 (m, 4H), 7.31-7.38 (m, 2H), 6.24-6.32 (m, 1H), 5.28-5.38 (m, 2H), 4.03-4.14 (m, 1H), 3.89-4.01 (m, 1H), 2.64-2.79 (m, 4H), 2.64 (s, 3H), 2.45-2.52 (m, 2H), 2.23-2.41 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 172.6, 161.6, 159.5, 158.2, 154.8, 154.5, 149.8, 139.4, 136.8, 129.0, 128.1, 126.6, 124.5, 123.6, 112.6, 79.9, 53.9, 52.2, 47.2, 32.0, 28.4, 22.3; ESI-MS m/z 480.2 (M + H) +; HRMS (ESI) calcd for C25H31N7OCl (M + H) + 480.2278, found 480.2278.
N-((R)-2-(5-chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18b) [α]D20 = -52.3 (c 1.8, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.82 (d, J = 4.5 Hz, 1H), 8.44 (d, J = 7.8 Hz, 1H), 7.89-7.76 (m, 2H), 7.43-7.28 (m, 6H), 6.1 (t, J = 6.0 Hz, 1H), 5.33-5.23 (m, 1H), 4.15-4.02 (m, 1H), 3.96-3.86 (m, 1H), 2.77 (t, J = 4.8 Hz, 4H), 2.60 (s, 3H), 2.33-2.11 (m, 10H), 1.76-1.64 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.4, 161.6, 159.4, 158.3, 154.7, 149.7, 139.8, 136.8, 128.8, 127.8, 126.6, 124.6, 123.5, 112.6, 57.6, 54.9, 52.8, 51.2, 46.8, 45.6, 34.0, 22.2; ESI-MS m/z 494.1 (M + H) +; HRMS (ESI) calcd for C26H33ClN7O (M + H) + 494.2430, found 494.2440.
N-((R)-2-(5-chloro-2-(6-methoxypyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18c) [α]D20 = -65.9 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.23 (d, J = 6 Hz, 1H), 8.08 (d, J = 7.2 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.37-7.30 (m, 5H), 6.88 (d, J = 8.1 Hz, 1H), 6.05 (t, J = 6.3 Hz, 1H), 5.19 (m, 1H), 4.24 (m, 1H), 4.12 (s, 3H), 3.72 (m, 1H), 2.75 (m, 4H), 2.57 (s, 3H), 2.35 (m, 1H), 2.20 (m, 4H), 1.99 (m, 4H), 1.58 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 169.0, 159.8, 157.5,155.1, 154.4, 147.9, 135.9, 135.1, 124.6, 123.6, 122.3, 112.8, 108.4, 108.0, 53.6, 52.1, 49.6, 49.2, 42.3, 41.5, 30.1, 18.1, 18.0. ESI-MS m/z 524.3 (M + H) +. HRMS (ESI) calcd for C27H35ClN7O2 (M + H) + 524.2535, found 524.2521.
N-((R)-2-(5-chloro-2-(6-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18d) [α]D20 = -68.1 (c 0.4, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.36 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 6.6 Hz, 1H), 7.81 (t, J = 7.8 Hz, 1H), 7.48-7.25 (m, 5H), 6.28-6.18 (m, 1H), 5.29-5.18 (m, 1H), 4.50-4.21 (m, 2H), 4.21-4.07 (m, 1H), 3.77-3.65 (m, 1H), 2.87-2.75 (m, 4H), 2.57 (s, 3H), 2.38-2.20 (m, 4H), 2.20-2.03 (m, 4H), 1.71-1.57 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.4, 161.4, 158.4, 157.9, 155.2, 151.4, 140.1, 139.4, 128.6, 127.6, 126.5, 125.3, 121.8, 112.7, 57.7, 55.3, 53.8, 46.7, 45.6, 34.0, 222, 22.0; ESI-MS m/z 528.1 (M + H) +. HRMS (ESI) calcd for C26H32Cl2N7O (M + H) + 528.2040, found 528.2036.
N-((R)-2-(5-chloro-2-(3-methoxypyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18e) 1H NMR (300 MHz, CDCl3) δ 8.34 (m, 1H), 7.78 (d, J = 6.9, 1H), 7.38 (m, 2H), 7.36-7.23 (m, 5H), 6.05 (m, 1H), 5.24 (m, 1H), 4.10 (m, 1H), 3.88 (s, 3H), 3.68 (m, 1H), 2.79 (m, 4H), 2.35-2.21 (m, 4H), 2.22-2.01 (m, 4H), 1.66 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 173.3, 161.3, 160.2, 158.2, 154.0, 146.0, 141.1, 139.9, 128.6, 127.6, 126.5, 124.8, 119.4, 111.9, 57.7, 55.7, 55.1, 53.5, 46.2, 45.5, 33.8, 22.2, 22.0. ESI-MS m/z 524.3 (M + H) +. HRMS (ESI) calcd for C27H35ClN7O2 (M + H) + 524.2535, found 524.2526.
N-((R)-2-(5-chloro-2-(3-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18f) [α]D20 = -49.0 (c 3.5, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.62 (d, J = 4.8 Hz, 1H), 7.88-7.82 (m, 1H), 7.42 (d, J = 7.2 Hz, 1H), 7.39-7.24 (m, 5H), 6.24 (t, J = 5.4 Hz, 1H), 5.29-5.19 (m, 1H), 4.10-3.97 (m, 1H), 3.85-3.73 (m, 1H), 2.84-2.72 (m, 4H), 2.57 (s, 3H), 2.37-2.09 (m, 8H), 1.78-1.64 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.8, 161.6, 160.1, 158.4, 154.0, 147.6, 139.8, 138.7, 130.7, 129.0, 128.1, 126.9, 124.9, 112.7, 57.8, 54.8, 53.9, 46.9, 45.7, 34.3, 22.4, 22.3. ESI-MS m/z 528.2 (M + H) +; HRMS (ESI) calcd for C26H32Cl2N7O (M + H) + 528.2040, found 528.2036.
N-((R)-2-(5-chloro-2-(5-methoxypyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18g) [α]D20 = -21.2 (c 0.4, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.48 (d, J = 2.1 Hz, 1H), 8.42 (d, J = 9 Hz, 1H), 7.83 (d, J = 6.6 Hz, 1H), 7.38-7.30 (m, 6H), 6.02 (t, J = 5.4 Hz, 1H), 5.30-5.23 (m, 1H), 4.10-4.02 (m, 1H), 3.93-3,86 (m, 4H), 2.78-2.75 (m, 4H), 2.58 (s, 3H), 2.36-2.13 (m, 8H), 1.73-1.65 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 173.4, 161.7, 159.3, 158.3, 156.7, 147.4, 139.9, 137.8, 128.9, 127.9, 126.7, 124.4, 120.5, 112.0, 57.7, 55.8, 55.0, 54.0, 46.8, 45.8, 34.2, 22.3; ESI-MS m/z 524.3(M + H)+. HRMS (ESI) calcd for C27H35ClN7O2 (M + H)+ 524.2535, found 524.2546.
N-((R)-2-(5-chloro-2-(5-fluoropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18h) [α]D20 = -39.1 (c 0.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.64 (d, J = 2.4 Hz, 1H), 8.49 (dd, J = 4.8, 8.7 Hz, 1H), 7.70 (d, J = 6.9 Hz, 1H), 7.54 (dt, J = 2.7 Hz, 8.1 Hz, 1H), 7.41-7.28 (m, 5H), 6.18 (t, J = 5.4 Hz, 1H), 5.28 (m, 1H), 4.05 (m, 1H), 3.88 (m, 1H), 2.76 (t, J = 4.5 Hz, 4H), 2.58 (s, 3H), 2.27-2.12 (m, 8H), 1.72 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.5, 161.6, 160.2 (d, J = 260 Hz), 158.5, 158.3, 151.0, 139.6, 138.0 (d, J = 24 Hz), 128.9, 127.9, 126.6, 125.0 (d, J = 5.1 Hz), 123.4 (d, J = 18 Hz), 112.6, 57.6, 54.6, 54.1, 46.9, 45.8, 34.1, 22.2. ESI-MS m/z 512.3 (M + H) +. HRMS(ESI) calcd for C26H32ClFN7O (M + H) + 512.2335, found 512.2344.
N-((R)-2-(5-chloro-2-(5-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18i) [α]D20 = -45.2 (c 1.9, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.73 (m, 1H), 8.41 (d, J = 8.4 Hz, 1H), 7.85-7.72 (m, 2H), 7.41-7.29 (m, 5H), 6.20 (m, 1H), 5.28 (m, 1H), 4.06 (m, 1H), 3.86 (m, 1H), 2.76 (m, 4H), 2.58 (s, 3H), 2.31-2.09 (m, 8H), 1.71 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.4, 161.5, 158.5, 158.1, 152.8, 148.5, 139.5, 136.4, 133.2, 128.8, 127.9, 126.5, 124.3, 112.7, 57.5, 54.5, 53.9, 46.8, 45.7, 34.0, 22.1. ESI-MS m/z 528.1 (M + H) +; HRMS (ESI) calcd for C26H32Cl2N7O (M + H) + 528.2040, found 528.2039.
