

Abstract
Quantitative Fluorescent Speckle Microscopy (QFSM) is a live cell imaging method to analyze the dynamics of macromolecular assemblies with high spatial and temporal resolution. Its greatest successes were in the analysis of actin filament and adhesion dynamics in the context of cell migration and microtubule dynamics in interphase and the meotic/mitotic spindle. Here, we focus on the former application to illustrate the procedures of FSM imaging and the computational image processing that extracts quantitative information from these experiments. QFSM is advantageous over other methods because it measures the movement and turnover kinetics of the actin filament (F-actin) network in living cells across the entire field of view. Experiments begin with microinjection of fluorophore-labeled actin into cells, which generate a low ratio of fluorescently-labeled:endogenous unlabeled actin monomers. Spinning disk confocal or wide-field imaging then visualizes fluorophore clusters (2–8 actin monomers) within the assembled F-actin network as speckles. QFSM software identifies and computationally tracks and utilizes the location, appearance, and disappearance of speckles to derive network flows and maps of the rate of filament assembly and disassembly.
Keywords: Live Cell Imaging, Microinjection, Image Processing, Actin, Speckles
Introduction
Quantitative Fluorescent Speckle Microscopy (QFSM) has been established as a powerful technique to assess the spatiotemporal regulation of macromolecular assemblies. In our lab it has been used extensively to study actin dynamics at the leading edge of migrating cells, where it revealed an unexpected complexity in structural dynamics, force generation, and response to regulatory signals [1–3]. QFSM turned out to be advantageous over other techniques for these experiments because it quantifies the rates of actin assembly, disassembly, and flow across the entire leading edge in constitutively motile cells with intrinsically balanced signaling programs. It therefore allows the study of spatial and temporal relations between the formation, turnover, and mechanical outputs of the filament network. For example, it can answer the question how assembly rate changes at the cell edge relate transiently to changes in filament flows 10 microns behind the cell edge; and how this relationship is affected by signaling events. In contrast, phalloidin and DNaseI actin staining in fixed cells yields information on the absolute levels of polymerized filamentous and unpolymerized globular (F- and G-actin) at specific time points, but subtle changes in actin assembly and disassembly rates cannot be detected. Laser photobleaching and photoactivation of labeled actin can yield measurements of actin flow and turnover rates, but only in specific marked regions of the cell. Actin QFSM is technically challenging, but is well worth the investment as the method detects subtle changes, provides spatial and temporal resolution, and is statistically robust.
This unit describes detailed methods for obtaining and computationally analyzing fluorescent actin speckle images in live cells. Fluorophore-labeled actin is microinjected into cells in order to generate a low ratio of labeled:endogenous unlabeled actin monomers (see Basic Protocol 1). Incorporation of the fluorescently-labeled actin into the F-actin network yields fluorophore clusters of 2–8 actin monomers that appear as local intensity maxima, or speckles. Confocal imaging visualizes the speckles over time (see Basic Protocol 2). QFSM software identifies and computationally tracks fluctuations in speckle intensity (see Basic Protocol 3). Speckle appearances and disappearances are stochastic indicators of F-actin assembly and disassembly, and speckle translocation is an indicator of F-actin flow. These data are then visualized via vectorial maps of flow, scalar maps of flow speed, and kinetic maps of polymer assembly and disassembly rates. The data is stored in convenient numerical formats to allow further modeling of the processes.
We present methods to analyze the effects of cellular perturbations, such as the inhibition of cellular kinases, on actin dynamics in real-time. Alternative methods to pre-treat and image control and experimental cells are described in the Commentary at the end of the chapter. Basic Protocols 1–3 can also be modified in order to visualize and track other macromolecular assemblies, such as microtubules, intermediate filaments, and adhesion complexes. However, the size and motion of these other molecules differ enough from actin that their computational analysis involves modification of the optimal input values used in QFSM analysis.
1. MICROINJECT FLUORESCENT ACTIN INTO CELL
Obtaining high-quality speckle images amenable to speckle detection and tracking with QFMS software depends on successful microinjection of fluorescently-labeled actin. For this reason, cell types that can be easily microinjected are the cells of choice for QFSM studies. The cells should be large (> 50 μm in diameter), should spread flat when plated, and should be naturally adherent to glass coverslips. It also helps if the cell’s cytosol occupies an area greater than its nucleus. Here, we provide a protocol for PtK1 cells, a rat kangaroo kidney epithelial cell line widely used for migration studies.
Prepare Cells for Analysis
Materials
35 mm glass-bottomed culture dishes (MatTek Corporation, 200 Homer Avenue, Ashland, MA 01721, USA, Tel (508) 881–6771, Fax (508) 879–1532, http://www.glass-bottom-dishes.com).
1 N Hydrochloric Acid (HCl, store at room temperature for 1 year)
Phosphate Buffered Saline (PBS, 20 mM phosphate, 150 mM sodium chloride, pH 7.4, autoclave to sterilize, store at room temperature for 2 months, pre-warm to 37 °C just before use)
70% ethanol (ETOH, v/v) in water (store at room temperature for 1 year)
PtK1 cells (American Type Culture Collection, P.O. Box 1549, Manassas, VA 20108, USA, Tel (703) 365–2700, Fax (703) 365–2750, http://www.atcc.org).
PtK1 growth media: F-12 media with 20 mM Hepes and 10% (v/v) FBS (store ≤ 1 month at 4 °C, pre-warm to 37 °C just before use)
Treat the inside of the glass-bottomed dishes with enough 1 N HCl to cover the glass for 5 min.
Wash the dishes with PBS
Sterilize the glass-bottomed dishes with 70% ETOH
Wash the dishes twice with PBS
Plate PtK1 cells on glass-bottomed dishes in PtK1 growth media, at low density so that the cells are in islands of 10–20 cells on the day of analysis.
Incubate cells at 37 °C, 5% CO2 for 24–28 hours.
Sterile tissue culture technique should be used while treating the dishes and plating the cells. PtK1 cells will secrete enough extracellular matrix in 24 hours so that they adhere and spread very flat. Standard substrate coating (fibronectin or collagen) can also be used to enable different cell types to adhere to the glass [4]. Coatings will not affect microinjection or the following QFSM analysis.
Prepare Fluorescent Actin
Materials
100 mM adenosine tri-phosphate dissolved in water (ATP, store in at −20 °C for 1 year)
100 mM dithiothretol, dissolved in water (DTT, store at −20 °C for 1 year)
G-buffer stock: 5 mM Tris-HCl, pH 8.0, 0.2 mM CaCl2 (filter with 0.2 micron filter to remove particulates, store at 4 °C)
Alexa568-conjugated actin (Molecular Probes, 5791 Van Allen Way, PO Box 6482, Carlsbad, California 92008, Tel (800) 955–6288, Fax (800) 331–2286 http://www.invitrogen.com/site/us/en/home/brands/Molecular-Probes.html)
opaque microcentrifuge tubes
double distilled water (ddH2O, filter with 0.2 micron filter to remove particulates and store at 4 °C)
microultracentrifuge tubes
microultracentrifuge capable of spinning at 75,000 × g, with rotor pre-cooled to 4 °C.
Add 1 μl of 100 mM ATP and 2.5 μl of 100 mM DTT to 500 μl G-buffer stock to make G buffer with 0.2 mM ATP and 0.5 mM DTT. Keep on ice.
Dilute Alexa-conjugated actin to about 4 mg/ml with 20–30 μl G buffer.
Aliquot 2 μl of 4 mg/ml labeled actin into opaque eppendorf tubes and immediately snap freeze with liquid nitrogen at −80 °C.
