

Abstract

This account focuses on the application of ω-transaminases, lyases, and oxidases for the preparation of amines considering mainly work from our own lab. Examples are given to access α-chiral primary amines from the corresponding ketones as well as terminal amines from primary alcohols via a two-step biocascade. 2,6-Disubstituted piperidines, as examples for secondary amines, are prepared by biocatalytical regioselective asymmetric monoamination of designated diketones followed by spontaneous ring closure and a subsequent diastereoselective reduction step. Optically pure tert-amines such as berbines and N-methyl benzylisoquinolines are obtained by kinetic resolution via an enantioselective aerobic oxidative C–C bond formation.
Introduction
Biocatalysis, thus applying enzymes for organic synthesis, has become a vivid alternative for established organic methods.1,2 Tailoring biocatalysts by protein engineering and design allows to adapt the catalyst for the desired reaction conditions.3 Several biocatalytic methods have been established to access optically pure amines and derivates which play a pivotal role as building blocks for pharmaceutical drugs, agrochemicals, natural products, and as chiral ligands.4 Whilst originally mainly hydrolases were broadly applied for the preparation of amines in optically pure form,5 other types of enzymes like ω-transaminases, lyases, and oxidases came into focus for this purpose more recently. This account discusses the application of these three types of enzymes for the preparation of optically pure amines with special emphasis on the work performed in our own laboratories. The account is divided into three main sections according to the type of the obtained final product, namely primary-, secondary-, and tertiary-amines.
Primary Amines
Within the past few years, the asymmetric synthesis of α-chiral prim-amines from the corresponding ketones was heavily investigated by employing ω-transaminases (ωTAs).6,7 The asymmetric amination of the prochiral ketone is favored over the reverse reaction, namely the kinetic resolution of racemic amines with these enzymes.8 ωTAs catalyze the reductive amination of the substrate ketone using an amine donor which serves as nitrogen source and provides the required electrons, thereby the amine donor gets oxidized to the corresponding ketone (Scheme 1). The required cofactor pyridoxal 5′-phosphate (PLP) acts thereby as electron and nitrogen shuttle and is added to the reaction mixture in general at a concentration of 0.1 to 1 mM for 50 mM substrate concentration.
Scheme 1. Asymmetric amination of ketones employing ω-transaminases.

Even though various amines can be applied as amine source, 2-propylamine seems to be an ideal, achiral, and cheap reagent, as was recently demonstrated on industrial scale for the preparation of the diabetes type 2 drug sitagliptin (performed on a multikilogram scale with a substrate concentration of ca. 200 g/L).9 Nevertheless, even for this privileged product the oxidized amine donor (acetone) had to be removed via gas flow at elevated temperature (45 °C); alternatively, the formed acetone can be removed at reduced pressure10 or reduced to the corresponding alcohol.11 On the other hand, using alanine as the amine donor, the formed coproduct pyruvate is commonly removed either by recycling it back to alanine6d,12 or by reduction to lactate.12b In the first case, an alanine dehydrogenase (AlaDH) is required, while in the second one a lactate dehydrogenase (LDH) is employed. Because NAD(P)H is consumed in stoichiometric amounts in both methods, established systems for NAD(P)H recycling are employed (formate dehydrogenase, glucose dehydrogenase, phosphite dehydrogenase). Notably, removal of the coproduct pyruvate is required due to the nonfavored equilibrium of the amination reaction.13d In the case of using 2-propylamine in excess as amine donor (e.g., 10 equivalents), full conversion is not reached in general, except for selected substrates, where the product amine is thermodynamically favored.9,10,14,15 Consequently, the approaches employing LDH or AlaDH might be preferred for general substrates because it allows full conversion; nevertheless, the search for ideal amine donors is still ongoing.16
Testing various substrates and ωTAs,17 the (S)-enantiomer as well as the (R)-enantiomer of each amine was accessible in most cases in optically pure form (>99% ee) at 50 mM substrate concentration (Table 1). A number of ω-TAs turned out to be rather useful in our own studies like the (S)-stereoselective ωTAs from Vibrio fluvialis (VF-ωTA),18Chromobacterium violaceum (CV-ωTA),19Bacillus megaterium (BM-ωTA),20Paracoccus denitrificans (PD-ωTA),21Pseudomonas fluorescens (PF-ωTA),22Alcaligenes denitrificans (AD-ωTA),23 as well as a variant from Arthrobacter citreus (ArS-ωTA).24 (R)-Selective ωTAs of interest originated from Aspergillus terreus (AT-ωTA),25Hyphomonas neptunium (HN-ωTA),25Arthrobacter sp. (ArR-ωTA)26 as well as its variant ArRmut11-ωTA.9a All enzymes were overexpressed in Escherichia coli and employed in general as permeabilized (freeze-dried) cell preparation; thus neither enzyme purification was not required nor cell disruption. In most cases, the ratio of weight of substrate to freeze-dried whole cell catalyst preparation was ∼1/2.
Table 1. Asymmetric amination of various ketones at 50 mM substrate concentration.
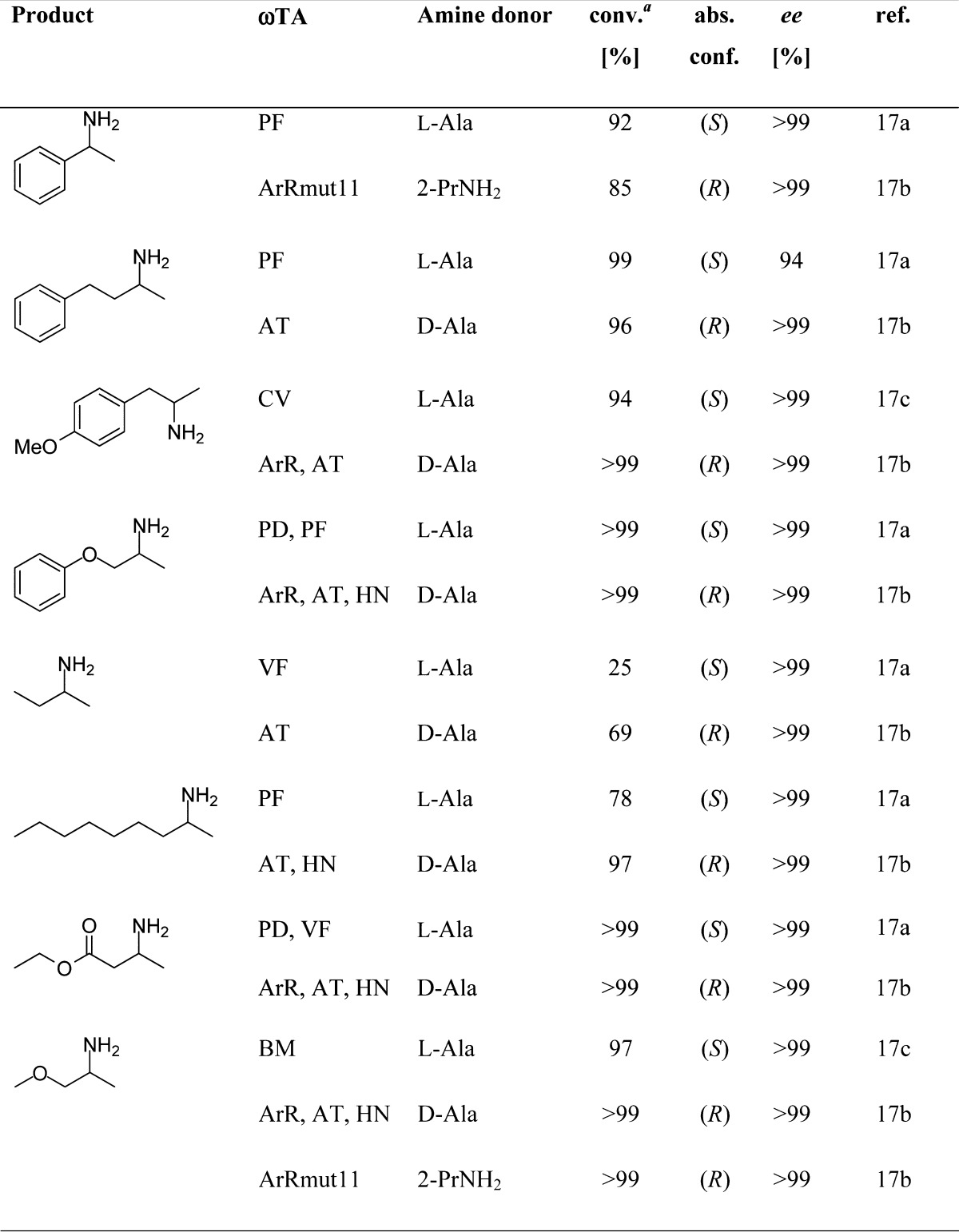
Conversion was measured by GC.
The applicability of the ωTAs was shown in a short chemoenzymatic synthesis of (S)-rivastigmine, which was prepared within a linear sequence of three steps employing either PD-ωTA or VF-ωTA in the asymmetric key step (45 mM substrate concentration; 100 mg scale) (Scheme 2).27 The primary (S)-amine was obtained thereby with perfect optical purity (>99% ee) and excellent conversions (up to >99%), which was subsequently dimethylated to a tertiary amine by chemical means. (S)-Rivastigmine is used for the symptomatic treatment of mild to moderate Alzheimer’s disease and dementia due to Parkinson’s disease.
Scheme 2. Chemoenzymatic asymmetric synthesis of (S)-rivastigmine within three steps.