N-((R)-2-(2-(5-bromopyridin-2-yl)-5-chloro-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18j) [α]D20 = -47.8 (c 2.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.83 (m, d, J = 2.1 Hz, 1H), 8.38-8.31 (m, 1H), 8.00-7.92 (m, 1H), 7.83 (d, J = 6.9 Hz, 1H), 7.41-7.25 (m, 5H), 6.30-6.20 (m, 1H), 5.31-5.21 (m, 1H), 4.12-3.96 (m, 1H), 3.92-3.79 (m, 1H), 2.80 (m, 4H), 2.57 (s, 3H), 2.43-2.09 (m, 8H), 1.81-1.62 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.5, 161.6, 158.7, 158.3, 153.3, 150.7, 139.6, 139.4, 128.9, 127.9, 126.6, 124.8, 122.3, 112.8, 57.6, 54.7, 54.0, 46.9, 45.7, 34.1, 22.21, 22.16. ESI-MS m/z 572.0 (M + H) +. HRMS (ESI) calcd for C26H32BrClN7O (M + H) + 572.1535, found 572.1541.
N-((R)-2-(5-chloro-6-methyl-2-(5-nitropyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18k) [α]D20 = -25.8 (c 1.3, CHCl3). 1H NMR (300 MHz, CDCl3) δ 9.58 (m, 1H), 8.68 (m, 1H), 8.60 (dd, J = 2.7 Hz, 8.7 Hz, 1H), 7.43-7.30 (m, 6H), 6.42 (t, J = 4.2 Hz, 1H), 5.31 (m, 1H), 4.08 (m, 1H), 3.90 (m, 1H), 2.75 (m, 4H), 2.62 (s, 3H), 2.35-2.17 (m, 8H), 1.77 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.7, 161.8, 159.7, 158.3, 157.6, 145.0, 144.4, 139.3, 131.9, 129.1, 128.2, 126.7, 123.9, 113.8, 57.4, 54.2, 54.0, 47.2, 45.8, 34.1, 22.2, 22.1. ESI-MS m/z 539.3 (M + H) +. HRMS (ESI) calcd for C26H32ClN8O2 (M + H) + 539.2280, found 539.2271.
N-((R)-2-(5-chloro-2-(5-(dimethylamino)pyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18l) [α]D20 = -52.0 (c 0.3, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.35-8.26 (m, 2H), 8.07 (m, 1H), 7.41-7.33 (m, 4H), 7.30 (m, 1H), 7.04 (dd, J = 2.7 Hz, 9.0 Hz, 1H), 5.88 (t, J = 5.7 Hz, 1H), 5.23 (m, 1H), 4.10 (m, 1H), 3.86 (m, 1H), 3.09 (s, 6H), 2.79 (t, J = 4.8 Hz, 4H), 2.27 (m, 4H), 2.13 (m, 4H), 1.66 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.4, 161.8, 160.0, 158.4, 146.8, 142.6, 140.3, 134.6,129.0, 127.9, 126.8, 124.2, 118.2, 111.3, 57.9, 55.7, 53.5, 46.8, 45.6, 40.2, 34.4, 22.5. ESI-MS m/z537.3 (M + H) +.
N-((R)-2-(5-chloro-6-methyl-2-(5-(pyrrolidin-1-yl)pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18m) [α]D20 = -66.3 (c 1.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.32 (d, J = 8.7 Hz, 1H), 8.16-8.05 (m, 2H), 7.40-7.29 (m, 5H), 6.89 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 5.86 (m, 1H), 5.22 (m, 1H), 4.09 (m, 1H), 3.86 (m, 1H), 3.40 (m, 4H), 2.76 (m, 4H), 2.56 (s, 3H), 2.23 (m, 4H), 2.16-2.00 (m, 8H), 1.65 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.3, 161.4, 159.8, 158.2, 144.2, 141.5, 140.1, 134.0, 128.7,127.6, 126.6, 124.2, 117.6, 110.8, 57.8, 55.4, 54.0, 47.4, 46.5, 45.7, 34.2, 25.4, 22.3, 22.2. ESI-MS m/z 563.3 (M + H) +. HRMS (ESI) calcd for C30H40ClN8O (M + H) + 563.3008, found 563.3033.
N-((R)-2-(5-chloro-2-(4-methoxypyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18n) [α]D20 = -48.5 (c 0.9, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.62 (d, J = 6.0 Hz, 1H), 8.00 (d, J = 2.1 Hz, 1H), 7.87 (d, J = 7.5 Hz, 1H), 7.41-7.28 (m, 5H), 6.91 (dd, J = 2.4 Hz, 6.0 Hz, 1H), 6.03 (t, J = 4.8 Hz, 1H), 5.31-5.22 (m, 1H), 4.18-4.06 (m, 1H), 3.99-3.84 (m, 4H), 2.82-2.74 (m, 4H), 2.59 (s, 3H), 2.32-2.12 (m, 8H), 1.74-1.63 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.3, 166.4, 161.4, 159.2, 158.2, 156.3, 150.8, 139.7, 128.7, 127.6, 126.5, 112.5, 110.7, 109.4, 57.4, 55.2, 54.8, 52.8, 51.1, 46.5, 45.1, 33.9, 22.1. ESI-MS m/z 524.3 (M + H) +. HRMS (ESI) calcd for C27H35ClN7O2 (M + H) + 524.2535, found 524.2552.
N-((R)-2-(5-chloro-2-(4-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-(piperazin-1-yl)butanamide (18o) [α]D20 = -36.0 (c 2.1, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.70 (d, J = 5.4 Hz, 1H), 8.47-8.43 (m, 1H), 7.65 (d, J = 6.9 Hz, 1H), 7.42-7.28 (m, 5H), 6.24-6.15 (m, 1H), 5.33-5.23 (m, 1H), 4.14-4.01 (m, 1H), 3.96-3.85 (m, 1H), 2.82-2.72 (m, 4H), 2.59 (s, 3H), 2.38-2.13 (m, 8H), 1.79-1.66 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 173.5, 161.6, 158.3, 156.3, 150.5, 145.0, 139.7, 128.9, 127.9, 126.6, 124.8 123.9, 113.0, 57.5, 54.6, 53.5, 46.9, 45.4, 34.0, 22.2. ESI-MS m/z 528.1 (M + H) +; HRMS (ESI) calcd for C26H32Cl2N7O (M + H) + 528.2040, found 528.2027.
5.1.5. Typical procedure for synthesizing compounds 20
5-chloro-6-methyl-N-((R)-2-phenyl-2-(3-(piperazin-1-yl)propylamino)ethyl)-2-(pyridin-2-yl)pyrimidin-4-amine (20a)
To a solution of 11a (115 mg, 0.34 mmol) and bromide 19a (156 mg, 0.51 mmol) in dry DMF (1 mL) were added NaI (152 mg, 1.02 mmol) and anhydrous K2CO3 (140 mg, 1.02 mmol). After being stirred overnight at 100 °C for 24 h, the reaction mixture was cooled to rt, and diluted with ethyl acetate, and washed with water and brine. The organic phase was dried (Na2SO4), filtered and concentrated. The crude product was treated at with TFA (0.3 mL) and CH2Cl2 (1 mL) for 1 h at rt. After diluting with ethyl acetate, the solution was washed with aq. NaHCO3 and brine, and dried over Na2SO4. Evaporation and column chromatography on silica gel (eluent: 5–10% EtOH—DCM or similar conditions) afforded 20 as the foamy solid (60 mg, 38% yield). [α]D20 = -23.1 (c 0.15, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.79 (d, J = 4.8 Hz, 1H), (8.42-8.36, m, 1H), 7.79 (dt, J = 1.8, 7.5 Hz, 1H), 7.42-7.29 (m, 6H), 6.18-6.10 (m, 1H), 3.97-3.86 (m, 2H), 3.72-3.60 (m, 1H), 2.91-2.82 (m, 4H), 2.68-2.52 (m, 9H), 2.48-2.31 (m, 4H), 1.74-1.60 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 161.4, 159.6, 157.8, 155.0, 149.8, 141.6, 136.6, 128.7, 127.7, 127.0, 124.3, 123.5, 112.7, 62.1, 57.5, 54.5, 47.0, 46.1, 46.0, 26.7, 22.3; ESI-MS m/z 466.2 (M + H) +; HRMS (ESI) calcd for C25H33ClN7 (M + H) + 466.2481, found 466.2496.
2-(4-(3-((R)-2-(5-chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethylamino)propyl)piperazin-1-yl)ethanol (20b)
Following a similar procedure from 19a to 20a, 20b was obtained from 19b. [α]D20 = -13.9 (c 0.65, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.80 (d, J = 4.8 Hz, 1H), 8.39 (d, J = 7.8 Hz, 1H), 7.83-7.75 (m, 1H), 7.43-7.28 (m, 5H), 6.15-6.08 (m, 1H), 3.98-3.87 (m, 2H), 3.70-3.56 (m, 3H), 2.68-2.57 (m, 6H), 2.56-2.35 (m, 9H), 1.76-1.61 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 161.4, 159.7, 157.8, 155.1, 149.9, 141.7, 136.6, 128.7, 127.7, 127.0, 124.3, 123.5, 112.7, 62.1, 59.2, 57.7, 56.9, 53.4, 52.9, 47.0, 46.2, 27.1, 22.3. ESI-MS m/z 510.2 (M + H) +; HRMS (ESI) calcd for C27H37ClN7O (M + H) + 510.2743, found 510.2751.