Just before microinjection, dilute 2 μl actin in 18 μl ice-cold ddH2O, so that the final actin injection concentration is 0.5 μg/μl. Transfer the actin to a microultracentrifuge tube.
Ultracentrifuge actin for 30 min at 67,000–85,000 × g, 4 °C.
Pipet off 15 μl of actin from the top without disturbing the pellet, and transfer to a new pre-chilled microcentrifuge tube. Keep prepared actin on ice in dark.
Successful actin microinjection and QFSM imaging requires purified actin labeled with an optimal fluorophore and devoid of actin aggregates. We recommend using actin labeled with the Alexa 568 fluorophore, as it is very bright and the orange to red wavelengths (560–630 nm) cause less auto-fluorescence and photo-damage. X-rhodamine-labeled actin from Cytoskeleton can also be used. These molecules are succinimidyl ester derivatives in which the dye is reacted with exposed lysine residues on the surface of polymerized actin. The polymerized actin is then de-polymerized to generate labeled actin monomers. Alexa568-actin from Molecular Probes has a labeling ratio is of 1–2 fluorophores per G-actin monomer. A much higher labeling ratio may affect the turnover kinetics (and binding to associated proteins) of the actin filament. A lower labeling ratio will cause a poor signal to noise ratio that results in un-analyzable speckles.
It is important to carry out all steps in the cold room at 4 °C, and to have all tubes pre-chilled on ice. This reduces actin aggregation and polymerization, which will clog the needle and prevent microinjection during the next step. Preparing 2 μL aliquots also reduces the generation of insoluble aggregates during repeated freezing-thawing. Using opaque microcentrifuge tubes or wrapping the tubes in foil prevents fluorophore photobleaching due to direct light exposure.
Microinject Cells with Fluorescently-labeled Actin
Materials
micropipette puller (Sutter Instrument Company, One Digital Drive, Novato, CA 94949, USA, Tel (415) 883–0128, Fax (415)883–0572, http://www.sutter.com)
10 cm length borosilicate glass with filament, 1.0 mm outside diameter, 0.78 mm inside diameter (Sutter Instrument Company)
Prepped fluorescent actin (see Prepare Fluorescent Actin)
Microsyringe or 2–10 μl pipette
microloaders pipette tips
inverted fluorescent microscope with a microinjector systems (phase ring, 40X phase contrast objective, fluorescence light source, 568-compatible excitation and emission filters, transjector capable of controlled backpressure, micromanipulator for injection of single cells, at room temperature)
prewarmed PtK1 growth media (see Materials under Prepare Cells for Analysis)
PtK1 cells ready for analysis (see Prepare Cells for Analysis)
Pull 5–10 needles with 2 cycles of “bee-stinger” protocol (Heat 780, Pull 100, Vel 10, Time 250, Pressure 500) and 1 cycle of “gradual taper” protocol (Heat 780, Pull 105, Vel 10, Time 220). The bee-stinger protocol generates tips with a small shoulder, similar to those sold by Eppendorf and generally advertised as ideal for injecting adherent cells. However, the shoulder can sometimes damage cells. A longer, more gradual taper is ideal for cell health, but smaller tips are more susceptible to clogging.
Backload 0.5 – 1.0 μL of actin solution (stored on ice in dark) into the needle tip using a microloader pipette tip. Gently dispense the solution to avoid air bubbles. Remove any bubbles and excess solution along the capillary by moving the pipette tip up and down in the needle and taking up excess liquid surrounding the bubble.
Place cells on microscope stage, open the shutter for transmitted white light, align the microscope for Kohler illumination, and adjust the z-focus on the microscope until the cells are in view.
Attach the needle to the injector holder without touching the needle tip. Position the holder so that the needle makes a 35–50 degree angle to the base of the microscope stage. These lower angles help reduce the chance of accidently breaking the needle tip on the glass coverslip when lowering it towards the cells.
Set the microinjector to range 1 and transfer pressure between 25–40 hPa, which ensures a constant outward flow of actin molecules and no influx of F-12 media. Using the micromanipulator, lower the tip of the needle until it breaks the surface of the media bathing the cells and position the tip directly above the center of the objective. The end of the needle tip should appear as a bright halo through the eyepiece.
Focus above the cells. Slowly lower the needle tip until the halo gradually constricts and you see the needle tip or needle shaft through the eyepiece. Re-focus on the cells and slowly lower the needle tip so that the end of the needle and the PtK1 cells are both in clear view. Re-adjust the phase ring to improve contrast if necessary.
Switch the illumination to TRITC or 568 nm fluorescence and confirm that the Alexa568-labeled actin is flowing out of the needle.
Switch back to transmitted white light and select a flat cell on the edge of an island to inject. When navigating, ensure the needle tip is well above your cells so that the needle does not hit the coverglass and break. The free edge of the chosen cell should face the needle tip. Position the needle so the tip is within the perinuclear region, above or below the nucleus in the x,y plane (see Figure 1). This is the thickest part of the cell, so injecting at this location reduces the chance that you will poke the needle through the other side of the cell and into the glass coverslip.
Slowly lower the needle until the tip depresses, then pierces the membrane. If the needle has fully pierced the membrane, the area around the hole will appear brighter. As soon as you see this brightening, raise the needle until it is no longer touching the cell. The actual piercing and injection process should take 0.5 – 2 seconds.
Confirm that cells have been injected by switching to TRITC or 568 nm fluorescence. The injected cells should appear fluorescent throughout the entire cell. Do not attempt to re-inject cells that did not get injected, as they will become damaged and unhealthy.
Remove free fluorescent actin from the dish by exchanging the cell media with fresh pre-warmed PtK1 media. Then incubate the cells at 37 °C, 5% CO2 for 30 min to allow the actin to integrate into the existing cytoskeleton network.
Figure 1.

Optimal site of microinjection. A flat cell with its cell edge opposite of the needle is chosen for injection. The needle is positioned directly above or below the nucleus, where the cell is widest and then slowly lowered into the cell.
The microinjection step of QFSM is the most difficult step with the biggest learning curve. The actin often polymerizes within the needle tip and clogs the needle. In addition, it is easy to over-inject and damage the cells.
If in step 10 of Microinject Cells with Fluorescently-labeled Actin, the “injected” cells do not contain fluorescent actin or the cells have only 1 patch of actin where the needle touched the cell, then you should check for flow out of the needle (repeat step 6). Lack of flow out of the needle tip could be due to either an air bubble inside the tip or polymerized actin clogging the tip. In order to regain flow, first attempt to push out the air bubble or polymerized actin using the “clear’ or “clean” function on the transjector. This briefly increases the pressure of the flow and can be observed through the eyepiece as ripples in the media and nearby debris dispersing. Second, if the problem is polymerized actin, you can also attempt to widen the needle tip by breaking the tip against the coverslip. In an area free of cells, gently lower the tip until it presses against the glass and slowly move the tip in the x/y position. The breakage area should be no more than a 1/5th of the area of the needle point (the portion of the needle that starts to converge to a point) and the transfer pressure will need to be reduced to 5–15 hPa so that the cells are not over-injected with the faster flow that comes out of a wider tip. Third, if clogging problems are consistently encountered you may need to replace the needle with a new one or re-ultracentrifuge the actin (repeat steps 5 and 6 of Prepare Fluorescent Actin). Lastly, you can use needles with larger hole sizes that better accommodate the delivery of unwanted polymerized actin filaments along with the monomeric actin (optimize step 1 of Microinject Cells with Fluorescently-labeled Actin). To create needles with a shorter taper and wider tip (up to 1 μm in diameter), adjust the micropipette puller’s last protocol step to decrease the pull in 5 unit increments and increase the time delay (cooling time) in 10 unit increments. Bee-stinger microinjection needles can also be purchased through commercial vendors, such as Eppendorf.