3-Aryl-GABA (3-aryl γ-aminobutyric acid) derivatives28 play an important role in several nervous system functions. The precursor (4-phenylpyrrolidin-2-one) was prepared in a dynamic kinetic resolution employing a ωTA on a 100 mg scale giving the desired product with 92% isolated yield (Scheme 3).29 Unfortunately, the stereoselectivity of the employed ωTA (ATA-117, which is almost identical to ArR-ωTA) led only to 68% ee in this particular case. Nevertheless, the reported general synthetic strategy allows obtaining optically enriched 4-arylpyrrolidin-2-one within only three synthetic steps with 54% overall yield, which represents a significant improvement compared to previous approaches.
Scheme 3. Dynamic kinetic resolution of a racemic α-chiral aldehyde to prepare a 3-phenyl-GABA precursor.

For some cases, the racemic amine is easier to access than the corresponding prochiral ketone. Consequently, deracemization30 of the racemic amine to yield the amine with theoretically quantitative yield and >99% ee is of interest. For example, deracemization of the drug mexiletine (an orally effective antiarrhythmic agent) was achieved by combining the reverse reaction, namely the enantioselective deamination with the stereoselective amination employing stereocomplementary enzymes up to a 100 mg scale (Scheme 4).31 The deracemization was successfully performed in a one-pot two-step process.32 By using appropriate enzymes, (R)- as well as (S)-mexiletine could be prepared in optically pure form (>99% ee) and 97% isolated yield. For the deamination, pyruvate was recycled using an amine oxidase.33 The efficiency of the process could be further improved by immobilizing the ωTA of the first step by encapsulation in a sol–gel/Celite matrix to remove it prior to the second step.34
Scheme 4. Deracemization of rac-mexiletine via a two-step procedure.

Preparation of amines in an aqueous environment at neutral pH turns the isolation of the product amine cumbersome because it requires additionally the adjustment of the pH to basic conditions prior to extraction. Consequently, it would be desirable to perform the transformation in an organic environment, similar to reactions with lipases and proteases which are extensively employed in nonaqueous solvents.35 A recent study suggested that ωTAs need to be engineered and immobilized to be able to employ them in a purely organic environment.36 However, we found that by employing an appropriate cell-free freeze-dried ωTA preparation and by careful tuning the water activity, every ωTA, as far as tested, can indeed be employed in organic solvents.15 The solvents methyl t-butyl ether (MTBE) and ethyl acetate (EtOAc)37 turned out to be best suited. The maximum enzymatic activity was found at the same water activity aw of 0.6 for both solvents, which corresponded to a water concentration cw of 2% v/v and 0.65% v/v in EtOAc and MTBE, respectively. The ωTAs showed increased stability in organic solvents compared to buffer; for instance, the enzyme was still active after incubation in EtOAc for 24 h at 60 °C. In contrast, the ωTA was completely inactive after 5 min of incubation in aqueous buffer at the same temperature. Furthermore, all nine enzymes investigated accepted efficiently 2-propylamine as amine donor. As an example, methoxy-acetone (50 mM) was quantitatively aminated to enantiopure (R)-methoxy-2-propylamine in just two hours in MTBE using 3 equiv of 2-propylamine as amine donor (Scheme 5). Conversely, the amination in aqueous buffer at pH 7 afforded just 30% conversion employing 3 equiv of 2-propylamine after one day. Especially substrates carrying an oxygen atom in the vicinity of the ketone moiety, enabling the formation of an internal hydrogen bond between NH2 and O in the product amine, were aminated in general with high conversion with just 3 equiv of amine. These products are probably thermodynamically favored. Another important observation was that no substrate inhibition was found in MTBE (aw 0.6) (up to 500 mM), while in aqueous buffer inhibition occurred typically at 10 mM substrate concentration. Finally, because the catalyst forms a solid phase, it could be easily separated by filtration and recycled several times without loss of activity.
Scheme 5. Asymmetric bio-amination of ketones in MTBE at defined water activity aw = 0.6.