5.1.6. Typical procedure for synthesizing compounds 22
5-chloro-6-methyl-N-((R)-2-phenyl-2-(4-(piperazin-1-yl)butylamino)ethyl)-2-(pyridin-2-yl)pyrimidin-4-amine (22a)
To a solution of 11a (85 mg, 0.25 mmol) and aldehyde 21a (79 mg, 0.25 mmol) in MeOH (0.7 mL) was added acetic acid (72 μL, 1.25 mmol) under argon. The reaction mixture was stirred for 10 min at rt, and then NaBH3CN (47 mg, 0.75 mmol) was added at 0 °C. After being stirred for 1 h, the reaction mixture was diluted with CH2Cl2, and washed with water. The organic phase was dried over Na2SO4 and concentrated. The crude product was treated at with TFA (0.3 mL) and CH2Cl2 (1 mL) for 1 h at rt. After diluting with ethyl acetate, the solution was washed with aq. NaHCO3 and brine, and dried over Na2SO4. Evaporation and column chromatography on silica gel (eluent: 5–10% EtOH—DCM or similar conditions) afforded 22a (58 mg, 49% yield). [α]D20 = -11.6 (c 1.95, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.80 (d, J = 4.2 Hz, 1H), 8.40 (d, J = 8.1 Hz, 1H), 7.79 (dt, J = 1.8, 7.5 Hz, 1H), 7.42-7.27 (m, 6H), 6.14-6.06 (m, 1H), 3.97-3.85 (m, 2H), 3.72-3.61 (m, 1H), 2.88 (t, J = 4.8 Hz, 4H), 2.61 (s, 3H), 2.60-2.50 (m, 2H), 2.46-2.24 (m, 6H), 1.60-1.41 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 161.2, 159.5, 157.7, 154.9, 149.7, 141.5, 136.5, 128.6, 127.6, 126.9, 124.3, 123.4, 112.6, 62.0, 58.8, 54.0, 47.1, 46.9, 45.6, 28.0, 24.2, 22.1. ESI-MS m/z 480.2 (M + H) +; HRMS (ESI) calcd for C26H34ClN7 (M + H) + 480.2637, found 480.2630.
2-(4-(4-((R)-2-(5-chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethylamino)butyl)piperazin-1-yl)ethanol (22b)
Following a similar procedure from 21a to 22a, 22b was obtained from 21b. [α]D20 = -28.1 (c 0.15, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.80 (d, J = 4.8 Hz, 1H), 8.39 (d, J = 7.8 Hz, 1H), 7.83-7.75 (m, 1H), 7.42-7.28 (m, 6H), 6.10-6.02 (m, 1H), 3.96-3.84 (m, 2H), 3.73-3.55 (m, 4H), 2.61 (s, 3H), 2.59-2.37 (m, 10H), 2.36-2.22 (m, 4H), 1.58-1.40 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 161.4, 159.6, 157.8, 149.8, 141.6, 136.6, 128.7, 127.7, 127.0, 124.3, 123.5, 112.7 62.0, 59.1, 58.4, 57.6, 53.2, 52.8, 47.2, 46.9, 28.2, 24.6. ESI-MS m/z 524.2 (M + H) +; HRMS (ESI) calcd for C28H39ClN7O (M + H) + 524.2899, found 524.2908.
5.1.7. Typical procedure for synthesizing compounds 24
N-((R)-2-(5-chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethyl)-2-(piperazin-1-yl)ethanesulfonamide (24a)
To a cooled solution (0 °C) of 11a (594 mg, 1.75 mmol) and Et3N (0.53 mL, 3.85 mmol) in CH2Cl2 (3 mL) was added 2-chloroethanesulfonyl chloride (627 mg, 3.85 mmol) with stirring. After addition, the mixture was stirred at rt for 5 h, and then reaction was quenched with ice water. The reaction mixture was extracted with CH2Cl2, and the combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was dissolved in EtOH before tert-butyl piperazine-1-carboxylate (171 mg, 0.4 mmol) was added. The resultant solution was heated at 50 °C for 16 h, and then concentrated in vacuo. The residue was treated at with TFA (0.3 mL) and CH2Cl2 (1 mL) for 1 h at rt. After diluting with ethyl acetate, the solution was washed with aq. NaHCO3 and brine, and dried over Na2SO4. Evaporation and column chromatography on silica gel (eluent: 5–10% EtOH—DCM or similar conditions) afforded 24a. [α]D20 = -38.9 (c 4.0, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.86-8.80 (m, 1H), 8.47-8.39 (m, 1H), 7.83 (t, J = 4.2 Hz, 1H), 7.42-7.27 (m, 5H), 5.79 (t, J = 5.7 Hz), 4.98-4.58 (m, 3H), 4.12-3.87 (m, 2H), 3.02-2.85 (m, 2H), 2.84-2.71 (m, 2H), 2.71-2.61 (m, 2H), 2.57 (s, 3H), 2.32-2.18 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 161.7, 159.4, 158.0, 154.4, 149.8, 139.4, 136.9, 128.9, 128.2, 126.9, 124.8, 123.5, 112.6, 58.0, 52.6, 52.4, 50.4, 47.6, 45.0, 22.2; ESI-MS m/z 516.1 (M + H) +; HRMS (ESI) calcd for C24H31N7O2SCl2 (M + H) + 516.1943, found 516.1955.
N-((R)-2-(5-chloro-6-methyl-2-(pyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethyl)-2-(4-(2-hydroxyethyl)piperazin-1-yl)ethane sulfonamide (24b)
Following a similar procedure from 11a to 24a, 24b was obtained from 11a. [α]D20 = -46.5 (c 0.8, CHCl3);1H NMR (300 MHz, CDCl3) δ 8.85 (d, J = 4.8 Hz, 1H), 8.45 (d, J = 7.8 Hz, 1H), 7.84 (t, J = 7.5 Hz, 1H) 7.42-7.28 (m, 6H), 5.79 (t, J = 5.7 Hz, 1H), 4.97-4.89 (m, 1H), 4.13-4.01 (m, 1H), 4.00-3.90 (m, 1H), 3.55 (t, J = 5.4 Hz, 2H), 3.03-2.91 (m, 2H), 2.74-2.65 (m, 2H), 2.58 (s, 3H), 2.47-2.15 (m, 12H). 13C NMR (75 MHz, CDCl3) δ 161.8, 159.4, 158.1, 154.4, 149.9, 139.3, 136.9, 128.9, 128.2, 126.9, 124.8, 123.5, 112.6, 59.1, 58.0, 57.7, 52.7, 52.4, 52.1, 50.4, 47.7, 22.2. ESI-MS m/z 560.3 (M + H) +; HRMS (ESI) calcd for C26H35N7O3SCl (M + H) + 560.2205, found 560.2199.
5.1.8. General procedure for preparing compounds 25
To a solution of acid (0.232 mmol), 11b (0.193 mmol), HOBt (0.232 mmol) and EDCI (0.232 mmol) in CH2Cl2 (1 mL) was added DIPEA (0.483 mmol). The resultant solution was stirred for 20 h at rt before it was concentrated. The residue was dissolved in ethyl acetate, and washed with saturated NaHCO3 and brine. The organic layer was dried over Na2SO4, concentrated and then purified by flash chromatography (eluent: 5–10% EtOH—DCM or similar conditions) to give the corresponding amide.
N-((R)-2-(5-chloro-2-(5-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-2-phenylacetamide (25a) [α]D20 = -4.2 (c 0.4, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.73 (d, J = 2.4 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.79 (dd, J = 2.1, 8.4 Hz, 1H), 7.40-7.29 (m, 3H), 7.29-7.22 (m, 2H), 7.18-7.10 (m, 3H), 7.08-6.97 (m, 3H), 5.89 (m, 1H), 5.27 (m, 1H), 4.02 (m, 1H), 3.85 (m, 1H), 3.50 (s, 2H), 2.61 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 171.3, 161.8, 158.6, 158.2, 152.9, 148.7, 139.4, 136.5, 134.7, 133.3, 128.91, 128.86, 128.7, 127.9, 127.0, 126.4, 124.3, 112.7, 54.9, 46.4, 43.6, 22.3. ESI-MS m/z 492.1 (M + H) +; HRMS (ESI) calcd for C26H24Cl2N5O (M + H) + 492.1352, found 492.1344.
N-((R)-2-(5-chloro-2-(5-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-3-phenylpropanamide (25b) white solid (70% yield): [α]D20 = -3.1 (c 1.5, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.64 (m, 1H), 8.40 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.62 (brs, 1H), 7.30 (m, 3H), 7.18 (m, 5H), 6.98 (m, 2H), 6.00 (brs, 1H), 5.18 (m, 1H), 3.91 (m, 1H), 3.79 (m, 1H), 2.82 (t, J = 7.8 Hz, 2H), 2.59 (s, 3H), 2.42 (t, J = 7.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 172.7, 161.7, 158.5, 158.3, 152.8, 148.6, 140.7, 139.6, 136.5, 133.4, 128.8, 128.2, 127.7, 126.5, 126.0, 124.3, 112.8, 55.3, 46.8, 38.0, 31.6, 22.2. ESIMS m/z 506.0 (M + H)+; HR-ESIMS (m/z): Calcd. For C27H26Cl2N5O (M + H)+ 506.1509, found 506.1504.
N-((R)-2-(5-chloro-2-(5-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-4-phenylbutanamide (25c) white solid (76% yield): [α]D20 = -57.1 (c 0.9, CHCl3). 1H NMR (300 MHz, CDCl3) δ 8.69 (d, J = 2.7 Hz, 1H), 8.41 (d, J = 8.4 Hz, 1H), 7.80 (m, 1H), 7.45-7.28 (m, 6H), 7.25-7.11 (m, 3H), 7.03 (m, 2H), 6.06 (brs, 1H), 5.26 (m, 1H), 4.04 (m, 1H), 3.84 (m, 1H), 2.57 (s, 3H), 2.46 (m, 2H), 2.12 (m, 2H), 1.83 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 173.3, 161.7, 158.4, 152.8, 148.6, 141.4, 139.7, 136.5, 133.4, 128.9, 127.9, 126.6, 125.9, 124.3, 112.8, 55.1, 46.8, 35.7, 35.1, 27.1, 22.2.