The prepped actin concentration and room temperature injection are optimized to reduce needle clogging. Injecting at higher concentrations (1–3 μg/μl) will increase the frequency of needle clogging. If the injected cells are too dim to image in the following Image Actin Dynamics section, then try injecting with increased transfer pressure (35–55 hPa) to increase the flow and amount of actin entering the cells, rather than increasing the stock actin concentration. Injecting at room temperature rather than 37°C reduces the amount of actin polymerization and needle clogging. Inject many cells in the center of the dish with each injection session, but do not spend more than 20 – 30 min on any one dish. Longer durations at ambient temperature and CO2 levels will stress the cells.
If your “injected” cells do not contain fluorescent label, but actin is flowing out of the needle tip, then the tip is likely too dull or broken to efficiently penetrate the cell. Load a new tip with actin and repeat the injections (repeat steps 2–11 of Microinject Cells with Fluorescently-labeled Actin).
The second major hurdle with actin microinjection is microinjecting too much volume or with too much force. When microinjection is performed correctly, the final cell shape will appear unchanged. Too much injection volume results in quick retraction of the cell edge and in some cases cell explosion. Too much injection volume and force can also be more subtle. The cells may appear fine initially, but later during the speckle imaging (see Image Actin Dynamis), the injected cells may be uniformly retracting while neighboring cells exhibit both protrusion and retraction dynamics. In this case, repeat the microinjection with reduced transfer pressure. Simple practice will also help reduce the injection volume, as you will get quicker at drawing up the needle tip after it pokes the cells, which reduces the volume of actin entering the cells. If you are encountering problems with microinjection and cell health, please view our video of the actin FSM technique[5]. It contains footage of proper microinjection in which cell health is maintained. Experiments using GFP-actin or Snap-tagged actin rather than microinjection are also possible, yet less-well established. These tagging methods bear the risk of significantly perturbing the actin dynamics and it is difficult to control the concentration of labeled actin. Importantly, the use of Lifeact is inappropriate for QFSM. Low concentrations of Lifeact can generate image patterns of random puncta resembling bona fide speckle patterns that allow tracking of actin filament flows. However, the intensity fluctuations and appearance and disappearance of individual puncta result from a convolution of actual turnover of actin filaments and the association/dissociation to the Lifeact peptide with the filaments.
2. IMAGE ACTIN DYNAMICS
Materials
Cells microinjected with fluorescent actin (see Microinject Cells with Fluorescently-labeled Actin)
L-15 Imaging Media: phenol red-free Leibovitz’s L-15 media + 10% FBS (v/v) (store for ≤1 month at 4 °C, pre-warm to 37 °C before use)
EC-Oxyrase or OxyFluor (store in 100μl aliquots for 1 year at −80 °C, Oxyrase, Inc., P.O. Box 1345, Mansfield, Ohio, 44901, Tel: (888) 699–3733, http://www.oxyrase.com)
mineral oil
inverted spinning disc confocal microscope (transmitted light source, fluorescence lamp, appropriate excitation laser, appropriate excitation and emission filters, motorized shutters, 0.4 numerical aperture 100X oil-immersion Plan-Apochromatic objective, cooled CCD camera with pixels < 7 μm, vibration table, imaging software)
ethanol (ETOH)
lens paper
glass-cleaner
tissues
double distilled water (ddH2O)
immersion oil for objective lens
Change cell media to L15 + 10% FBS+ 20 mM Hepes.
Add 30 μl freshly thawed Oxyrase per ml of media to the cells. Oxyrase helps prevent photo-bleaching and photo-damage by reducing the dissolved oxygen to water.
Cover the media with mineral oil to reduce media evaporation and oxygen exchange, which would saturate the Oxyrase.
Pre-warm the microscope stage to 37 °C.
Clean the objective lens with a drop of ETOH and lens paper. Clean the bottom of the coverglass of the injected cells with glass cleaner and tissues.
Apply immersion oil to the objective lens and place a Matek dish with microinjected cells onto the stage. Wait 10–15 min. for the cells’ temperature to stabilize before imaging (step 10). For new speckle microscopists, steps 7 – 9 can take 15 min, so the wait time is not necessary.
Remove all prisms and analyzers from the optical path. Focus on cells using transmitted light.
Find the injected cells by using widefield 568/TRITC fluorescence. Use neutral density filters, keep the shutter closed whenever you are not looking at the cells, and generally minimize the time of fluorescence exposure to reduce photobleaching. Look for injected cells that have healthy curved leading edges. Over-injected cells will have retracted and jagged edges with numerous filopodia (Figure 2). Do not image over-injected cells. Focus on the leading edge of the cell by snapping confocal images. Do not focus with a “live” view, as this will photobleach your sample.
Set the Acquisition settings. For PtK1 cells injected with Alexa568-labeled actin, and imaged with a Nikon Ti inverted microscope with a Yokagawa CSU-X1 spinning disk confocal with Spectral Borealis modification, a 561nm (200mW) solid state laser, and a Hamamatsu ORCA-AG cooled CCD camera, we use an exposure time of 700–900 ms, laser intensity of 18–19, and a time interval of 10 sec for a 15 min time series. In general, the exposure setting should not be greater than 1 sec, the time interval between frames should be 5–10 sec, and photobleaching limits the time series to 15–30 min. Gain settings on the camera can be turned up to increase the signal to noise. Do not bin the image to increase the signal to noise, as this reduces resolution rendering later speckle detection impossible.
Acquire time-series.
Figure 2.
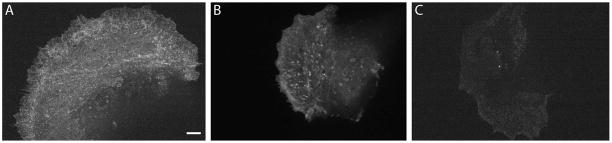
Choosing a cell for imaging. A) Example of an optimally-microinjected cell that will yield analyzable speckles. B) Example of an over-injected cell in which speckles are indistinguishable from background fluorescence and stress fibers are not broken. Although filopodia are not evident, the edge is uniformly retracting. C) Example of an under-injected cell. The speckles are dim are spaced too far apart. Scale bar: 5 microns.
An ideal cell and exposure setting for QFSM analysis will exhibit actin speckles spaced by ~2 speckle diameters to the next neighboring speckle. For epithelial cells, the speckled actin network should appear homogeneous and salt and pepper-like. Dense stress fibers or lamellipodia may be visible, but will have a dotted/speckled appearance (see Figure 2A). Over-microinjected cells will have densely packed speckles that cannot be distinguished from each other. Stress fibers and the lamellipodia will appear as solid lines (see Figure 2B). Under-microinjected cells or cells injected with poorly-labeled or photo-bleached actin will appear very dim through the eyepiece. The cells may contain speckles that are far apart (>10 speckles apart) and appear randomly scattered. Stress fibers and the lamellipodia will be indistinguishable (see Figure 2C).
It is critical that focus is maintained throughout the duration of the time-lapse sequence. This can be particularly challenging since the imaging plane is quite thin. We use Nikon’s near-infrared Perfect Focus focusing system, which provides real-time focus adjustments. Other microscope manufacturers offer similar real-time focus systems.
3. QUANTIFY ACTIN DYNAMICS
Materials
Computer with 64-bit processing capabilities and at least 4GB RAM.
Matlab (Mathworks, version R2010b and above) with the following toolboxes: Image Processing, Curve Fitting and Statistics.