In contrast to the amination of ketones, the direct amination of alcohols would not require any reducing agents in the ideal case due to the same oxidation state of alcohol and amine, as already demonstrated with metal catalysts via the “borrowing hydrogen” approach.38 Because no enzyme is known, to the best of our knowledge, which aminates an alcohol, we designed an artificial biocatalyst cascade/network to accomplish this task (Scheme 6).39 In the first step, the alcohol was oxidized by an alcohol dehydrogenase (ADH) consuming NAD+, leading to the formation of the aldehyde and NADH. In the second sequential step, a ωTA aminated the intermediate aldehyde at the expense of l-alanine. For the regeneration of l-alanine from pyruvate, a l-alanine dehydrogenase (AlaDH) was employed, consuming ammonia and NADH. The latter was provided from the oxidation step where NADH was liberated. Thus, the AlaDH connected the oxidation with the reductive amination by transferring the hydride from NADH, the byproduct of the oxidation, to the amination step by regenerating the amine donor alanine from pyruvate. Careful optimization of temperature and cosolvent allowed the diamination of 1,ω-alkanediols, leading to 99% conversion to the diamines. For example, the diamination of 1,10-decanediol on preparative scale (174 mg substrate) led to 94% conversion and 70% isolated yield. Alternatively, the alcohol can be oxidized by employing an oxidase followed by amination with a ωTA,40 however, in this case, additional redox reagents are required.
Scheme 6. Biocatalyst network for the amination of alcohols in water.

Conditions: water 10% v/v 1,2-dimethoxyethane, 0.35 mM PLP, pH 8.5, 25 °C, 20 h.
Other options employing oxidases or lyases to prepare optically pure primary amines established by other groups include (i) the deracemization employing a monoamine oxidase (MAO)41 and an achiral reducing agent or (ii) via addition of ammonia to alkenes. The deracemization employing MAO, will be described with examples in the chapters below.
The asymmetric addition of ammonia to a C=C-double bond is catalyzed by various lyases,42 whereby the addition of NH3 to unsaturated acids can be utilized for the synthesis of chiral α-amino acids. For example, the closely related methylaspartate and phenyl ammonium lyase (MAL and PAL) are used on an industrial scale for production of amino acides like l-phenyl alanine and l-aspartate on a multiton scale since the 1980s. Furthermore, MAL has been coupled with an aspartate-4-decarboxylase in a continuous process to yield l-alanine.43 Whilst the substrate spectrum of the wild type MAL is narrow, engineered methylaspartate ammonia lyase (MAL) accepted a broader spectrum of nucleophiles beside ammonia44 compared to its wild type.45 A single variant (Q73A) accepted various nucleophiles, including linear and cyclic alkylamines, giving also access to secondary amines (Scheme 7) (see also chapter below).
Scheme 7. Ammonia lyases catalyze the reversible addition of ammonia to α,β-unsaturated acids.

Secondary Amines
Because ωTAs have not been described so far to transfer any substituted amine group to a ketone to yield the secondary amine directly, we designed special substrates leading to amino ketones which spontaneously cyclized to yield, after reduction, a cyclic secondary amine. The interesting question was whether the ωTA will be capable of discriminating between two ketone moieties within the same molecule. Indeed, in transforming various 1,5-diketones, it was found that the asymmetric reductive amination proceeds with perfect regio- as well as with perfect stereoselectivity and high conversion at 50 mM substrate concentration (Schemes 8 and 9). Exclusively the sterically less demanding (ω-1)-ketone-moiety within each molecule of the investigated 1,5-diketones was aminated, leading to spontaneous ring-closure to afford the optically pure Δ1-piperideine (ee > 99%), which was diasteroselectively reduced to the corresponding cis- and trans-piperidines, respectively.46 Notably, most of the investigated amino-transferases were able to discriminate between very small differences in size (e.g., methyl vs cyclopropyl), yielding one regioisomer in optically pure form.
Scheme 8. Chemoenzymatic synthesis of optically pure cis-2,6-disubstituted piperidines via regio- and stereoselective mono-amination of 1,5-diketones yielding Δ1-piperideines followed by diastereoselective reduction.

Scheme 9. Chemoenzymatic synthesis of all four stereoisomers of the alkaloids dihydropinidine and epi-dihydropinidine.