N-((R)-2-(5-chloro-2-(5-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-5-phenylpentanamide (25d) white solid (80% yield): 1H NMR (300 MHz, CDCl3) δ 8.73 (d, J = 2.4 Hz, 1H), 8.42 (d, J = 7.5 Hz, 1H), 7.80 (m, 1H), 7.44-7.28 (m, 6H), 7.24 (m, 2H), 7.16 (m, 1H), 7.08 (m, 2H), 6.05 (brs, 1H), 5.25 (m, 1H), 4.02 (m, 1H), 3.84 (m, 1H), 2.58 (s, 3H), 2.48 (m, 2H), 2.12 (m, 2H), 1.62-1.39 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 173.4, 161.6, 158.3, 152.7, 148.5, 142.0, 139.6, 136.5, 133.3, 128.8, 128.18, 128.16, 127.8, 126.4, 125.9, 124.3, 112.7, 55.0, 46.7, 36.2, 35.5, 30.9, 25.3, 22.1. ESIMS m/z 556.1 (M + H)+; HR-ESIMS (m/z): Calcd. For C29H30Cl2N5O (M + H)+ 534.1822, found 534.1819.
N-((R)-2-(5-chloro-2-(5-chloropyridin-2-yl)-6-methylpyrimidin-4-ylamino)-1-phenylethyl)-6-phenylhexanamide (25e) white solid (75% yield): 1H NMR (300 MHz, CDCl3) δ 8.72 (m, 1H), 8.41 (d, J = 7.8 Hz, 1H), 7.79 (m, 1H), 7.43-7.30 (m, 5H), 7.30-7.21 (m, 3H), 7.20-7.08 (m, 3H), 6.06 (brs, 1H), 5.26 (m, 1H), 4.04 (m, 1H), 3.84 (m, 1H), 2.59 (s, 3H), 2.50 (t, J = 7.5 Hz, 2H), 2.08 (m, 2H), 1.49 (m, 4H), 1.17 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 173.7, 161.7, 158.4, 152.8, 148.6, 142.4, 139.7, 136.6, 133.4, 128.9, 128.3, 128.2, 127.9, 126.5, 125.6, 124.4, 112.8, 55.1, 46.8, 36.5, 35.7, 31.1, 28.8, 25.6. 22.2.
5.1.9. General procedure for preparing compounds 26 and 27
To a solution of 1 1f (0.401 mmol) and aldehyde (0.401 mmol) in MeOH (3 mL) was added acetic acid (0.441 mmol) under argon. The reaction mixture was stirred for 10 min at rt, and then NaBH3CN (26 mg, 0.401 mmol) was added at 0 °C. After being stirred for 1 h, the reaction mixture was diluted with CH2Cl2, and washed with saturated NaHCO3. The organic phase was dried over Na2SO4 and concentrated. Purification by flash chromatography (eluent: 5–10% EtOH—DCM or similar conditions) gave the corresponding amine.
N-((R)-2-(benzylamino)-2-phenylethyl)-5-chloro-2-(5-chloropyridin-2-yl)-6-methylpyrimidin-4-amine (26a) [α]D20 = -15.0 (c 2.5, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.70 (d, J = 2.4 Hz, 1H), 8.29 (d, J = 8.4 Hz, 1H), 7.73 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 7.23-7.44 (m, 11H), 6.13 (t, J = 4.8 Hz, 1H), 3.86-3.97 (m, 2H), 3.80 (d, J = 13.2 Hz, 1H), 3.60-3.69 (m, 2H), 2.61 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 161.4, 157.7, 153.1, 148.6, 141.2, 140.0, 136.3, 133.0, 128.9, 128.5, 128.2, 127.9, 127.1, 127.1, 124.3, 61.0, 51.1, 47.0, 22.3. ESI-MS m/z 464.1 (M + H) +; HRMS (ESI) calcd for C25H24N5Cl2 (M + H) + 464.1403, found 464.1393.
5-chloro-2-(5-chloropyridin-2-yl)-6-methyl-N-((R)-2-(phenethylamino)-2-phenylethyl)pyrimidin-4-amine (26b) foamy solid, [α]D20 = -16.5 (c 1.7, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.4 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.70 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 7.13-7.39 (m, 11H), 6.00 (t, J = 5.1 Hz, 1H), 3.81-3.96 (m, 2H), 3.58-3.65 (m, 1H), 2.76-3.87 (m, 4H), 2.60 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 161.4, 158.7, 157.7, 153.1, 148.6, 141.3, 139.8, 136.4, 133.0, 128.8, 128.6, 128.4, 127.8, 127.0, 126.2, 124.4, 112.8, 61.9, 48.3, 47.0, 36.3, 22.3. ESI-MS m/z 478.0 (M + H) +; HRMS (ESI) calcd for C26H26N5Cl2 (M + H) + 478.1560, found 478.1544.
5-chloro-2-(5-chloropyridin-2-yl)-6-methyl-N-((R)-2-phenyl-2-(3-phenylpropylamino)ethyl)pyrimidin-4-amine (26c) foamy solid, [α]D20 = -13.8 (c 2.4, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.4 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.72 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 7.08-7.41 (m, 11H), 6.16 (t, J = 5.1 Hz, 1H), 3.85-3.94 (m, 2H), 3.58-3.68 (m, 1H), 2.49-2.70 (m, 7H), 1.75-1.85 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.4, 158.7, 157.7, 153.1, 148.6, 142.0, 141.5, 136.3, 133.0, 128.7, 128.3, 128.2, 127.8, 126.9, 125.8, 124.4, 112.8, 61.9, 46.9, 46.6, 33.4, 31.8, 22.3. ESI-MS m/z 492.0 (M + H) +; HRMS (ESI) calcd for C27H28N5Cl2 (M + H) + 492.1716, found 492.1708.
5-chloro-2-(5-chloropyridin-2-yl)-6-methyl-N-((R)-2-phenyl-2-(4-phenylbutylamino)ethyl)pyrimidin-4-amine (26d) foamy solid, [α]D20 = -20.8 (c 0.3, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.1 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 2.1 Hz, 8.4 Hz, 1H), 7.12-7.41 (m, 11H), 6.15 (d, J = 5.1 Hz, 1H), 3.85-3.96 (m, 2H), 3.67-3.72 (m, 1H), 2.50-2.66 (m, 7H), 1.60-1.70 (m, 2H), 1.48-1.58 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.4, 158.7, 157.7, 153.1, 148.6, 142.3, 136.4, 133.1, 128.8, 128.4, 128.3, 127.8, 127.0, 125.7, 124.4, 112.9, 62.1, 47.1, 46.9, 35.7, 29.6, 29.0, 22.3. ESI-MS m/z 506.1 (M + H) +; HRMS (ESI) calcd for C28H30N5Cl2 (M + H) + 506.1873, found 506.1879.
5-chloro-2-(5-chloropyridin-2-yl)-6-methyl-N-((R)-2-phenyl-2-(5-phenylpentylamino)ethyl)pyrimidin-4-amine (26e) foamy solid, [α]D20 = -9.9 (c 1.2, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.73 (d, J = 1.5 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 1.5 Hz, 8.4 Hz, 1H), 7.13-7.41 (m, 10H), 6.16 (s, 1H), 3.85-3.95 (m, 2H), 3.62-3.70 (m, 1H), 2.49-2.60 (m, 7H), 1.47-1.64 (m, 4H), 1.33-1.40 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.5, 158.8, 157.8, 153.3, 148.8, 142.6, 136.5, 133.2, 128.9, 128.5, 128.4, 127.9, 127.1, 125.8, 124.5, 113.0, 62.1, 47.3, 47.0, 36.0, 31.4, 30.0, 27.0, 22.4. ESI-MS m/z 520.1 (M + H) +; HRMS (ESI) calcd for C29H32N5Cl2 (M + H) + 520.2029, found 520.2034.
5-chloro-2-(5-chloropyridin-2-yl)-6-methyl-N-((R)-2-phenyl-2-(6-phenylhexylamino)ethyl)pyrimidin-4-amine (26f) foamy solid, [α]D20 = -12.3 (c 1.4, CHCl3). 1H NMR (300 MHz, CDCl3): δ 8.73 (d, J = 2.4 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 7.14-7.43 (m, 10H), 6.15 (t, J = 4.8 Hz, 1H), 3.84-3.94 (m, 2H), 3.61-3.70 (m, 1H), 2.46-2.60 (m, 7H), 1.53-1.63 (m, 2H), 1.42-1.47 (m, 2H), 1.25-1.33 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 161.5, 158.8, 157.8, 153.3, 148.8, 142.8, 136.5, 133.2, 128.9, 128.5, 128.4, 127.9, 127.1, 125.7, 124.5, 62.1, 47.4, 47.0, 36.0, 31.6, 30.1, 29.3, 27.2, 22.4. ESI-MS m/z 556.1 (M + H) +; HRMS (ESI) calcd for C30H34N5Cl2 (M + H) + 534.2186, found 534.2175.
4-(4-((R)-2-(5-chloro-2-(5-chloropyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethylamino)butyl)phenol (27a) 1H NMR (300 MHz, CDCl3): δ 8.68 (d, J = 1.2 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.75 (m, 1H), 7.26-7.40 (m, 5H), 6.94 (d, J = 7.6 Hz, 2H), 6.71 (d, J = 8.1 Hz, 2H), 6.02 (t, J = 5.1 Hz, 1H), 3.85-3.92 (m, 1H), 3.71-3.83 (m, 2H), 2.93 (s, 3H), 2.46-2.55 (m, 7H), 1.56-1.60 (m, 2H), 1.48-1.53 (m, 2H). ESI-MS m/z 522.3 (M + H) +; HRMS (ESI) calcd for C28H30N5Cl2O (M + H) + 522.1835, found 522.18219.