Obtain the QFSM Software
Download the latest version of the QFSM analysis software from http://lccb.hms.harvard.edu/software.html.
Extract all the files from the zip file you downloaded on the disk.
Start Matlab and add the directory containing the code to the Matlab path (see installation instructions).
Setup a Database for the Speckle Movie and Load it for Processing
The first step in any analysis is to set up a database for the movie. This database contains links to the directories with the raw image files, links to all the processed and stored result files, and the parameters used for image processing. Prepare the image files for the database by storing each channel (wavelength) of each time-series in a separate directory (folder), with one file per frame (time point) of the movie. To indicate the time point, the software package expects the following filename convention: MyMovieXXX.tif. MyMovie is a placeholder for any string, including underscores and dashes that specifies the generic name of the movie. XXX is a placeholder for a numeric value identifying the time point of that particular frame, e.g. 007, 008, 009, etc. In addition to organizing the raw image sequences, gather information on the camera (pixel size, acquisition time, bit depth), the objective lens (numerical aperture), and the fluorescent channels (emission wavelength) before launching the software. The QFSM analysis software supports multi-channel FSM movies. Here, we limit our description of the image analysis to single-channel data.
-
From the Matlab command prompt, launch the movie selector interface by typing movieSelectorGUI or qfsmPackageGUI.
This command will bring up the movie selection panel (Figure 3A). The left panel displays all of the movies to be processed. The buttons next to the list allow the user to modify the list by creating, opening, and removing movies. The right panel displays the available software packages that can be used to process the movies. -
Create a new movie database by clicking on New.
This will bring up the movie edition interface (Figure 3B). Click on Add Channel. Select the folder containing the images to be analyzed. Repeat the Add Channel operation for all of the speckle channels of your FSM movie that you want to analyze.
Click on Advanced Channel Settings. In the new window, fill out the emission wavelength (in nm) for each channel.
-
In the movie edition interface, fill in the pixel size (in nm), time interval (in s), numerical aperture and camera bit depth of the movie.
Note: once saved, these fields cannot be further modified. In case of an erroneous input a new movie database has to be generated by repeating steps 2 – 5. Optionally, enter additional notes specific to your movie.
In the output directory panel, click Select Path and choose a location on the disk where all results of the analysis should be saved.
-
Click on Save.
This will open a pop-up window asking where to save the movie database. The operation will save a MAT file (Matlab format) containing all the movie information including all results from the processing. This MAT file can be later reused for loading the movie database (see next section). -
In the movie selection panel (Figure 3A), load the new movie by clicking Open. Select the MAT file saved when creating the movie database.
If the movie database file has been relocated on the disk, the software will ask for relocation of all its components by comparing the new path of the movie database file to the old path. -
Repeat steps 2–10 until all of the FSM movies you want to process are listed in the left panel.
A list of movie databases can be saved as a list using the Save as Movie List button. The resulting movie list is also saved as a MAT file on the disk. To load all of the movies in a list, repeat step 9 and select the MAT file containing the movie list. Select QFSM package on the right panel and click Continue at the bottom right of the movie selection window.
Figure 3.
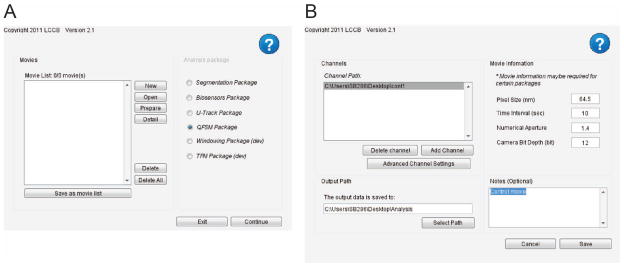
Movie selection user interfaces. A) Interface for selecting FSM movies to process. B) Interface for creating a new FSM movie.
Run the QFSM software
The QFSM analysis follows a series of steps illustrated in Figure 4. Two steps are optional: the Noise Model Calibration and the Flow Tracking. In accordance with this work flow, the main panel of the QFSM analysis software (Figure 5) consists of the following components:
Figure 4.

Schematic representation of the workflow of the QFSM analysis software. The processing encompasses essential steps (speckle detection and tracking) and optional but recommended steps.
Figure 5.

Main QFSM analysis user interface. A) This drop down menu contains the FSM movies to be analyzed. B) The lower part of the window lists all the processing steps of a QFSM analysis. Each step can be setup using the Setting button C), scheduled for running using the checkbox next to its name D). After clicking the Run button E), the outputs of the individual processes can be visualized using the corresponding Result button F). G) Icons on the left of each process indicate the status of the process. The presence of an icon next to a process indicates that the process has currently stored results in the movie database. In this example, step 8 has never been run. The green check box items next to step 1 and 2 indicate that results of these processes are updated in the movie database. The exclamation point triangle next to steps 3 – 7 indicate that the results are no longer updated, requiring rerunning of these processes. This can happen, for instance, when a parameter in step 3 has been changed. (H) Specific help can be queried using the icons on the far right of the process.
A top panel that displays the current processed movie. If you are processing multiple movies, use the drop-down menu/arrows to switch between movies.
A sequence of processes that can be run either individually or in a complete flow. Each process needs to be setup before it can be run. To setup a process, click on its associated Setting button. For all process setting interfaces, a help button is located in the top right corner of the window. Once setup, processes appear in bold characters.
A checkbox next to each process marks it as scheduled for processing. To run a process, check the respective box and click Run. Once successfully run, the output of the process can be visualized by clicking the Result button.
Icons next to each process indicate the current processing status. Only processes that have been run at least once successfully have an associated icon. To get more information, click on an icon.
A Run button. Only checked processes with non-green icons will be run when hitting Run. If you want to re-run a process that has been run before, check the Force Run checkbox.
In the case of processing multiple movies through multiple steps, the processes that are to be run need to be individually set up and checked. In each setting interface, check the Apply to all movies checkbox to set up processes in batch. Check Apply Check/Uncheck to All Movies to schedule processes to run for all movies. Finally, click Run All Movies to run scheduled processes of all movies in the list.
When running QFSM processes, all results will be saved in a subfolder of the movie database output directory (see step 7 of Setup a Database for a Speckle Movie and Load it for Processing) called QFSMPackage. All paths described below are relative to this main path. In addition, each process generates a log-file summarizing specific numerical values related to the process.
Noise Model Calibration - Optional (Figure 6A)
Figure 6.

User interfaces to select QFSM processing steps. A) Noise model calibration. B) Thresholding. C) Mask refinement. D) Speckle Detection.
The calibration of a noise model needs only be performed once per experimental set-up (camera at a given exposure time with a given laser intensity). If a successful noise model has been created for given experimental conditions, you can reuse it during speckle detection for other movies. We recommend however to calibrate models on individual movies when it is possible.
Setup the noise model calibration step by clicking on its Settings button.
Select the channels you want to use to calculate a noise model.
-
Select a background region of the movie by clicking on Select Window of Interest.
A pop-up window should open showing the channel and the frame chosen in the setting interface. Adjust the rectangular Region of Interest (ROI) to select a region containing only background. Use the frame slider to make sure no parts of a cell move into the ROI during the movie. Adjust the depth of the stack to be used to calibrate the noise model.
-
Click on Apply.
The results of the noise model calibration as well as the cropped images are stored in a folder called “noise”. There is no visualization of the noise model calibration output when clicking on Result. However, the values of the noise model will appear in the speckle detection setting interface.
Mask Creation – Thresholding and Mask Refinement (Figure 6B & C)
This step segments the cell area from the background and stores the result as a sequence of masks.
Thresholding
Set up the Thresholding process by clicking on its Setting button.