Reaction conditions: (a) diketone (78 mg, 0.5 mmol, 50 mM), CV-ωTA, PLP (1 mM), NAD+ (1 mM), l-alanine (10 equiv), NH4HCOO (150 mM), 11 U FDH, 12 U AlaDH; 26 h, 30 °C, 120 rpm; (b) analogue to (a) but with ArR-ωTA and d-alanine; (c) Pd/C, H2, 4 h, 22 °C; (d) Et3Al in hexane (5.0 mmol), LiAlH4 in THF (2.5 mmol), THF, −78 °C, 2 h.
This strategy was employed for the shortest and highest yielding route to all four diastereomers of the natural alkaloids dihydropinine (cis) and epi-dihydropinidine (anti) (Scheme 9). Starting from nonane-2,6-dione, which can be obtained within one chemical step from commercial compounds, the (R)- and (S)-Δ1-piperideine, respectively, were obtained under perfect regio- and enantiocontrol at full conversion using enantiocomplementary ωTAs (50 mM substrate concentraiton; 78 mg). The cis-isomers were obtained via subsequent Pd/C catalyzed hydrogenation whereas the anti-isomers were prepared by reduction of a Lewis-acid-mediated conformationally changed Δ1-piperideine.47
Approaches to sec-amines from other groups employing oxidases and lyases include for instance the application of the already mentioned monoamine oxidase (MAO) as well as lyases, such as the norcoclaurine or strictosidine synthase. The norcoclaurine synthase (NCS) catalyses an asymmetric Pictet–Spengler condensation48 and is located at the beginning of the secondary metabolic pathway of plants, yielding benzylisoquinoline alkaloids. It condenses stereoselectively a tyrosine derived aldehyde with dopamine to give (S)-norcoclaurine (Scheme 10).49 Recently, the substrate tolerance was shown to be rather broad, especially concerning the aldehyde50 and preparative scale experiments with non-natural substrates were demonstrated (up to 2.2 g product).51 A related enzyme catalyzing a Pictet–Spengler reaction is the strictosidine synthase, which seems to be more substrate specific especially regarding the aldehyde.52
Scheme 10. Stereoselective enzymatic condensation of dopamine and 4-hydroxyphenyl acetaldehyde to (S)-norcoclaurine.

Reaction conditions taken from ref (48).
The MAO of Aspergillus niger (MAO-N) was subjected to random mutagenesis to broaden the substrate specificity. Variants were obtained which oxidized compounds such as rac-1-methyl-tetrahydroisquinoline, allowing its deracemization (Scheme 11),53 as well as the desymmetrization of meso-pyrrolidines.54
Scheme 11. Examples for the deracemization of a sec- and tert-amines employing the monoamine oxidase from Aspergillus niger and borohydride.

Tertiary Amines
By employing a monoamine oxidase, tert-amines like crispine A could also be successfully deracemized (Scheme 11).55 In our group, a completely different approach was applied to obtain optically pure tert-amines: in the asymmetric key step, an enzyme catalyzed C–C bond was formed in an aerobic oxidative enantioselective reaction by employing the berberine bridge enzyme (BBE) (Scheme 12). BBE transforms the natural substrate (S)-reticuline to (S)-scoulerine at the expense of molecular oxygen.
Scheme 12. Enantioselective oxidative C–C bond formation catalysed by BBE at the expense of O2.

BBE originating from California poppy (Eschscholzia californica)56 can efficiently be expressed in Pichia pastoris.57 Just recently, the mechanism was revised,58 and it was found that BBE possesses a biocovalently linked FAD.59 We showed that BBE can be employed in a kinetic resolution starting with racemic N-methyl 1,2,3,4-tetrahydrobenzylisoquinolines, leading to the formation of optically pure 9-hydroxy 1,2,3,4-tetrahydroprotoberbines as well as remaining optically pure benzylisoquinolines (Scheme 13).60 The enzyme accepts modifications at the isoquinoline ring at position 6 and 7, allowing there in either position various substituents such as methoxy, hydroxy, or just hydrogen or a bridging methylendioxy functionality. Additionally, the presence of a methoxy group in position 8 is tolerated.
Scheme 13. Racemic substrates resolved in a kinetic resolution by BBE.