5-chloro-N-((R)-2-(4-(4-chlorophenyl)butylamino)-2-phenylethyl)-2-(5-chloropyridin-2-yl)pyrimidin-4-amine (27b) foamy solid (53% yield): 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.1 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.73 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 7.28-7.40 (m, 5H), 7.22 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 6.09 (t, J = 5.4 Hz, 1H), 3.83-3.92 (m, 2H), 3.62-3.70 (m, 1H), 2.59 (s, 3H), 2.47-2.56 (m, 4H), 1.57-1.67 (m, 2H), 1.43-1.53 (m, 2H). 13 (100 MHz, CDCl3): δ 161.5, 158.8, 157.9, 153.2, 148.7, 141.6, 140.8, 136.5, 133.2, 131.5, 129.8, 129.7, 128.9, 128.4, 127.9, 127.1, 124.5, 112.9, 62.2, 47.2, 47.0, 35.1, 29.7, 29.0, 22.3. ESI-MS m/z 540.3 (M + H) +; HRMS (ESI) calcd for C28H29N5Cl3 (M + H) + 540.1500, found 540.1483.
4-(4-((R)-2-(5-chloro-2-(5-chloropyridin-2-yl)pyrimidin-4-ylamino)-1-phenylethylamino)butyl)benzonitrile (27c) 1H NMR (300 MHz, CDCl3): δ 8.71 (d, J = 1.2 Hz, 1H), 8.36 (d, J = 8.7 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 7.8 Hz, 2H), 7.29-7.42 (m, 5H), 7.21 (d, J = 7.8 Hz, 2H), 6.21 (s, 1H), 3.87-3.98 (m, 2H), 3.69-3.76 (m, 1H), 2.51-2.65 (m, 7H), 1.61-1.71 (m, 2H), 1.51-1.57 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.1, 157.8, 157.7, 152.3, 148.3, 147.7, 139.3, 133.6, 132.1, 129.1,129.0, 128.3, 127.1, 124.5, 118.9, 109.6, 62.1, 35.6, 28.7, 28.3, 22.1. ESI-MS m/z 531.4 (M + H) +;HRMS (ESI) calcd for C29H29N6Cl2 (M + H) + 531.1844, found 531.18253.
5-chloro-2-(5-chloropyridin-2-yl)-N-((R)-2-phenyl-2-(4-(4-propoxyphenyl)butylamino)ethyl)pyrimidin-4-amine (27d) 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.1 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.72 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 7.26-7.40 (m, 5H), 7.03 (d, J = 8.4 Hz, 2H), 6.80 (d, J = 8.4 Hz, 2H), 6.10 (t, J = 5.4 Hz, 1H), 3.82-3.93 (m, 4H), 3.62-3.70 (m, 1H), 2.59 (s, 3H), 2.49-2.56 (m, 4H), 1.75-1.85 (m, 2H), 1.57-1.66 (m, 2H), 1.47-1.54 (m, 2H), 1.02 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 161.5, 158.8, 157.8, 157.3, 153.2, 148.7, 141.7, 136.4, 134.3, 133.1, 129.3, 128.8, 127.8, 127.1, 124.5, 114.4, 112.9, 69.6, 62.1, 47.3, 47.0, 34.9, 29.8, 29.4, 22.7, 22.3, 10.6. ESI-MS m/z 564.5 (M + H) +; HRMS (ESI) calcd for C31H36N5OCl2 (M + H) + 564.2307, found 564.2291.
5-chloro-2-(5-chloropyridin-2-yl)-N-((R)-2-(4-(furan-2-yl)butylamino)-2-phenylethyl)-6-methylpyrimidin-4-amine (27e) [α]D20 = -9.0 (c 1.50, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.1 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 2.1 Hz, 8.4 Hz, 1H), 7.27-7.41 (m, 5H), 6.26 (t, J = 2.1 Hz, 1H), 6.12 (m, 1H), 6.73 (d, J = 5.1 Hz, 1H), 5.94 (d, J = 3.3 Hz, 1H), 3.85-3.96 (m, 2H), 3.66-3.75 (m, 1H), 2.50-2.64 (m, 7H), 2.13 (s, 1H), 1.63-1.73 (m, 2H), 1.48-1.58 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.5, 158.5, 157.9, 156.1, 153.2, 148.7, 141.2, 140.9, 136.5, 133.2, 128.9, 128.0, 127.2, 124.5, 113.0, 110.2,104.9, 62.2, 47.0, 46.9, 29.5, 27.8, 25.8, 22.4. ESI-MS m/z 496.4 (M + H) +; HRMS (ESI) calcd forC26H28N5OCl2 (M + H) + 496.1666, found 496.1665.
5-chloro-2-(5-chloropyridin-2-yl)-6-methyl-N-((R)-2-phenyl-2-(4-(thiophen-2-yl)butylamino)ethyl)pyrimidin-4-amine (27f) [α]D20 = -10.9 (c 1.35, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.1 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 2.1 Hz, 8.4 Hz, 1H), 7.26-7.42 (m, 5H), 7.09 (dd, J = 0.9 Hz, 5.1 Hz, 1H), 6.87-6.90 (m, 1H), 6.73 (d, J = 3 Hz), 6.15 (t, J = 4.8 Hz, 1H), 3.86-3.98 (m, 2H), 3.69-3.77 (m, 1H), 2.79 (t, J = 7.5 Hz, 2H), 2.53-2.67 (m, 5H), 2.30 (s, 1H), 1.66-1.76 (m, 2H), 1.52-1.62 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.6, 158.8, 157.9, 153.2, 148.7, 145.2, 136.5, 133.2, 129.0, 128.9, 128.1, 127.2, 126.8, 124.5, 124.2, 123.0, 113.1, 62.2, 47.0, 46.9, 29.8, 29.5, 29.3, 22.4. ESI-MS m/z 512.1 (M + H) +; HRMS (ESI) calcd for C26H28N5SCl2 (M + H) + 512.1447, found 512.1437.
5-chloro-2-(5-chloropyridin-2-yl)-6-methyl-N-((R)-2-phenyl-2-(4-(quinolin-2-yl)butylamino)ethyl)pyrimidin-4-amine (27g) foamy solid (32% yield): [α]D20 = -14.4 (c 1.50, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.1 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 8.04 (t, J = 8.7 Hz, 2H), 7.65-7.78 (m, 3H), 7.48 (t, J = 7.8 Hz, 1H), 7.23-7.37 (m, 6H), 6.13 (t, J = 5.1 Hz, 1H), 3.83-3.95 (m, 2H), 3.64-3.72 (m, 1H), 2.96 (t, J = 7.5 Hz, 2H), 2.60-2.67 (m, 2H), 2.57 (s, 3H), 2.14 (s, 1H), 1.82-1.92 (m, 2H), 1.56-1.66 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 162.6, 161.5, 158.8, 157.9, 153.3, 148.7, 148.0, 141.4, 136.5, 136.4, 133.2, 129.5, 128.9, 128.8, 127.9, 127.6, 127.1, 126.8, 125.9, 124.5, 121.4, 113.0, 62.1, 47.2, 47.0, 39.0, 29.9, 27.6, 22.3. ESI-MS m/z 557.3 (M + H) +; HRMS (ESI) calcd for C31H31N6Cl2 (M + H) + 557.1992, found 557.1982.
N-((R)-2-(2-(1H-pyrrol-1-yl)ethylamino)-2-phenylethyl)-5-chloro-2-(5-chloropyridin-2-yl)-6-methylpyrimidin-4-amine (27h) [α]D20 = -13.7 (c 1.45, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 2.4 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 2.4 Hz, 8.4 Hz, 1H), 7.31-7.41 (m, 5H), 6.60 (m, 2H), 6.12 (m, 2H), 6.08 (t, J = 5.4 Hz, 1H), 3.82-3.93 (m, 4H), 3.65-3.73 (m, 1H), 2.59 (s, 3H), 2.45-2.57 (m, 2H), 2.10 (s, 1H), 1.74-1.84 (m, 2H), 1.41-1.51 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 160.6, 157.8, 156.9, 152.2, 147.7, 140.2, 135.5, 132.2, 127.9, 127.0, 126.1, 123.5, 119.5, 112.0, 107.0, 61.2, 48.5, 46.0, 45.8, 28.3, 26.3, 21.3. ESI-MS m/z 495.3 (M + H) +; HRMS (ESI) calcd for C26H29N6Cl2 (M + H) + 495.1826, found 495.1825.
5.1.10. General procedure for preparing compounds 30
Phenylacetonitrile (11 mmol) was added dropwise to a suspension solution of NaH (11 mmol) in anhydrous DMF (20 mL) at 0 °C under nitrogen. After the reaction mixture was stirred for 30 min at rt, bromide (10 mmol) was added in a dropwise manner. The resultant solution was stirred overnight at rt before water was added to quench the reaction. After the mixture was extracted with ethyl acetate, the organic phase was washed with brine, and dried over Na2SO4. Evaporation and column chromatography on silica gel afforded the crude alkylation product, which was dissolved in anhydrous Et2O. The resultant solution was added dropwise to a suspension of LiAlH4 (4.4 mmol) in anhydrous Et2O at rt under nitrogen. After being stirred for 1 h, the reaction was quenched by adding water. Ether extract work up followed by chromatography provided crude amine, which was dissolved in THF. To this solution were added 11b and TEA before it was refluxed for 24 h. Ethyl acetate work up followed by purification by flash chromatography (eluent: 5–10% EtOH—DCM or similar conditions) gave the corresponding condensation product.