Select the channels you want to threshold.
-
Adjust the standard deviation of the low-pass Gaussian filter if necessary.
By default, each image is low-pass filtered with a Gaussian kernel of standard deviation equal to 1. Adjust the automatic thresholding method if necessary.
Click on Apply.
Setup the Mask Refinement process by clicking on its Setting button.
Select the channels whose masks should be refined.
Adjust the closure radius. Keep the fill holes option checked.
-
Click on Apply.
The output of the thresholding (respectively mask refinement) step is a series of binary masks for each channel stored in a folder called “masks” (respectively “refined_masks”). Clicking on Result will open a window in which the boundaries of these masks are computed and overlaid onto the raw FSM images. Check that the thresholding and mask refinement steps accurately mask the cell. If there are large areas of the cell or frames in which the cell is inappropriately included or excluded from the mask, repeat the thresholding with an increased standard deviation for the Gaussian filter (1.5, step 3) and/or repeat the thresholding with a different method (step 4). If small areas of edge protrusions are inappropriately excluded from the mask, repeat the mast refinement with an increased closure radius. This will, however, smoothen the entire edge of the mask.
Speckle Detection (Figure 6D)
Setup the speckle detection process by clicking on its Setting button.
Select the channels containing speckles.
-
Adjust the standard deviation of the low-pass Gaussian filtering if necessary.
The default standard deviation of the Gaussian low-pass filter is set to the theoretical value of the microscope point-spread function. -
Select the mask channels to be used during speckle detection.
The software will automatically use the output of the mask refinement when detecting the speckles. If several mask channels are selected (in multi-channel FSM movies), the intersection of these masks will be computed and used. If a noise model calibration was set up for this movie, the software will use the output of the noise model calibration for detecting speckles. If the calibration has been run successfully, the values of background mean, standard deviation and Gauss ratio (a measure for the spatial correlation of background noise) will be shown in the corresponding edit boxes.
-
If a noise model calibration was not setup for this movie, load a noise calibration model performed on an appropriate background movie. Click on Load noise model parameters. Select the MAT file created by running the noise model calibration step on the background movie.
The loaded values for the background mean, standard deviation and Gauss ratio will be displayed in the corresponding boxes. Adjust the confidence interval for the statistical selection of speckles by setting the alpha value. The default value is 0.01. Increasing the value to 0.1 will reduce the stringency of speckle selection so that more speckles are detected, but will also increase the detection of false positives.
-
Select the maximum number of iterations to be performed using the drop-down menu.
Generally 3 iterations are sufficient to detect all or nearly all of the speckles. -
If the maximum number of iterations is greater than 1, set the minimal fraction of speckles to be detected with each iteration.
High-order speckles will be detected iteratively until the maximum number of iterations is reached or fewer than the minimum fraction of speckles is detected by a given iteration. Using at least 0.01 (1%) will ensure repeating the hierarchical detection is useful. -
Click on Apply.
The output of speckle detection is a series of MAT files for each channel stored in a folder called “speckles”. Clicking on Result will yield a pop-up window in which detected speckles are overlaid to the FSM raw images (Figure 8A). Individual speckles are marker coded according to their hierarchical order (i.e. the iteration during which they were detected). Speckles of first, second, third and fourth order will respectively be displayed as circles, triangles, squares and diamonds. Generally, 3–5 speckles per micron2 should be detected in each frame. Speckle density can be assessed by adding a scale bar to the image, which is an option within the Result pop-up window.
Figure 8.

Typical output of four processes of the QFSM package. Scale bar: 5 microns. A) Detected speckles (Speckle detection). Inset: close-up of a region with a second-order speckle. B) Speckles tracks (Speckle Tracking). C) Speed map (Flow analysis) (D) Interpolated flow field (Flow Analysis) E) Polymerization map (Kinetic Analysis). F) Depolymerization maps (Kinetic Analysis). G) Combined polymerization map, where polymerization rates are in red, and depolymerization rates are in green.
Flow Tracking (Figure 7A)
Figure 7.

User interfaces to select QFSM processing steps. A) Flow tracking. B) Speckle tracking. C) Kinetic analysis. D) Flow analysis.
Flow tracking determines the coarse movement of the speckle field. It can be applied to image time lapse sequences with low speckle contrast but some texture. The algorithms and results of the QFSM flow tracking are comparable to a Spatiotemporal Image Correlation Spectroscopy (STICS) analysis [6]. Details of the underlying method, especially of the time integration, are discussed in [7]. In the case of high-speckle contrast, flow tracking will help initialize subsequent single particle tracking of individual speckles. Whether this initiation is necessary depends on the flow speed and the temporal sampling of the image sequence, together determining the displacement of speckles between two consecutive images. As a rule of thumb, flow tracking should be used when the speckle displacement is greater than ~1/3 of the distance between speckles. Otherwise, the displacement is small enough to warrant a robust assignment of speckle correspondences between consecutive images without prior information of the coarse movement of the speckle flow.
Initial optimization of parameter settings
Set up the flow tracking process by clicking on its Setting button.
-
Select the channels for which flow is going to be calculated.
Only channels with detected speckles can be processed by the flow tracking. Correlation-based flow tracking will be performed with templates centered at the position of each detected speckle. -
Set the frame step size to 0 for optimizing the flow tracking.
Flow will be calculated at only one time point for the whole movie. Set an initial integration window between 5–10 frames.
-
Set an initial size range of the correlation templates.
A better and more spatially homogenous speckle contrast makes a wide size range less necessary. Also, under these conditions the template size can be chosen relatively small, i.e. 11 – 13 pixels. If using a range of template sizes instead of a single size, the algorithm will determine the optimal template size for each image location. Using a range will significantly increase the processing time of the flow tracking. Choose a number of stationary frames to average for background subtraction.
Adjust the threshold for detecting vector field outliers.
Click on Apply.
Run the flow tracking process using the Run button. Adjust steps 4–7 until obtaining an accurate flow field with few outliers.
-
Set the frame step size to a value between 1 and 5 to track the flow through the whole movie. Run the flow tracking process using the Run button.
The output of flow tracking is a series of MAT files for each channel and each starting frame stored in a folder called “flow” Additionally, a summary file in the “flow” folder will give information on the total number of tracked templates as well as a statistics of the corresponding template sizes. Clicking on Result will yield a pop-up window in which the calculated flow vectors are overlaid onto the raw FSM images.
Speckle Tracking (Figure 7B)
Setup the speckle tracking process by clicking on its Setting button.
Select the channels in which speckles should be tracked.
-
Adjust the search radius used for linking speckles from frame to frame.
A search radius of 3–4 pixels should be sufficient for tracking speckles in PtK1 cells, especially when a flow field is available for initialization. Increasing the search radius is more permissive, so will link more speckles at the cost of false positive links. -
Set the correlation length of the actin flow in pixels.
The correlation length is used during hierarchical tracking and/or during flow interpolation with a Gaussian weighting method. Its physical value in microns is indicated. Recommended values for actin networks range from 1 – 2 μm. -
Select/unselect the hierarchical tracking option.
If selected the software uses the interpolated flow field from an initial speckle tracking run to propagate speckle movements between frames, allowing the assignment of speckle correspondences over longer distances. If no flow tracking was performed, this option is highly recommended. Otherwise hierarchical tracking is not needed. -
Using the drop-down menu, select a method to interpolate positions where no flow was estimated during flow tracking.