Concerning modifications at the phenol moiety, it was observed that the hydroxyl group in position 3′ is essential to maintain activity. To allow efficient transformation of the barely water-soluble benzylisoquinolines at 50–65 mM substrate concentration, cosolvents were required. The enzyme tolerated various organic solvents at remarkable high concentrations. Optimum conditions were found in the presence of 70% v/v toluene at pH 9 and 40 °C, allowing substrate concentrations up to 20 g/L.61 By employing these conditions, various substrates were successfully transformed on a 500 mg scale. For instance, this asymmetric key step enabled the first asymmetric total synthesis of (S)-scoulerine, a sedative and muscle-relaxing agent. For selected non-natural substrates, the formation of a minor regioisomer side product was observed, namely the 11-hydroxy berbines. The ratio of the main product to regioisomer can be influenced by the organic solvent used as well as by the substitution pattern at the aromatic isoquinoline part. Moreover, it was demonstrated that by introducing a fluoro atom in 6′ position and therefore blocking the C–C–bond forming position for the 11-hydroxy regioisomer, the 9-hydroxy berbines were formed exclusively as the main product.62
Conclusion
Biocatalysis has become a competitive method for asymmetric synthesis. The here presented approaches to various α-chiral prim-amines, as well as α-chiral sec- and tert-amines, demonstrate the power of enzymes with respect to outstanding stereoselectivities and mild reaction conditions. It clearly shows that the potential of biocatalytic methods awaits its exploitation.
Acknowledgments
Numerous funding agencies are thanked for supporting our research. Funding from the European Union’s Seventh Framework Programme FP7/2007-2013 was received under grant agreement no. 245144 (AmBioCas) for J.H.S. and F.G.M., within Marie Curie Networks for Initial Training fellowship in the project BIOTRAINS (FP7-PEOPLE-ITN-2008-238531) for A.R. and E.S. and under grant agreement no. 289646 (KyroBio) for E.F.; H.L. acknowledges DK “Molecular Enzymology” and financial support by the FWF (Project W9). R.C.S., D.P., and C.S.F. acknowledges the Federal Ministry of Economy, Family, and Youth (BMWFJ), the Federal Ministry of Traffic, Innovation and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol and ZIT—Technology Agency of the City of Vienna through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG.
The authors declare no competing financial interest.
References
- For selected books see:; a Practical Methods for Biocatalysis and Biotransformations 2; Whittall J., Sutton P., Eds.; Wiley & Sons: Chichester, UK, 2012. [Google Scholar]; b Gamenara D.; Seoane G.; Saenz Mendez P.; Domínguez de María P.. Redox Biocatalysis: Fundamentals and Applications; Wiley & Sons: Hoboken, NJ, 2013. [Google Scholar]; c Enzyme Catalysis in Organic Synthesis; Drauz K., Groeger H., May O., Eds.; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]; d Faber K.Biotransformations in Organic Chemistry 6th ed.; Springer: Heidelberg, 2011. [Google Scholar]; e Grunwald P.Biocatalysis: Biochemical Fundamentals and Applications; Imperial College Press: London, 2009. [Google Scholar]; f Modern Biocatalysis; Fessner W.-D., Anthonson T., Eds.; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]; g Liese A. K.; Seelbach K.; Wandrey C.. Industrial Biotransformations, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]; h Asymmetric Organic Synthesis with Enzymes; Gotor V., Alfonso I., Garcia-Urdiales E., Eds.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- For selected reviews see:; a Clouthier C. M.; Pelletier J. N. Chem. Soc. Rev. 2012, 41, 1585–1605. [DOI] [PubMed] [Google Scholar]; b Turner N. J. Catal. Sci. Technol. 2012, 2, 1523–1523. [Google Scholar]; c Duchek J.; Adams D. R.; Hudlicky T. Chem. Rev. 2011, 111, 4223–4258. [DOI] [PubMed] [Google Scholar]; d Hall M.; Bommarius A. S. Chem. Rev. 2011, 111, 4088–4110. [DOI] [PubMed] [Google Scholar]; e Hollmann F.; Arends I.; Buehler K.; Schallmey A.; Bühler B. Green Chem. 2011, 13, 226–265. [Google Scholar]; f Hollmann F.; Arends I.; Holtmann D. Green Chem. 2011, 13, 2285–2313. [Google Scholar]; g Pollard D. J.; Woodley J. Trends Biotechnol. 2007, 25, 66–73. [DOI] [PubMed] [Google Scholar]
- Bornscheuer U. T.; Huisman G. W.; Kazlauskas R. J.; Lutz S.; Moore J. C.; Robins K. Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- a Nugent T. C.; Marinova S. M. Synthesis 2013, 45, 153–166. [Google Scholar]; b Chiral Amine Synthesis: Methods, Developments and Applications; Nugent T. C., Ed.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Busto E.; Gotor-Fernández V.; Gotor V. Chem. Rev. 2011, 111, 3998–4035. [DOI] [PubMed] [Google Scholar]