5-chloro-2-(5-chloropyridin-2-yl)-N-(2,4-diphenylbutyl)-6-methylpyrimidin-4-amine (30a) 1H NMR (300 MHz, CDCl3): δ 8.74 (d, J = 2.1 Hz, 1H), 8.30 (d, J = 8.7 Hz, 1H), 7.76 (dd, J = 2.1 Hz, 8.7 Hz, 1H), 7.10-7.40 (m, 10H), 5.32 (t, J = 6.3 Hz, 1H), 3.96-4.05 (m, 1H), 3.59-3.68 (m, 1H), 2.94-3.00 (m, 1H), 2.50-2.61 (m, 5H), 2.04-2.14 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.4, 158.8, 157.7, 153.3, 148.7, 142.0, 141.8, 136.4, 133.1, 129.0, 128.4, 128.3, 127.9, 127.1, 125.9, 124.4, 112.7, 47.0, 45.0, 35.0, 33.4, 22.3. ESI-MS m/z 463.1 (M + H) +; HRMS (ESI) calcd for C26H25N4Cl2 (M + H) + 463.1457, found 463.1451.
5-chloro-2-(5-chloropyridin-2-yl)-N-(2,5-diphenylpentyl)-6-methylpyrimidin-4-amine (30b) foamy solid : 1H NMR (300 MHz, CDCl3): δ 8.73 (d, J = 1.8 Hz, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.78 (dd, J = 1.8 Hz, 8.4 Hz, 1H), 7.08-7.35 (m, 10H), 5.32 (m, 1H), 3.97-4.06 (m, 1H), 3.54-3.63 (m, 1H), 2.92-2.97 (m, 1H), 2.55-2.62 (m, 5H), 1.76-1.85 (m, 2H), 1.57-1.75 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.5, 158.9, 157.7, 153.8, 142.5, 142.1, 136.5, 133.2, 133.1, 128.9, 128.5, 128.4, 127.9, 127.1, 125.9, 124.5, 112.9, 45.9, 45.7, 35.9, 33.0, 29.3, 22.4. ESI-MS m/z 477.4 (M + H) +; HRMS (ESI) calcd for C27H27N4Cl2 (M + H) + 477.1618, found 477.1607.
5-chloro-2-(5-chloropyridin-2-yl)-N-(2,6-diphenylhexyl)-6-methylpyrimidin-4-amine (30c) foamy solid : 1H NMR (300 MHz, CDCl3): δ 8.74 (d, J = 1.8 Hz, 1H), 8.36 (d, J = 8.4 Hz, 1H), 7.74 (dd, J = 1.8 Hz, 8.4 Hz, 1H), 7.07-7.37 (m, 10H), 5.33 (t, J = 5.4 Hz, 1H), 3.97-4.05 (m, 1H), 3.55-3.64 (m, 1H), 2.90-3.00 (m, 1H), 2.51-2.60 (m, 5H), 1.72-1.83 (m, 2H), 1.57-1.69 (m, 2H), 1.26-1.37 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.5, 158.9, 157.7, 153.4, 148.8, 142.6, 142.5, 136.5, 133.2, 128.9, 128.4, 128.3, 127.9, 127.0, 126.8, 125.8, 124.4, 112.8, 47.0, 45.6, 35.8, 33.5, 31.5, 27.2, 22.3. ESI-MS m/z 491.4 (M + H)+; HRMS (ESI) calcd for C28H29N4Cl2 (M + H)+ 491.1767, found 491.1764.
5-chloro-2-(5-chloropyridin-2-yl)-N-(2,7-diphenylheptyl)-6-methylpyrimidin-4-amine (30d) foamy solid : 1H NMR (300 MHz, CDCl3): δ 8.75 (d, J = 2.7 Hz, 1H), 8.36 (d, J = 8.7 Hz, 1H), 7.75 (dd, J = 2.7 Hz, 8.7 Hz, 1H), 7.10-7.37 (m, 10H), 5.32 (t, J = 5.1 Hz, 1H), 3.96-4.05 (m, 1H), 3.56-3.63 (m, 1H), 2.88-2.93 (m, 1H), 2.51-2.57 (m, 5H), 1.70-1.76 (m, 2H), 1.54-1.59 (m, 2H), 1.29-1.31 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 161.3, 158.6, 157.7, 153.3, 148.7, 142.6, 142.5, 136.4, 133.2, 128.8, 128.4, 128.3, 127.8, 127.0, 125.6, 124.4, 112.8, 46.9, 45.6, 35.8, 33.4, 31.2, 29.2, 27.3, 22.2. ESI-MS m/z 505.5 (M + H)+; HRMS (ESI) calcd for C298H31N4Cl2 (M + H)+ 505.19267, found 505.19203.
5.2 Biological assays
5.2.1. Materials
30% acrylamide/bis solution (37.5:1) (161-0158), ammonium persulfate (161-0700) and N,N,N′,N′-tetramethylethlenediamine (TEMED) (161-0800) for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was purchased from Bio-Rad Laboratories (Hercules, CA). CoCl2·6H2O (C-3169) was purchased from Sigma-Aldrich (St. Louis, MO). Other commercially available chemicals were purchased from Sigma-Aldrich and Fisher Scientific (Hampton, NH). Restriction enzymes were purchased from New England BioLabs (Ipswich, MA).
5.2.2. Cell lines and cell culture
High Five (BTI-TN-5B1-4) cells (Life Technologies B855-02, Carlsbad, CA) used for the expression of recombinant HsMetAP2 were grown in Grace's insect cell medium (Life Technologies 11605-094) supplemented with 10% FBS and were maintained in a nonhumidified incubator at 27°C as per manufacturer's instructions.
5.2.3. ProIP-coupled MetAP activity assay
MetAP is known to have substrate specificity completely orthogonal to that of ProIP. The BcProIP-coupled MetAP activity assay was adapted from [47] with modifications. In this assay, MetAP activity was measured by continuously monitoring the production of pNA with BcProIP. Aliquots of purified recombinant human MetAP1 and MetAP2 proteins stored at -80 °C were thawed on ice. Repeated freeze-thaw cycles were avoided. The coupling enzyme BcProIP was taken from the stock at -20 °C. The substrate methionylprolyl-p-nitroanilide (Met-Pro-pNA) and the test compounds were dissolved in 100% DMSO. The total reaction volume was 100 μL and the reaction was carried out in 96-well plates at room temperature. 0.15 μM of HsMetAP1 or 0.1 μM of HsMetAP2 was incubated with either vehicle (DMSO) or test compounds in MetAP assay buffer containing 15 U/mL of BcProIP and 10 μM of CoCl2 at room temperature for 20 minutes. The enzymatic reaction was initiated by the addition of 600 μM of Met-Pro-pNA. Absorbance at 405 nm (ε = 1.06×104 M-1cm-1) was recorded every 30 seconds within the first 30 minutes of reaction by the FLUOstar OPTIMA microplate reader. After subtracting the background hydrolysis determined in the absence of MetAP, the net increases in the absorbance were plotted versus the reaction time and the slope of the early linear portion (usually between 2 minutes and 15 minutes after the reaction started) of the curve was determined by the function “linear regression analysis” in GraphPad Prism 5. The initial rate was then calculated from the slope. In addition, to rule out the possibility that any inhibitor found by the BcProIP-coupled MetAP assay might work simply because it inhibited the coupling enzyme, compounds showing inhibitory effects were counter screened against 0.1 U/mL of BcProIP, 600 μM of Pro-pNA and 10 μM of CoCl2 in the MetAP assay buffer. Although the initial rates of 0.1 U/mL of BcProIP were greatly decreased by 100 μM of 18m and 18n, the remaining activity of 15 U/mL of BcProIP was still enough to saturate the coupling reaction in MetAP activity assays (data no shown).
5.2.4. Determination of the IC50 or EC50 values of the inhibitors
For in vitro MetAP activity assays, the initial rates of the reactions determined by the function “linear regression analysis” in GraphPad Prism 5 were first normalized to the initial rates of the vehicle control, and then plotted versus the concentrations of the compound (in a logarithmic scale). For cell proliferation assays, the [3H]-thymidine counts determined by the MicroBeta plate reader were first normalized to the counts of the vehicle control, and then plotted versus the concentrations of the compound (in a logarithmic scale). The inhibitory dose-response curve was generated by the nonlinear regression function “log(inhibitor) vs. response-Variable slope” in GraphPad Prism 5, using the model “Y=Bottom+(Top-Bottom)/{1+10×[(LogEC50-X)×HillSlope]}” and the fitting method “least squares”. Top and Bottom are plateaus in the units of the Y axis. The Top values of the dose-response curve were always constrained to 1 (100% of the vehicle control). The Bottom values of the dose-response curve were constrained to zero (a complete inhibition) when the calculated Bottom values were smaller than zero. Otherwise, the Bottom values were not constrained and will be reported when they were larger than 0.1 (10% of the vehicle control). EC50 was used to define the concentration of an inhibitor that gave a response half way between Bottom and Top. When Top=1 and Bottom=zero, IC50 was used to replace EC50.
Supplementary Material
Acknowledgments
We thank Drs. Daisuke Tsuru and Ana Kitzono for providing us with pPI-20 plasmid containing BcProIP gene. This work was supported in part by a grant under National Institutes of Health (NIH) R01 CA078743. Molecular graphics and analyses were performed in part with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, funded by grants from the NIH National Center for Research Resources (2P41RR001081) and National Institute of General Medical Sciences (9P41GM103311).