This option is only valid if flow tracking has been completed in the previous step. The following three interpolation methods are available: 1) No interpolation in which speckles with no estimated flow will be considered as speckles with zero-magnitude flow, 2) Nearest-neighbor flow interpolation which speckles with no estimated flow will use the nearest finite flow, and 3) Gaussian weighting interpolation in which speckles with no estimated flow will integrate the flow of all neighboring speckles using a Gaussian weighted interpolation. The value defined in step 4 will be used to as the scaling parameter (1 σ) for the Gaussian interpolation kernel. -
Click on Apply.
The output of speckle tracking is a single MAT file for each channel stored in a folder called “speckleTracks.” Clicking on Result will yield a pop-up window in which three types of tracking outputs can be overlaid onto the FSM raw images: 1) all trajectories of valid speckles present in the current frame (Figure 8B). Trajectory length can be controlled as a time-integrating drag-tail. 2) Vectors showing the instantaneous frame-to-frame displacement of all valid speckles present in the current frame. For visualization, the vector magnitude can be scales. 3) Combination of vectors used for initialization of the single speckle tracking in the current frame and the actual displacement of the speckle between the current frame and the next frame. This view is used to assess the quality of the initialization flow field for single speckle tracking.
Kinetic Analysis (Figure 7C)
Setup the kinetic analysis process by clicking on its Setting button.
-
Adjust the value of the time window over which polymerization and depolymerization events should be integrated.
For PtK1 cells, integrating the values over 3 – 5 minutes should yield kinetic maps with clear bands of high rates of polyermization/depolymerization at the edge (lamellipodium) and lower rates of random polymerization/depolymerization just inside the edge (lamella) [1]. Lower averaging in time can be afforded at the expense of higher spatial averaging (see next item). To follow the non-steady state dynamics in actin polymerization/depolymerization, for instance during protrusion and retraction events, shorter time windows on the order of 2 or 3 frames (generally 20 – 30 s) will be required [2]. Naturally, this data will be noisier and different methods of averaging are required to extract meaningful information about the functional behaviors of actin turnover (see e.g. [8]) -
Adjust the value of the Gaussian standard deviation to be applied to the images to calculate interpolated polymerization or depolymerization maps and to display these events as pseudo-color movies.
A value of 5 pixels is generally suitable for viewing kinetic maps of PtK1 cells. -
Click on Apply.
The output of the kinetic analysis is a series of MAT files for each channel stored in a folder called “kineticAnalysis.” Clicking on Result will yield a pop-up window that can display four types of kinetic maps: 1) map of net filament turnover (local rate of polymerization minus depolymerization), 2) polymerization map (Figure 8E), 3) depolymerization map (Figure 8F) and 4) combined bichannel polymerization/depolymerization maps (Figure 8G).
Flow Analysis (Figure 7D)
Set up the flow analysis process by clicking on its Setting button.
-
Adjust the value of the time window over which flow should be interpolated.
Generally, flow values should be interpolated over a smaller time window than that used for the kinetic analysis, as each frame contains more flow information than polymerization/depolymerization events. For PtK1 cells, 1– 2 minutes should yield flow speed maps in which a narrow band of flow (500–2000 nm/min) at the cell edge (lamellipodium) is followed by a wider band of slower retrograde flow (200–500 nm/min) [1]. Adjust the value of the grid size used to create speed maps.
Set the correlation length in pixels.
-
Click on Apply.
The output of flow analysis is a series of MAT files for each channel stored in a folder called “flowAnalysis.” Clicking on Result will yield a pop-up window that can display three types of flow maps: 1) speed maps that display the magnitude of the flow interpolated on a grid (Figure 8C), 2) error, and 3) signal to noise ratio maps that display the fluctuation of the tracked flow over the interpolated flows. Vector fields showing the interpolated flow and error flow can also be overlaid onto the error maps (Figure 8D) and the noise and SNR of the local flow can be displayed as circles scaling with the intensity.
COMMENTARY
Background information
The injection and imaging of very low concentrations of fluorescent actin and microtubule subunits was originally developed to distinguish the polymers from out-of-focus fluorescence [9, 10]. The speckles are diffraction-limited regions that contain more labeled subunits incorporated into the polymers than neighboring regions. By high-resolution imaging they are visible as local intensity maxima. While FSM enabled the visualization of steady-state polymer translocation, its full power was only exploited with the development of software to detect and analyze the hundreds of thousand speckles in an FSM time-lapse sequence.
Quantitative analysis of the speckle data can be performed using the QFSM analysis software. The processing framework is divided into individual steps that allow the user to regularly assess the quality of their analysis (Figure 4). Full analysis of speckle movies consists of three steps: 1) setting up a movie database for analysis, 2) setting up and running individual analysis processes and 3) visualizing and assessing the quality of the results.
We developed the first version of QFSM software in 2003 and validated its ability to track actin assembly and disassembly events in living cells with a spatially stationary cortex, i.e. contact inhibited cells [11]. This publication introduced early models for the statistical distinction of speckles from noise and for the computation of assembly and disassembly rates from the foreground and background intensity changes that lead to speckle appearance and disappearance – birth and death events, respectively. Importantly, as discussed in detail in [11], and in summary form in [12], a speckle birth can occur both due to actual assembly or the disassembly of nearby polymer. Equivalently, a speckle death can occur due to actual disassembly or the assembly of nearby polymer. The kinetic analysis module of the QFSM software contains algorithms to distinguish these scenarios. Critically, the analysis relies on complete speckle tracks. Incomplete tracks introduce erroneous birth and death events. To some extent the software can detect those, yet it does not relax the requirement for optimized tracking. Therefore, it took us a while to get the QFSM software ready for the analysis of non-stationary polymer networks. Key papers describing speckle flow field and single speckle tracking methods that enable the extraction of complete speckle trajectories from true birth to true death include [7, 13].
Enabled by these technical advances in the QFSM analysis, we applied the method to characterize actin dynamics in newt lung epithelial and PtK1 cells migrating in their normal growth medium. The speckles delivered an unprecedented detail of information, since a speckle field samples the dynamics of a polymer at “densest packaging” afforded by diffraction, i.e. at ~500 nm between data points. Their analysis led to the discovery that the cell edge contains two kinetically, i.e. in terms of actin polymer turnover, and kinematically, i.e. in terms of actin polymer movement, distinct actin networks [1]. The network at the very edge was termed the lamellipodium. It completely disassembles within 1–3 μm of the edge. This 3 μm band also undergoes rapid retrograde flow (> 300 nm/min). A much wider area of slower retrograde flow (100–250 nm/min) abutting the lamellipodium was termed the lamella.
More recently, we have combined QFSM with edge windowing to locally compare actin dynamics in protruding versus retracting edges [8]. Cross-correlation scores were calculated for different time lags between the edge protrusion and actin turnover and flow maps in order to determine functional relationships between the local dynamics of the actin polymer networks and edge movement. We found that dependent on the position of the cell in an island vs at the edge of a wound, and the function of a cell as a so-called leader or follower, these relationships vary. The relationships also vary when regulatory signals or actin accessory factors are perturbed. Together, these data indicate that QFSM is a powerful method to investigate the regulation of cytoskeleton dynamics by cell-external and cell-internal cues.
We anticipate continuing to improve upon the QFSM software as new scientific questions necessitate new approaches. For example, future improvements may include methods to directly compare actin turnover rates and integrate actin analyses with edge windowing and protrusion/retraction analyses. The speckle methodology (injecting low concentrations of monomeric cytoskeleton proteins) has also been successfully applied to microtubules and adhesions and combined with actin QFSM [14, 15].