- For recent reviews see:; a Malik M. S.; Park E.-S; Shin J.-S. Appl. Microbiol. Biotechnol. 2012, 94, 1163–1171. [DOI] [PubMed] [Google Scholar]; b Mathew S.; Yun H. ACS Catal. 2012, 2, 993–1001. [Google Scholar]; c Rudat J.; Brucher B. R.; Syldatk C. AMB Express 2012, 2, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tufvesson P.; Lima-Ramos J.; Jensen J. S.; Al-Haque N.; Neto W.; Woodley J. M. Biotechnol. Bioeng. 2011, 108, 1479–1493. [DOI] [PubMed] [Google Scholar]; e Koszelewski D.; Tauber K.; Faber K.; Kroutil W. Trends Biotechnol. 2010, 28, 324–332. [DOI] [PubMed] [Google Scholar]; f Turner N. J.; Truppo M. in Chiral Amine Synthesis: Methods, Developments and Applications; Nugent T. C., Ed.; Wiley-VCH, Weinheim, Germany, 2010, pp 431–459. [Google Scholar]; g Hailes H. C.; Dalby P. A.; Lye G. J.; Baganz F.; Micheletti M.; Szita N.; Ward J. M. Curr. Org. Chem. 2010, 14, 1883–1893. [Google Scholar]; h Ward J.; Wohlgemuth R. Curr. Org. Chem. 2010, 14, 1914–1927. [Google Scholar]; i Höhne M.; Bornscheuer U. T. ChemCatChem 2009, 1, 42–51. [Google Scholar]; j Zhu D.; Hua L. Biotechnol. J. 2009, 4, 1420–1431. [DOI] [PubMed] [Google Scholar]
- For selected recent papers on ω-transaminases see:; a Steffen-Munsberg F.; Vickers C.; Thontowi A.; Schätzle S.; Tumlirsch T.; Humble M. S.; Land H.; Berglund P.; Bornscheuer U. T.; Höhne M. ChemCatChem 2013, 5, 150–153. [Google Scholar]; b Humble M. S.; Cassimjee K. E.; Abedi V.; Federsel H.-J.; Berglund P. ChemCatChem 2012, 4, 1167–1172. [Google Scholar]; c Truppo M.; Janey J. M.; Grau B.; Morley K.; Pollack S.; Hughes G.; Davies I. Catal. Sci. Technol. 2012, 2, 1556–1559. [Google Scholar]; d Shaheer Malik M.; Park E.-S.; Shin J.-S. Green Chem. 2012, 14, 2137–2140. [Google Scholar]; e Cassimjee K. E.; Svedendahl-Humble M.; Land H.; Abedi V.; Berglund P. Org. Biomol.Chem. 2012, 10, 5466–5470. [DOI] [PubMed] [Google Scholar]; f Seo Y.-M.; Mathew S.; Bea H.-S.; Khang Y.-H.; Lee S.-H.; Kim B.-G.; Yun H. Org. Biomol. Chem. 2012, 10, 2482–2485. [DOI] [PubMed] [Google Scholar]; g Humble M. S.; Cassimjee K. E.; Håkansson M.; Kimbung Y. R.; Walse B.; Abedi V.; Federsel H.-J.; Berglund P.; Logan D. T. FEBS J. 2012, 279, 779–792. [DOI] [PubMed] [Google Scholar]; h Schätzle S.; Steffen-Munsberg F.; Thontowi A.; Höhne M.; Robins K.; Bornscheuer U. T. Adv. Synth. Catal. 2011, 353, 2439–2445. [Google Scholar]; i Bea H.-S.; Park H.-J.; Lee S.-H.; Yun H. Chem. Commun. 2011, 47, 5894–5896. [DOI] [PubMed] [Google Scholar]; j Matosevic S.; Lee G. J.; Baganz F. J. Biotechnol. 2011, 155, 320–329. [DOI] [PubMed] [Google Scholar]
- a Hwang B.-Y.; Chob B.-K.; Yun H.; Koteshwar K.; Kim B.-G. J. Mol. Catal. B: Enzym. 2005, 37, 47–55. [Google Scholar]; b Stewart J. D. Curr. Opin. Chem. Biol. 2001, 5, 120–129. [DOI] [PubMed] [Google Scholar]
- a Savile C. K.; Janey J. M.; Mundorff E. C.; Moore J. C.; Tam S.; Jarvis W. R.; Colbeck J. C.; Krebber A.; Fleitz F. J.; Brands J.; Devine P. N.; Huisman G. W.; Hughes G. J. Science 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]; b Mutti F. G.; Sattler J.; Tauber K.; Kroutil W. ChemCatChem 2011, 3, 109–111. [Google Scholar]
- Matcham W.; Bhatia M.; Lang W.; Lewis C.; Nelson R.; Wang A.; Lu W. Chimia 1999, 53, 584–589. [Google Scholar]
- a Cassimjee K. E.; Branneby C.; Abedi V.; Wells A.; Berglund P. Chem. Commun. 2010, 46, 5569–5571. [DOI] [PubMed] [Google Scholar]; b Yun H.; Yang Y.-H.; Cho B.-K.; Hwang B.-Y.; Kim B.-G. Biotechnol. Lett. 2003, 25, 809–814. [DOI] [PubMed] [Google Scholar]
- a Koszelewski D.; Lavandera I.; Clay D.; Guebitz G. M.; Rozzell D.; Kroutil W. Angew. Chem., Int. Ed. 2008, 47, 9337–9340. [DOI] [PubMed] [Google Scholar]; b Truppo M. D.; Rozzell J.; Moore J. C.; Turner N. J. Org. Biomol. Chem. 2009, 7, 395–398. [DOI] [PubMed] [Google Scholar]
- a Truppo M. D.; Rozzell J. D.; Turner N. J. Org. Process Res. Dev. 2010, 14, 234–237. [Google Scholar]; b Koszelewski D.; Clay D.; Rozzell D.; Kroutil W. Eur. J. Org. Chem. 2009, 2289–2292. [Google Scholar]; c Koszelewski D.; Lavandera I.; Clay D.; Rozzell D.; Kroutil W. Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar]; d Shin J.-S.; Kim B.-G. Biotechnol. Bioeng. 1999, 65, 206–211. [PubMed] [Google Scholar]
- a Smith E. B.; Chen B. H.; Hibbert E. G.; Kaulmann U.; Smithies K.; Galman J. L.; Baganz F.; Dalby P. A.; Hailes H. C.; Lye G. J.; Ward J. M.; Woodley J. M.; Micheletti M. Org. Process Res. Dev. 2010, 14, 99–107. [Google Scholar]; b Smithies K.; Smith M. E. B.; Kaulmann U.; Galman J. L.; Ward J. M.; Hailes H. C. Tetrahedron: Asymmetry 2009, 20, 570–574. [Google Scholar]; c Ingram C. U.; Bommer M.; Smith M. E. B.; Dalby P. A.; Ward J. M.; Hailes H. C.; Lye G. J. Biotechnol. Bioeng. 2006, 96, 559–569. [DOI] [PubMed] [Google Scholar]; d Iwasaki A.; Yamada Y.; Kizaki N.; Ikenaka Y.; Hasegawa J. Appl. Microbiol. Biotechnol. 2006, 69, 499–505. [DOI] [PubMed] [Google Scholar]
- Mutti F. G.; Kroutil W. Adv. Synth. Catal. 2012, 354, 3409–3413. [Google Scholar]
- Wang B.; Land H.; Berglund P. Chem. Commun. 2013, 49, 161–163. [DOI] [PubMed] [Google Scholar]