Abbreviations
- BcProIP
Bacillus coagulans proline iminopeptidase
- DMSO
dimethyl sulfoxide
- EC50
half maximal effective concentration
- HEPES
2-[4-(2-hydroxylethyl)-1-piperazinyl]ethanesulfonic acid
- HRP
horseradish peroxidase
- HsMetAP1
human cytosolic type 1 methionine aminopeptidase
- HsMetAP2
human cytosolic type 2 methionine aminopeptidase
- IC50
half maximal inhibitory concentration (IC50 equals to EC50 when the bottom value of an inhibitory dose-response curve is zero)
- Ki
dissociation constant for inhibitor binding
- map
gene encoding prokaryotic methionine aminopeptidase
- map1
gene encoding eukaryotic cytosolic type 1 methionine aminopeptidase
- map2
gene encoding eukaryotic cytosolic type 2 methionine aminopeptidase
- MetAP
methionine aminopeptidase
- MetAP1
eukaryotic cytosolic type 1 methionine aminopeptidase
- MetAP2
eukaryotic cytosolic type 2 methionine aminopeptidase
- NME
N-terminal methionine excision
- SAR
structure-activity relationship
- tHsMetAP1
N-terminally truncated human methionine aminopeptidase
Footnotes
Atomic coordinates for the N-terminally truncated human MetAP1 in complex with 26d and Co(II) have been deposited in the Protein Data Bank (www.pdb.org) under the access code 4HXX
Notes: The authors report no conflicts of interest.
Author contributions: All authors were involved in designing experiments and interpreting data. P. Z. and X. Y. synthesized most of the new compounds for this study and F. Z. carried out most of the biological experiments. They contributed equally to this work. P. Z., X. Y., R. W., Y. Z. and S. B. synthesized the new compounds. F. Z. and X. C. characterized the inhibitory activities of the compounds with human MetAPs. S. B. G., M. F. and F. Z. performed the crystallographic analysis. F. Z., S. B. G., J. O. L. and D. M. wrote the manuscript.
Supplementary data: Supplementary data associated with this article include: the expression and purification of HsMetAP1 and HsMetAP2; the cloning, expression and purification of BcProIP and tHsMetAP1; the determination of the activity of Bacillus coagulans ProIP and the coupling condition; the crystallization of tHsMetAP1; the collection and processing of X-ray diffraction data; and the structure determination and refinement.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kozak M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol Rev. 1983;47:1. doi: 10.1128/mr.47.1.1-45.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waller JP. The NH2-terminal residue of the proteins from cell-free extract of E. coli. J Mol Biol. 1963;7:483. doi: 10.1016/s0022-2836(63)80096-0. [DOI] [PubMed] [Google Scholar]
- 3.Meinnel T, Mechulam Y, Blanquet S. Methionine as translation start signal: A review of the enzymes of the pathway in Escherichia coli. Biochimie. 199375:1061. doi: 10.1016/0300-9084(93)90005-d. [DOI] [PubMed] [Google Scholar]
- 4.Solbiati J, Chapman-Smith A, Miller JL, Miller CG, Cronan JE., Jr Processing of the N termini of nascent polypeptide chains requires deformylation prior to methionine removal. J Mol Biol. 1999;290:607. doi: 10.1006/jmbi.1999.2913. [DOI] [PubMed] [Google Scholar]
- 5.Boissel JP, Kasper TJ, Bunn HF. Cotranslational amino-terminal processing of cytosolic proteins. Cell-free expression of site-directed mutants of human hemoglobin. J Biol Chem. 1988;263:8443. [PubMed] [Google Scholar]
- 6.Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: the methionine aminopeptidase and Nα-acetyl transferase families. Trends in Biochem Sci. 1998;23:263. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 7.(a) D'souza VM, Bennett B, Copik AJ, Holz RC. Divalent metal binding properties of the methionyl aminopeptidase from Escherichia coli. Biochemistry. 2000;39:3817. doi: 10.1021/bi9925827. [DOI] [PubMed] [Google Scholar]; (b) D'souza VM, Swierczek SI, Cosper NJ, Meng L, Ruebush S, Copik AJ, Scott RA, Holz RC. Kinetic and structural characterization of manganese(II)-loaded methionyl aminopeptidases. Biochemistry. 2002;41:13096. doi: 10.1021/bi020395u. [DOI] [PubMed] [Google Scholar]; (c) Ye QZ, Xie SX, Ma ZQ, Huang M, Hanzlik RP. Structural basis of catalysis by monometalated methionine aminopeptidase. Proc Natl Acad Sci U S A. 2006;103:9470. doi: 10.1073/pnas.0602433103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chai SC, Ye QZ. Analysis of the stoichiometric metal activation of methionine aminopeptidase. BMC Biochem. 2009;10:32. doi: 10.1186/1471-2091-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chai SC, Lu JP, Ye QZ. Determination of binding affinity of metal cofactor to the active site of methionine aminopeptidase based on quantitation of functional enzyme. Anal Biochem. 2009;395:263. doi: 10.1016/j.ab.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Flinta C, Persson B, Jornvall H, von Heijne G. Sequence determinants of cytosolic N-terminal protein processing. Eur J Biochem. 1986;154:193. doi: 10.1111/j.1432-1033.1986.tb09378.x. [DOI] [PubMed] [Google Scholar]; (b) Hirel PH, Schmitter JM, Dessen P, Fayat G, Blanquet S. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc Natl Acad Sci U S A. 1989;86:8247. doi: 10.1073/pnas.86.21.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chang YH, Teicher U, Smith JA. Purification and characterization of a methionine aminopeptidase from Saccharomyces cerevisiae. J Biol Chem. 1990;265:19892. [PubMed] [Google Scholar]; (d) Yang G, Kirkpatrick RB, Ho T, Zhang GF, Liang PH, Johanson KO, Casper DJ, Doyle ML, Marino JP, Jr, Thompson SK, Chen W, Tew DG, Meek TD. Steady-state kinetic characterization of substrates and metal-ion specificities of the full-length and N-terminally truncated recombinant human methionine aminopeptidases(type 2) Biochemistry. 2001;40:10645. doi: 10.1021/bi010806r. [DOI] [PubMed] [Google Scholar]; (e) Frottin F, Martinez A, Peynot P, Mitra S, Holz RC, Giglione C, Meinnel T. The proteomics of N-terminal methionine cleavage. Mol Cell Proteomics. 2006;5:2336. doi: 10.1074/mcp.M600225-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Xiao Q, Zhang F, Nacev BA, Liu JO, Pei D. Protein N-terminal processing: substrate specificity of Escherichia coli and human methionine aminopeptidases. Biochemistry. 2010;49:5588. doi: 10.1021/bi1005464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang SY, McGary EC, Chang S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J Bacteriol. 1989;171:4071. doi: 10.1128/jb.171.7.4071-4072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller CG, Kukral AM, Miller JL, Movva NR. pepM is an essential gene in Salmonella typhimurium. J Bacteriol. 1989;171:5215. doi: 10.1128/jb.171.9.5215-5217.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Chang YH. Amino-terminal protein processing in Saccharomyces cerevisiae is an essential function that requires two distinct methionine aminopeptidases. Proc Natl Acad Sci U S A. 1995;92:12357. doi: 10.1073/pnas.92.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arfin SM, Kendall RL, Hall L, Weaver LH, Stewart AE, Matthews BW, Bradshaw RA. Eukaryotic methionyl aminopeptidases: two classes of cobalt-dependent enzymes. Proc Natl Acad Sci U S A. 1995;92:7714. doi: 10.1073/pnas.92.17.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Addlagatta A, Hu X, Liu JO, Matthews BW. Structural basis for the functional differences between type I and type II human methionine aminopeptidases. Biochemistry. 2005;44:14741–14749. doi: 10.1021/bi051691k. [DOI] [PubMed] [Google Scholar]