Critical parameters and troubleshooting
Obtaining high-quality FSM images
The most critical steps to obtaining analyzable FSM images are removing polymerized actin from monomeric actin used for microinjection, gently microinjecting the cells with a small volume, and preventing photobleaching and maintaining focus during image acquisition. We highly recommend scientists new to microinjection practice the injection procedure with fluorescently-labeled dextran, which does not polymerize, and monitor cell health over the course of 1–2 hours. Microinjection should then be practiced with fluorescently-labeled actin to ensure the actin preparation and needle tip size are suitably optimized to prevent needle clogging. A successful microinjection and FSM experiment can be viewed in[5].
In this unit, we describe a method that uses a layer of mineral oil to reduce media evaporation and oxygen exchange. Oxygen exchange with the air would quickly saturate the Oxyrase and result in rapid photo-bleaching and photo-damage. Using an open dish scenario permits acute changes of the medium during the experiment, e.g. to expose cells to small molecule inhibitors of particular components of actin-regulating pathways. Midway through image acquisition, buffer with chemical inhibitors can be slowly flowed into the Matek dish while imaging in order to monitor the effects of drug exposure in real-time. The new media will sink to the bottom of the dish, while the mineral oil will continue to float. Control cells must be treated with their original imaging media to ensure the buffer addition process does not shear or otherwise stimulate the cells. A perfusion chamber can also be used to ensure introduction of the new buffer occurs rapidly and without disturbing the cells being imaged.
An alternative method to prepare cells for QFSM imaging is detailed in[5, 16]. With this method, cells are plated and microinjected on square glass coverslips contained in 35 mm petri dishes. The coverslips are then assembled into a closed chamber with a glass slide, using double-sided tape as spacers and VALAP (a 1:1:1 mixture of Vaseline, Lanolin, and Paraffin) as a sealant. The closed chamber does an excellent job at preventing gas exchange and photo-bleaching. In order to assess the role of specific molecules in regulating actin dynamics, microinjected cells can also be treated with chemical inhibitors before assembling the chamber. However, in contrast to the oil-sealed open chambers we propose in this protocol, closed chambers preclude the monitoring of transient perturbations.
QFSM Analysis
Successful utilization of the QFSM software requires a time investment in that one must check that each step has been carried out successfully before proceeding to the next step. The flow tracking step, in particular, requires significant optimization when using a cell line or condition for the first time. When checking the results of each QFSM analysis step, refer to Table 1 to troubleshoot problems with the analysis. The critical parameters for each step are discussed below.
Table 1.
Troubleshooting Guide for QFSM analysis software
| Problem | Possible Cause | Solution |
|---|---|---|
| Noise Model Calibration | ||
| Low Gauss ratio (typically <3) | Cropped region contains parts of cell. | Modify region of interest along the stack depth. |
| Spatial and/or temporal range of the background region is too small. | Acquire a movie containing background only. Run the noise model calibration on this background movie. (a) | |
| Thresholding/Mask Refinement | ||
| Cell mask is not determined properly | Automatic thresholding fails | Use alternate thresholding methods/Set threshold value manually |
| Inhomogeneous background | Crop region of the movie for analysis. | |
| Multiple cells in the movie | Adjust the object number in mask refinement Crop each cell as a separate movie |
|
| Speckle detection | ||
| Misdetected speckles | Low Gauss ratio | See Noise Model Calibration above |
| Low signal to noise ratio | Increase alpha value | |
| Dense speckles are not resolved | Increase number of iterations | |
| Flow Tracking | ||
| Inhomogenous flow | Template size too short/big | Use variable template range to determine adapted size. |
| Integration window too short | Optimize integration window. | |
| Background is perturbing correlation | Use stationary background subtraction | |
| High number of outliers | Decrease outlier threshold | |
| Speckle Tracking | ||
| Short tracks | Track lifetime is too short | Use iterative speckle detection |
| Random flow directionality | Initialization problem |
|
| Kinetic Analysis/Flow Analysis | ||
| Maps | Sparse kinetic events cannot generate sufficient information | Increase time windows for averaging |
If none of these help then the camera noise is electronically correlated (can happen especially with first generation EM-CCD or sCMOS cameras); or there is a high level of stray light in the microscope.
Noise Model Calibration
The calibration of a valid noise model is critical for the statistical detection of speckles. From a cropped stack of background, the calibration step computes the mean and the standard deviation of the background intensity. To validate the Gaussian noise assumption, the noise model calibration calculates an additional value called Gauss ratio. The theoretical value of the Gauss ratio for perfect Gaussian noise is . The conversion between numerical aperture and σPSF is discussed in [17]. For high NA lenses and small pixels as required for FSM, experimental values between 3 and 5 indicate good noise models. Models with a ratio close to 1 indicate spatial correlation in the noise patterns. This can either be caused by a defective camera or high amounts of stray-light in the microscope.
Mask Creation – Thresholding and Mask Refinement
Initial masks are created by first low-pass filtering each image using a Gaussian kernel. The threshold value is then determined based on the intensity histograms of the filtered images. Assuming the region of lowest intensity corresponds to the background, the default thresholding algorithm uses a spline interpolation of the histogram to measure the first minimum of intensity following the background. Alternative popular thresholding methods are proposed for selecting the threshold such as the Otsu and Rosin algorithms [18, 19]. Once individual images are thresholded, masks are post-processed using a series of refinement operations. Small, unconnected regions are discarded, holes within the cell are filled and the mask border is smoothened.
Speckle Detection
Speckles are detected as local intensity maxima [11, 13]. First, the image is low-pass filtered using a Gaussian kernel of standard deviation equal to the standard deviation of the theoretical point-spread function. Local maxima are then identified within a window of 5×5 pixels and compared to the mean intensity of the three nearest local intensity minima. For each speckle, a statistical test is applied to determine the significance of local maxima to local minima intensity differences using the noise model parameters and the confidence level (α-value) specified by the user. If performing more than one iteration during speckle detection, peaks corresponding to existing speckles are subtracted from the image and new speckles are detected using the same procedure as described above. The validity of each newly detected speckle is determined using its relative position and intensity to neighboring speckles of lower order [13]
Flow Tracking
Quasi-stationary flow is tracked using image cross-correlation [7]. The particular feature of the implemented algorithm is an integration of the correlation scores over multiple consecutive frame pairs. This permits the successful tracking of very weak contrast signals, typical for FSM, assuming that locally the flow direction and speed alters substantially slower than the frame rate. Flow outliers are identified and removed using a modified version of the median test [20]. Flow tracking is the most time-consuming of the whole QFSM analysis workflow (see Table 2). It is, however, a highly recommended step as it greatly improves the quality of the speckle tracking.
Table 2. Estimated processing times.
Tests have been performed on the example movie using Ubuntu 10.04, 64 bit 3,2Ghz 4 Cores and Matlab R2011a. The movie used for benchmarking the software is a single channel FSM movie with 91 frames of 656 by 1118 pixels.
| Step | Processing time | Notes |
|---|---|---|
| Noise estimation | 3 s | Region of interest: 300×650 pixels. |
| Thresholding | 15 s | Default parameters |
| Mask Refinement | 7 s | Default parameters |
| Speckle detection | 2 min | Maximum number of iterations: 3 Total number of speckles: ~2500/frame |
| Flow tracking | 5 hr 30 min | Template size: 11 pixels Average time: 0.3 s/per speckle per frame |
| Speckle tracking | 5min | Gaussian weighted interpolation |
| Kinetic analysis | 8 min | |
| Flow analysis | 10 min | Number of frames for time averaging: 15 |
Speckle tracking
If flow tracking has been performed, speckle movements are interpolated using the tracked flow. Then, for each frame, the candidate links between projected speckles from the previous frame and detected speckles in the current frame are established within the specified search radius. Note one projected speckle may have multiple candidate links to speckles in the current frame, while speckles in the current frame may have candidate links from multiple candidate links. While in an early version of the QFSM software we resolved this ambiguity using a neural network, the currently downloadable version casts this association step as a linear assignment problem as discussed in [21]. Once speckles have been linked for the entire movie, single-frame gaps are closed. The gap between two tracks is closed if an insignificant speckle, i.e. a rejected local intensity maximum, is found within the search radius of the propagated position in the missing frame.