- a Mutti F. G.; Fuchs C. S.; Pressnitz D.; Turrini N. G.; Sattler J. H.; Lerchner A.; Skerra A.; Kroutil W. Eur. J. Org. Chem. 2012, 1003–1007. [Google Scholar]; b Mutti F. G.; Fuchs C. S.; Pressnitz D.; Sattler J. H.; Kroutil W. Adv. Synth. Catal. 2011, 353, 3227–3233. [Google Scholar]; c Koszelewski D.; Göritzer M.; Clay D.; Seisser B.; Kroutil W. ChemCatChem 2010, 2, 73–77. [Google Scholar]; d Andrade L. H.; Silva A. V.; Milani P.; Koszelewski D.; Kroutil W. Org. Biomol. Chem. 2010, 8, 2043–2051. [DOI] [PubMed] [Google Scholar]; e Clay D.; Koszelewski D.; Grischek B.; Gross J.; Lavandera I.; Kroutil W. Tetrahedron: Asymmetry 2010, 21, 2005–2009. [Google Scholar]; f Koszelewski D.; Lavandera I.; Clay D.; Rozzell D.; Kroutil W. Adv. Synth. Catal. 2008, 350, 2761–2766. [Google Scholar]
- Shin J.-S.; Yun H.; Jang J.-W.; Park I.; Kim B.-G. Appl. Microbiol. Biotechnol. 2003, 61, 463–471. [DOI] [PubMed] [Google Scholar]
- Kaulmann U.; Smithies K.; Smith M. E. B.; Hailes H. C.; Ward J. M. Enzyme Microb. Technol. 2007, 41, 628–637. [Google Scholar]
- Hanson R. L.; Davis B. L.; Chen Y.; Goldberg S. L.; Parker W. L.; Tully T. P.; Montana M. A.; Patel R. N. Adv. Synth. Catal. 2008, 350, 1367–1375. [Google Scholar]
- Park E.; Kim M.; Shin J.-S. Adv. Synth. Catal. 2010, 352, 3391–3398. [Google Scholar]
- Kawano S.; Ito N.; Yasohara Y. (Kaneka Corporation, Japan: ) WO 2007/139055 A1 20071206, 2007.
- Yun H.; Lim S.; Cho B.-K.; Kim B.-G. Appl. Environ. Microbiol. 2004, 70, 2529–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannuri S.; Kamat S. V.; Garcia A. R. M. (Cambrex North Brunswick Inc.) WO 2006/063336 A2 20060615, 2006.
- Höhne M.; Schätzle S.; Jochens H.; Robins K.; Bornscheuer U. T. Nature Chem. Biol. 2010, 6, 807–813. [DOI] [PubMed] [Google Scholar]
- Yamada Y.; Iwasaki A.; Kizaki N. (Kaneka Corporation; ) EP 0987332 A1, 2000.
- a Fuchs M.; Koszelewski D.; Tauber K.; Sattler J. H.; Banko W.; Holzer A. K.; Pickl M.; Kroutil W.; Faber K. Tetrahedron 2012, 68, 7691–7694. [Google Scholar]; b Fuchs M.; Koszelewski D.; Tauber K.; Kroutil W.; Faber K. Chem. Commun. 2010, 46, 5500–5502. [DOI] [PubMed] [Google Scholar]
- a Abdel-Hafez A. A.; Abdel-Wahab B. A. Bioorg. Med. Chem. 2008, 16, 7983–7991. [DOI] [PubMed] [Google Scholar]; b Colonge J.; Pouchol J.-M. Bull. Soc. Chim. Fr. 1962, 598–603. [Google Scholar]
- Koszelewski D.; Clay D.; Faber K.; Kroutil W. J. Mol. Catal. B: Enzym. 2009, 60, 191–194. [Google Scholar]
- a Turner N. J. Curr. Opin. Chem. Biol. 2010, 14, 115–121. [DOI] [PubMed] [Google Scholar]; b Parvulescu A. N.; De Vos P. A. J. D. E. Chem.—Eur. J. 2007, 13, 2034–2043. [DOI] [PubMed] [Google Scholar]; c Gruber C. C.; Lavandera I.; Faber K.; Kroutil W. Adv. Synth. Catal. 2006, 348, 1789–1805. [Google Scholar]
- Koszelewski D.; Clay D.; Rozzell D.; Kroutil W. Eur. J. Org. Chem. 2009, 2289–2292. [Google Scholar]
- Koszelewski D.; Pressnitz D.; Clay D.; Kroutil W. Org. Lett. 2009, 11, 4810–4812. [DOI] [PubMed] [Google Scholar]
- Truppo M. D.; Turner N. J.; Rozzell J. D. Chem. Commun. 2009, 2127–2129. [DOI] [PubMed] [Google Scholar]
- Koszelewski D.; Müller N.; Schrittwieser J. H.; Faber K.; Kroutil W. J. Mol. Catal. B: Enzym. 2010, 63, 39–44. [Google Scholar]
- a Organic Synthesis with Enzymes in Non-aqueous Media; Carrea G., Riva S., Eds.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]; b Serdakowski A. L.; Dordick J. S. Trends Biotechnol. 2008, 26, 48–54. [DOI] [PubMed] [Google Scholar]; c Klibanov A. M. Nature 2001, 409, 241–246. [DOI] [PubMed] [Google Scholar]
- Truppo M. D.; Strotman H.; Hughes G. ChemCatChem 2012, 4, 1071–1074. [Google Scholar]
- Buffer saturated EtOAc was previously employed in: Yun H.; Kim J.; Kinnera K.; Kim B.-G.. Biotechnol. Bioeng. 2006, 93, 391–395. [DOI] [PubMed] [Google Scholar]
- Selected recent examples:; a Lafrance M.; Roggen M.; Carreira E. M. Angew. Chem., Int. Ed. 2012, 51, 3470–3473. [DOI] [PubMed] [Google Scholar]; b Imm S.; Bähn S.; Zhang M.; Neubert L.; Neumann H.; Klasovsky F.; Pfeffer J.; Haas T.; Beller M. Angew. Chem., Int. Ed. 2011, 50, 7599–7603. [DOI] [PubMed] [Google Scholar]; c Pingen D.; Müller C.; Vogt D. Angew. Chem., Int. Ed. 2010, 49, 8130–8133. [DOI] [PubMed] [Google Scholar]; d Imm S.; Bähn S.; Neubert L.; Neumann H.; Beller M. Angew. Chem., Int. Ed. 2010, 49, 8126–8129. [DOI] [PubMed] [Google Scholar]; e Kawahara R.; Fujita K.-i.; Yamaguchi R. J. Am. Chem. Soc. 2010, 132, 15108–15111. [DOI] [PubMed] [Google Scholar]; f Saidi O.; Blacker A. J.; Farah M. M.; Marsden S. P.; Williams J. M. J. Angew. Chem., Int. Ed. 2009, 48, 7375–7378. [DOI] [PubMed] [Google Scholar]; g Blank B.; Michlik S.; Kempe R. Chem.—Eur. J. 2009, 15, 3790–3799. [DOI] [PubMed] [Google Scholar]; h Gunanathan C.; Milstein D. Angew. Chem., Int. Ed. 2008, 47, 8661–8664. [DOI] [PubMed] [Google Scholar]
- Sattler J. H.; Fuchs M.; Tauber K.; Mutti F. G.; Faber K.; Pfeffer J.; Haas T.; Kroutil W. Angew. Chem., Int. Ed. 2012, 51, 9156–9159. [DOI] [PubMed] [Google Scholar]
- Fuchs M.; Tauber K.; Sattler J. H.; Lechner H.; Pfeffer J.; Kroutil W.; Faber K. RSC Adv. 2012, 2, 6262–6265. [Google Scholar]
- a Alexeeva M.; Enright A.; Dawson M. J.; Mahmoudian M.; Turner N. J. Angew. Chem., Int. Ed. 2002, 41, 3177–3180. [DOI] [PubMed] [Google Scholar]; b Carr R.; Alexeeva M.; Enright A.; Eve T. S. C.; Dawson M. J.; Turner N. J. Angew. Chem., Int. Ed. 2003, 42, 4807–4810. [DOI] [PubMed] [Google Scholar]; c Turner N. J. Chem. Rev. 2011, 111, 4073–4087. [DOI] [PubMed] [Google Scholar]