- 15.Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, Hutchison CA, 3rd, Smith HO, Venter JC. Essential genes of a minimal bacterium. Proc Natl Acad Sci U S A. 2006;103:425. doi: 10.1073/pnas.0510013103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross S, Giglione C, Pierre M, Espagne C, Meinnel T. Functional and developmental impact of cytosolic protein N-terminal methionine excision in Arabidopsis. Plant Physiol. 2005;137:623. doi: 10.1104/pp.104.056861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Cutforth T, Gaul U. A methionine aminopeptidase and putative regulator of translation initiation is required for cell growth and patterning in Drosophila. Mech Dev. 1999;82:23. doi: 10.1016/s0925-4773(99)00006-4. [DOI] [PubMed] [Google Scholar]; (b) Wernert N, Stanjek A, Kiriakidis S, Hügel A, Jha HC, Mazitschek R, Giannis A. Inhibition of Angiogenesis In Vivo by ets-1 Antisense Oligo nucleotides-Inhibition of Ets-1 Transcription Factor Expression by the Antibiotic Fumagillin. Angew Chem Int Ed Engl. 1999;38:3228. doi: 10.1002/(sici)1521-3773(19991102)38:21<3228::aid-anie3228>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (c) Boxem M, Tsai CW, Zhang Y, Saito RM, Liu JO. The C. elegans methionine aminopeptidase 2 analog map-2 is required for germ cell proliferation. FEBS Lett. 2004;576:245. doi: 10.1016/j.febslet.2004.08.077. [DOI] [PubMed] [Google Scholar]; (d) Zhang Y, Yeh JR, Mara A, Ju R, Hines JF, Cirone P, Griesbach HL, Schneider I, Slusarski DC, Holley SA, Crews CM. A chemical and genetic approach to the mode of action of fumagillin. Chem Biol. 2006;13:1001. doi: 10.1016/j.chembiol.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yeh JR, Ju R, Brdlik CM, Zhang W, Zhang Y, Matyskiela ME, Shotwell JD, Crews CM. Targeted gene disruption of methionine aminopeptidase 2 results in an embryonic gastrulation defect and endothelial cell growth arrest. Proc Natl Acad Sci U S A. 2006;103:10379. doi: 10.1073/pnas.0511313103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ma AC, Fung TK, Lin RH, Chung MI, Yang D, Ekker SC, Leung AY. Methionine aminopeptidase 2 is required for HSC initiation and proliferation. Blood. 2011;118:544. doi: 10.1182/blood-2011-04-350173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bernier SG, Taghizadeh N, Thompson CD, Westlin WF, Hannig G. Methionine aminopeptidases type I and type II are essential to control cell proliferation. J Cell Biochem. 2005;95:1191. doi: 10.1002/jcb.20493. [DOI] [PubMed] [Google Scholar]
- 19.Hu X, Addlagatta A, Lu J, Matthews BW, Liu JO. Elucidation of the function of type 1 human methionine aminopeptidase during cell cycle progression. Proc Natl Acad Sci U S A. 2006;103:18148. doi: 10.1073/pnas.0608389103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quiiiol E, Adamczeski M, Bakus GJ, Crews P. Bengamides, heterocyclic anthelmintics from a Jaspidae marine sponge. J Org Chem. 1986;51:4494. [Google Scholar]
- 21.Thale Z, Kinder FR, Bair KW, Bontempo J, Czuchta AM, Versace RW, Phillips PE, Sanders ML, Wattanasin S, Crews P. Bengamides revisited: new structures and antitumor studies. J Org Chem. 2001;66:1733. doi: 10.1021/jo001380+. [DOI] [PubMed] [Google Scholar]
- 22.Towbin H, Bair KW, DeCaprio JA, Eck MJ, Kim S, Kinder FR, Morollo A, Mueller DR, Schindler P, Song HK, van Oostrum J, Versace RW, Voshol H, Wood J, Zabludoff S, Phillips PE. Proteomics-based target identification: bengamides as a new class of methionine aminopeptidase inhibitors. J Biol Chem. 2003;278:52964. doi: 10.1074/jbc.M309039200. [DOI] [PubMed] [Google Scholar]
- 23.Kinder FR, Jr, Versace RW, Bair KW, Bontempo JM, Cesarz D, Chen S, Crews P, Czuchta AM, Jagoe CT, Mou Y, Nemzek R, Phillips PE, Tran LD, Wang RM, Weltchek S, Zabludoff S. Synthesis and antitumor activity of ester-modified analogues of bengamide B. J Med Chem. 2001;44:3692. doi: 10.1021/jm010188c. [DOI] [PubMed] [Google Scholar]
- 24.Ingber D, Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H, Folkman J. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature. 1990;348:555. doi: 10.1038/348555a0. [DOI] [PubMed] [Google Scholar]
- 25.Kusaka M, Sudo K, Fujita T, Marui S, Itoh F, Ingber D, Folkman J. Potent anti-angiogenic action of AGM-1470: comparison to the fumagillin parent. Biochem Biophys Res Commun. 1991;174:1070. doi: 10.1016/0006-291x(91)91529-l. [DOI] [PubMed] [Google Scholar]
- 26.(a) Griffith EC, Su Z, Turk BE, Chen S, Chang YH, Wu Z, Biemann K, Liu JO. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem Biol. 1997;4:461. doi: 10.1016/s1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]; (b) Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci U S A. 1997;94:6099. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffith EC, Su Z, Niwayama S, Ramsay CA, Chang YH, Liu JO. Molecular recognition of angiogenesis inhibitors fumagillin and ovalicin by methionine aminopeptidase 2. Proc Natl Acad Sci U S A. 1998;95:15183. doi: 10.1073/pnas.95.26.15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu S, Widom J, Kemp CW, Crews CM, Clardy J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science. 1998;282:1324. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- 29.Turk BE, Griffith EC, Wolf S, Biemann K, Chang YH, Liu JO. Selective inhibition of amino-terminal methionine processing by TNP-470 and ovalicin in endothelial cells. Chem Biol. 1999;6:823. doi: 10.1016/s1074-5521(99)80129-x. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Lou P, Henkin J. Selective inhibition of endothelial cell proliferation by fumagillin is not due to differential expression of methionine aminopeptidases. J Cell Biochem. 2000;77:465. [PubMed] [Google Scholar]
- 31.Gervaz P, Fontolliet C. Therapeutic potential of the anti-angiogenesis drug TNP-470. Int J Exp Pathol. 1998;79:359. doi: 10.1046/j.1365-2613.1998.00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kruger EA, Figg WD. TNP-470: an angiogenesis inhibitor in clinical development for cancer. Expert Opin Investig Drugs. 2000;9:1383. doi: 10.1517/13543784.9.6.1383. [DOI] [PubMed] [Google Scholar]
- 33.Lijnen HR, Frederix L, Van Hoef B. Fumagillin reduces adipose tissue formation in murine models of nutritionally induced obesity. Obesity. 2010;18:2241. doi: 10.1038/oby.2009.503. [DOI] [PubMed] [Google Scholar]
- 34.Kass DJ, Rattigan E, Kahloon R, Loh K, Yu L, Savir A, Markowski M, Saqi A, Rajkumar R, Ahmad F, Champion HC. Early treatment with fumagillin, an inhibitor of methionine aminopeptidase-2, prevents Pulmonary Hypertension in monocrotaline-injured rats. PLoS One. 2012;7:e35388. doi: 10.1371/journal.pone.0035388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brahn E, Schoettler N, Lee S, Banquerigo ML. Involution of collagen-induced arthritis with anangiogenesis inhibitor, PPI-2458. J Pharmacol Exp Ther. 2009;329:615. doi: 10.1124/jpet.108.148478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu X, Zhu J, Srivathsan S, Pei D. Peptidyl hydroxamic acids as methionine aminopeptidase inhibitors. Bio org Med Chem Lett. 2004;14:77. doi: 10.1016/j.bmcl.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 37.Haldar MK, Scott MD, Sule N, Srivastava DK, Mallik S. Synthesis of barbiturate-based methionine aminopeptidase-1 inhibitors. Bioorg Med Chem Lett. 2008;18:2373. doi: 10.1016/j.bmcl.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo QL, Li JY, Liu ZY, Chen LL, Li J, Qian Z, Shen Q, Li Y, Lushington GH, Ye QZ, Nan FJ. Discovery and structural modification of inhibitors of methionine aminopeptidases from Escherichia coli and Saccharomyces cerevisiae. J Med Chem. 2003;46:2631. doi: 10.1021/jm0300532. [DOI] [PubMed] [Google Scholar]
- 39.Li JY, Chen LL, Cui YM, Luo QL, Gu M, Nan FJ, Ye QZ. Characterization of full length and truncated type I human methionine aminopeptidases expressed from Escherichia coli. Biochemistry. 2004;43:7892. doi: 10.1021/bi0360859. [DOI] [PubMed] [Google Scholar]
- 40.Hu X, Addlagatta A, Matthews BW, Liu JO. Identification of pyridinylpyrimidines as inhibitors of human methionine aminopeptidases. Angew Chem Int Ed Engl. 2006;45:3772. doi: 10.1002/anie.200600757. [DOI] [PubMed] [Google Scholar]
- 41.Chai SC, Ye QZ. Metal-mediated inhibition is a viable approach for inhibiting cellular methionine aminopeptidase. Bioorg Med Chem Lett. 2009;19:6862. doi: 10.1016/j.bmcl.2009.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Medwid JB, Paul R, Baker JS, Brockman JA, Du MT, Hallett WA, Hanifin JW, Hardy RA, Jr, Tarrant ME, Torley LW, Wrenn S. Preparation of triazolo[1,5-c]pyrimidines as potential antiasthma agents. J Med Chem. 1990;33:1230. doi: 10.1021/jm00166a023. [DOI] [PubMed] [Google Scholar]
- 43.Chen X, Chong CR, Shi L, Yoshimoto T, Sullivan DJ, Jr, Liu JO. Inhibitors of Plasmodium falciparum methionine aminopeptidase 1b possess antimalarial activity. Proc Natl Acad Sci U S A. 2006;103:14548. doi: 10.1073/pnas.0604101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nonato MC, Widom J, Clardy J. Human methionine aminopeptidase type 2 in complex with L- and D-methionine. Bioorg Med Chem Lett. 2006;16:2580. doi: 10.1016/j.bmcl.2006.02.047. [DOI] [PubMed] [Google Scholar]
- 45.Medwid JB, Paul R, Baker JS, Brockman JA, Du MT, Hallett WA, Hanifin JW, Hardy RA, Jr, Tarrant ME, Torley LW, Wrenn S. Preparation of triazolo[1,5-c]pyrimidines as potential antiasthma agents. J Med Chem. 1990;33:1230. doi: 10.1021/jm00166a023. [DOI] [PubMed] [Google Scholar]
- 46.Haap W, Hoelzl W, Petzold K. Eur Pat Appl. 2002 EP1254903. [Google Scholar]
- 47.Zhou Y, Guo XC, Yi T, Yoshimoto T, Pei D. Two continuous spectrophotometric assays for methionine aminopeptidase. Anal Biochem. 2000;280:159. doi: 10.1006/abio.2000.4513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.