Kinetic analysis
Kinetic analysis is performed in two steps. First, speckle birth/death events are classified by comparing their intensity variation relative to the background intensity variation [11]. Through a series of statistical tests of the significance of the foreground and background intensity changes about birth and death events, insignificant events are excluded and significant event are associated with either local polymerization or depolymerization. Kinetic maps are then generated by integrating the significant kinetic events over the designated time window and using Gaussian spatial blurring.
Flow analysis
Flow analysis interpolates the frame-to-frame displacements computed from speckle trajectories using Gaussian filtering with a set correlation length. Interpolated displacements are then averaged over a time window to remove noise fluctuations. Scalar speed maps are generated as a function of time.
Anticipated Results
The injection of live epithelial cells with low levels of Alexa568-actin should yield cells with homogeneous and salt and pepper-like actin speckles spaced by ~2 speckle diameters when imaged with a 1.4 N.A. objective. Dense stress fibers or lamellipodia will appear with breaks or as dots rather than as solid lines. When analyzed with QFSM software with an appropriate correlation length for the flow vectors and temporal duration over which values are averaged, actin speckles should exhibit retrograde flow from the leading edge toward the cell center at a rate of 0.1–2 μm/min. Under control conditions the actin turnover maps tend to exhibit a clear band of polymerization at the cell edge, followed immediately by a band of de-polymerization and then a wide area of reduced and randomly assembly/disassembly behavior.
Time Considerations
Obtaining high-quality time-series images for QFSM analysis takes a significant amount of practice. However, once one becomes proficient with gently microinjecting actin, the procedure and analysis can be carried out in a matter of days and yield a wealth of information that cannot be obtained through any other method. For example, QFSM is still the only method delivering non-steady state dynamics of polymer turnover across an entire field of view. It takes about half of day to prep, inject, and image the actin, depending on your comfort with the technique and the number of movies desired. The cells first need to be plated 1 – 2 days prior to the experiment and this requires ~30 min. On the day of the experiment, the actin needs to be ultracentrifuged (~1 h), needles pulled (~20 min), and several sets of cells injected and recovered (2–3 h). While later sets of cells are recovering, the first set dish of injected cells can be imaged. Imaging usually takes 30 min per 10–15 min time series, as the cells have to be equilibrated to the microscope’s temperature and humidity, the injected cells must be located, and their health evaluated. Determining the optimal QFSM parameters for analysis of the time-series can take several days, as different values must be empirically tested. Once the QFSM work flow is optimized, the analysis can be performed in batch mode. Analysis will take one to several days, depending on the number of FSM movies, but does not require hands-on manipulation of the software.
Footnotes
Internet resources with annotations
http://lccb.hms.harvard.edu/software.html
Web site for downloading the most recent version of the QFSM analysis software.
References
- 1.Ponti A, et al. Two distinct actin networks drive the protrusion of migrating cells. Science. 2004;305(5691):1782–6. doi: 10.1126/science.1100533. [DOI] [PubMed] [Google Scholar]
- 2.Ji L, Lim J, Danuser G. Fluctuations of intracellular forces during cell protrusion. Nat Cell Biol. 2008;10(12):1393–400. doi: 10.1038/ncb1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delorme V, et al. Cofilin activity downstream of Pak1 regulates cell protrusion efficiency by organizing lamellipodium and lamella actin networks. Developmental Cell. 2007;13(5):646–662. doi: 10.1016/j.devcel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell. 2006;125(7):1361–1374. doi: 10.1016/j.cell.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 5.Lim J, Danuser G. Live cell imaging of F-actin dynamics via Fluorescent Speckle Microscopy (FSM) J Vis Exp. 2009;(30) doi: 10.3791/1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hebert B, Costantino S, Wiseman PW. Spatiotemporal image correlation Spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophysical Journal. 2005;88(5):3601–3614. doi: 10.1529/biophysj.104.054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ji L, Danuser G. Tracking quasi-stationary flow of weak fluorescent signals by adaptive multi-frame correlation. Journal of Microscopy-Oxford. 2005;220:150–167. doi: 10.1111/j.1365-2818.2005.01522.x. [DOI] [PubMed] [Google Scholar]
- 8.Lim JI, et al. Protrusion and actin assembly are coupled to the organization of lamellar contractile structures. Exp Cell Res. 2010;316(13):2027–41. doi: 10.1016/j.yexcr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waterman-Storer CM, et al. Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr Biol. 1998;8(22):1227–30. doi: 10.1016/s0960-9822(07)00515-5. [DOI] [PubMed] [Google Scholar]
- 10.Salmon ED, Waterman CM. How we discovered fluorescent speckle microscopy. Molecular Biology of the Cell. 2011;22(21):3940–3942. doi: 10.1091/mbc.E11-07-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ponti A, et al. Computational analysis of F-actin turnover in cortical actin meshworks using fluorescent speckle microscopy. Biophys J. 2003;84(5):3336–52. doi: 10.1016/S0006-3495(03)70058-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danuser G, Waterman-Storer CM. Quantitative fluorescent speckle Microscopy of cytoskeleton dynamics. Annual Review of Biophysics and Biomolecular Structure. 2006:361–387. doi: 10.1146/annurev.biophys.35.040405.102114. [DOI] [PubMed] [Google Scholar]
- 13.Ponti A, et al. Periodic patterns of actin turnover in Lamellipodia and lamellae of migrating epithelial cells analyzed by quantitative Fluorescent Speckle Microscopy. Biophysical Journal. 2005;89(5):3456–3469. doi: 10.1529/biophysj.104.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salmon WC, Adams MC, Waterman-Storer CM. Dual-wavelength fluorescent speckle microscopy reveals coupling of microtubule and actin movements in migrating cells. J Cell Biol. 2002;158(1):31–7. doi: 10.1083/jcb.200203022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu K, et al. Differential transmission of actin motion within focal adhesions. Science. 2007;315(5808):111–115. doi: 10.1126/science.1135085. [DOI] [PubMed] [Google Scholar]
- 16.Waterman-Storer C. Fluorescent speckle microscopy (FSM) of microtubules and actin in living cells. Curr Protoc Cell Biol. 2002;Chapter 4(Unit 4–10) doi: 10.1002/0471143030.cb0410s13. [DOI] [PubMed] [Google Scholar]
- 17.Thomann D, et al. Automatic fluorescent tag detection in 3D with super-resolution: application to the analysis of chromosome movement. Journal of Microscopy-Oxford. 2002;208:49–64. doi: 10.1046/j.1365-2818.2002.01066.x. [DOI] [PubMed] [Google Scholar]
- 18.Otsu N. Threshold Selection Method from Gray-Level Histograms. IEEE Transactions on Systems Man and Cybernetics. 1979;9(1):62–66. [Google Scholar]
- 19.Rosin PL. Unimodal thresholding. Pattern Recognition. 2001;34(11):2083–2096. [Google Scholar]
- 20.Westerweel J, Scarano F. Universal outlier detection for PIV data. Experiments in Fluids. 2005;39(6):1096–1100. [Google Scholar]
- 21.Jaqaman K, et al. Robust single-particle tracking in live-cell time-lapse sequences. Nature Methods. 2008;5(8):695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]