- For reviews on ammonia lyases, see:; a Turner N. J. Curr. Opin. Chem. Biol. 2011, 15, 234–240. [DOI] [PubMed] [Google Scholar]; b de Villiers M.; Puthan Veetil V.; Raj H.; de Villiers J.; Poelarends G. J. ACS Chem. Biol. 2012, 7, 1618–1628. [DOI] [PubMed] [Google Scholar]; c MacDonald M. J.; D’Cunha G. B. Biochem. Cell. Biol. 2007, 85, 273–282. [DOI] [PubMed] [Google Scholar]; d Cooke H. A.; Christianson C. V.; Bruner S. D. Curr. Opin. Chem. Biol. 2009, 13, 460–468. [DOI] [PubMed] [Google Scholar]; e Asano Y.; Kato Y.; Levy C.; Baker P.; Rice D. Biocatal. Biotransform. 2004, 22, 131–138. [Google Scholar]; f Poppe L.; Rétey J. Curr. Org. Chem. 2003, 7, 1297–1315. [Google Scholar]; g Poppe L.; Paizs C.; Kovács K.; Irimie F.-D.; Vértessy B. Methods Mol. Biol. 2012, 794, 3–19. [DOI] [PubMed] [Google Scholar]
- a Pohl M., Liese A.. Industrial processes using lyases for C–C, C–N and C–O bond formation. In Patel R. N., Ed. Biocatalysis in the Pharmaceutical and Biotechnology Industries. CRC Press: Boca Raton, FL, 2007; pp 661–676. [Google Scholar]
- Raj H.; Szymański W.; de Villiers J.; Rozeboom H. J.; Puthan Veetil V.; Reis C. R.; de Villiers M.; Dekker F. J.; de Wildeman S.; Quax W. J.; Thunnissen A.-M. W. H.; Feringa B. L.; Janssen D. B.; Poelarends G. J. Nature Chem. 2012, 4, 478–484. [DOI] [PubMed] [Google Scholar]
- a Akhtar M.; Botting N. P.; Cohen M. A.; Gani D. Tetrahedron 1987, 43, 5899–5908. [Google Scholar]; b Akhtar M.; Cohen M. A.; Gani D. Tetrahedron Lett. 1987, 28, 2413–2416. [Google Scholar]; c Gulzar M. S.; Akhtar M.; Gani D. J. Chem. Soc., Perkin Trans. 1 1997, 649–655. [Google Scholar]
- Simon R. C.; Grischek B.; Zepeck F.; Steinreiber A.; Belaj F.; Kroutil W. Angew. Chem., Int. Ed. 2012, 51, 6713–6716. [DOI] [PubMed] [Google Scholar]
- Simon R. C.; Zepeck F.; Kroutil W. Chem.—Eur. J. 2013, 19, 2859–2865. [DOI] [PubMed] [Google Scholar]
- Stöckigt J.; Antonchick A. P.; Wu F.; Waldmann H. Angew. Chem., Int. Ed. 2011, 50, 8538–8564. [DOI] [PubMed] [Google Scholar]
- a Bonamore A.; Rovardi I.; Gasparrini F.; Baiocco P.; Barba M.; Molinaro C.; Botta B.; Boffia A.; Macone A. Green Chem. 2010, 12, 1623–1627. [Google Scholar]; b Samanai N.; Facchini P. J. Planta 2001, 213, 898–906. [DOI] [PubMed] [Google Scholar]
- Ruff B. M.; Bräse S.; O’Connor S. E. Tetrahedron Lett. 2012, 53, 1071–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesnot T.; Gershater M. C.; Ward J. M.; Hailes H. C. Adv. Synth. Catal. 2012, 354, 2997–3008. [Google Scholar]
- a Stöckigt J.; Barleben L.; Panjikar S.; Loris E. A. Plant Physiol. Biochem. 2008, 46, 340–355. [DOI] [PubMed] [Google Scholar]; b Yang L.; Zou H.; Zhu H.; Ruppert M.; Gong J.; Stöckigt J. Chem. Biodiversity 2010, 7, 860–870. [DOI] [PubMed] [Google Scholar]; c Wu F.; Zhu H.; Sun L.; Rajendran C.; Wang M.; Ren X.; Panjikar S.; Cherkasov A.; Zou H.; Stöckigt J. J. Am. Chem. Soc. 2012, 134, 1498–1500. [DOI] [PubMed] [Google Scholar]
- Carr R.; Alexeeva M.; Dawson M. J.; Gotor-Fernandez V.; Humphrey C. E.; Turner N. J. ChemBioChem 2005, 6, 637–639. [DOI] [PubMed] [Google Scholar]
- a Köhler V.; Bailey K. R.; Znabet A.; Raftery J.; Helliwell M.; Turner N. J. Angew. Chem., Int. Ed. 2010, 49, 2182–2184. [DOI] [PubMed] [Google Scholar]; b Znabet A.; Polak M. M.; Janssen E.; de Kanter F. J. J.; Turner N. J.; Orru R. V. A.; Ruijter E. Chem. Commun. 2010, 46, 7918–7920. [DOI] [PubMed] [Google Scholar]
- Bailey K. R.; Ellis A. J.; Reiss R.; Snape T. J.; Turner N. J. Chem Commun. 2007, 3640–3642. [DOI] [PubMed] [Google Scholar]
- Dittrich H.; Kutchan T. M. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 9969–9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler A.; Hartner F.; Kutchan T. M.; Glieder A.; Macheroux P. J. Biol. Chem. 2006, 281, 21276–21285. [DOI] [PubMed] [Google Scholar]
- a Winkler A.; Łyskowski A.; Riedl S.; Puhl M.; Kutchan T. M.; Macheroux P.; Gruber K. Nature Chem. Biol. 2008, 4, 739–741. [DOI] [PubMed] [Google Scholar]; b Gaweska H. M.; Roberts K. M.; Fitzpatrick P. F. Biochemistry 2012, 51, 7342–7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler A.; Motz K.; Riedl S.; Puhl M.; Macheroux P.; Gruber K. J. Biol. Chem. 2009, 284, 19993–20001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schrittwieser J. H.; Resch V.; Sattler J. H.; Lienhart W.-D.; Durchschein K.; Winkler A.; Gruber K.; Macheroux P.; Kroutil W. Angew. Chem., Int. Ed. 2011, 50, 1068–1071. [DOI] [PubMed] [Google Scholar]; b Schrittwieser J. H.; Resch V.; Wallner S.; Lienhart W.-D.; Sattler J. H.; Resch J.; Macheroux P.; Kroutil W. J. Org. Chem. 2011, 76, 6703–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resch V.; Schrittwieser J. H.; Wallner S.; Macheroux P.; Kroutil W. Adv. Synth. Catal. 2011, 13, 2377–2383. [Google Scholar]
- Resch V.; Lechner H.; Schrittwieser J. H.; Wallner S.; Gruber K.; Macheroux P.; Kroutil W. Chem.—Eur. J. 2012, 18, 13173–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]