

Abstract
Objectives
To define the audiologic and otologic phenotype of Hutchinson-Gilford Progeria syndrome (HGPS).
Study Design
Prospective case series.
Methods
Fifteen patients with HGPS were enrolled in a prospective natural history study; fourteen were evaluated in the neurotology clinic and eleven received audiologic evaluations. The physical exam and audiologic findings of these patients were reviewed to define an otologic and audiologic phenotype for HGPS in the largest series of subjects in the literature.
Results
All patients were noted to have stiff auricular cartilages, small or absent lobules and hypoplasia of the lateral soft tissue portion of the external ear canal leading to a shortened canal. Ten of 14 patients (71%) had dry cerumen impaction and four of 14 patients (29%) reported a history of recurrent otitis media. Nineteen of 22 ears (86.4%) demonstrated low frequency conductive hearing loss in the 250 Hz to 500 Hz range. Sixteen of 22 ears (73%) had type A tympanograms; three of 22 ears (14%) displayed bimodal or "W" peaked tympanograms; two of 22 ears (9%) had type B tympanograms; one of 22 ears (4%) had a type C tympanogram. Nine of 10 patients had distortion product otoacoustic emissions consistent with normal peripheral hearing sensitivity.
Conclusions
HGPS is caused by a mutation in the LMNA gene resulting in the production of an abnormal nuclear protein; this in turn affects nuclear structure and function. Patients with HGPS have characteristic otologic features due to cartilaginous and subcutaneous tissue abnormalities and typically demonstrate low frequency conductive hearing loss despite largely normal tympanometry. It is important to be aware of these conditions in managing these patients.
Keywords: Hutchinson-Gilford Progeria Syndrome, hearing loss, Progeria, middle ear, external ear
Introduction
Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare entity, occurring in approximately 1 in 4 million people.1 It is characterized by features that are often associated with advanced age. The condition was initially described by Jonathan Hutchinson in 1886 as “Congenital Absence of the Hair and Mammary Glands” and further characterized by Hastings Gilford in 1897. The name ‘progeria’, first coined by Gilford, literally means ‘prematurely old’ and is derived from ancient Greek.1 Children afflicted by HGPS have many distinctive features including alopecia, failure to thrive, short stature, lipodystrophy, joint abnormalities and facial features resembling elderly persons.1, 2 Infants are typically normal at birth, but rapidly develop characteristic features of the disease. Diagnosis is usually made by two to three years of age.1 Death occurs at an average age of 13 years, and is most often secondary to progressive cardiovascular disease.2, 3 HGPS is a sporadic autosomal dominant condition now known to be caused by a de novo point mutation in the lamin A (LMNA) gene4; most frequently, this is a nucleotide substitution of GGC to GGT in codon 608 of exon 11 of the LMNA gene.4, 5 This does not result in an amino acid change, but activates a cryptic splice site resulting in the production of abnormal lamin A (termed “progerin” or LAΔ50).4, 5 Lamin A plays an important role in the structural integrity and shape of the inner nuclear membrane; therefore, accumulation of the mutant protein leads to defects in nuclear architecture and function. This is hypothesized to be the cause of the premature aging phenotype of HGPS patients.5
Audiologic findings in HGPS have been primarily limited to isolated case reports. Some speculation has focused on whether or not presbycusis is part of the phenotype, and some authors have reported normal hearing in HGPS.1, 6
We systematically evaluated 15 patients as part of a larger scale natural history study at the National Institutes of Health in Bethesda, MD. This evaluation included otologic physical examination as well as audiologic assessment including pure tone audiometry, middle ear immittance testing, and otoacoustic emissions. Partial results of this study were published in a multidisciplinary review by Merideth et al.2
Materials and Methods
Fifteen patients were enrolled in a prospective natural history study approved by the institutional review board of the National Human Genome Research Institute. Of these patients, 14 (6 males and 8 females), aged 1–12 years (median, 7 years) had a neurotologic examination. Six males and five females aged 5–16 years (median 8 years) had comprehensive audiologic evaluations. Full hearing assessment could not be completed on the remaining four patients due to intolerance or non-cooperation during test procedures.
Audiometric evaluation consisted of pure-tone thresholds by air and bone conduction, speech audiometry, 226 Hz tympanometry, acoustic reflexes when tolerated, and distortion product otoacoustic emissions (DPOAE). When more than one audiogram was available, the more comprehensive of the two, or the one done in the absence of known pathology (e.g., otitis media) was included in the data analysis. Hearing thresholds ≤15 dB HL were considered within normal limits. Thresholds between 16 and 25 dB HL were classified as slight hearing loss, thresholds between 26 and 40 dB HL as mild hearing loss, and thresholds of 41–55 dB HL as moderate hearing loss.7 Air-bone gaps >10 dB were considered significant, even for cases with normal air conduction thresholds.
Tympanograms were classified according to Jerger’s criteria as type A, As, B, or C using age-appropriate normative data.8, 9 When more than one compliance peak was present, the tympanogram was described as notched (“W-shaped”).
DPOAEs were recorded in one-quarter octave bands using an Otodynamics Echoport Otoacoustic Emission System (Herts, United Kingdom). DPOAEs were interpreted as present when the signal-to-noise ratio exceeded 6 dB, and absent for lower signal-to-noise ratios.
Results
Clinical features
All of our patients’ pinnae had stiff cartilages and small or shortened lobules (Figure 1). All also exhibited hypoplasia of the lateral soft tissue portion of the external auditory canal (EAC) leading to a shortened canal. Ten of fourteen patients (71%) had some degree of firm cerumen impaction obstructing visualization of the tympanic membrane that often required manual removal or moistening drops. Table I provides a summary of physical exam findings. Four of fourteen patients (29%) reported a history of recurrent otitis media and three patients had evidence of middle ear effusion at the time of examination. In addition to their characteristic otologic features, patients displayed a lack of subcutaneous fat resulting in prominent scalp veins, narrowed nasal dorsa and stiffened nasal cartilages. The patients’ facies resembled an ‘inverted triangle’ due to a disproportionately hypoplastic midface and mandible, prominent eyes and thin lips (Figure 2).
Figure 1.

A typical Hutchinson-Gilford progeria syndrome pinna with stiff cartilages and a small lobule.
Table I.
Physical Characteristics of HGPS Patients
| Patient | Age | Absent/small lobule | Stiff cartilage | Shortened EAC | Cerumen impaction |
|---|---|---|---|---|---|
| 1 | 8 | yes | yes | yes | yes |
| 2 | 8 | yes | yes | yes | yes |
| 3 | 6 | yes | yes | yes | yes |
| 4 | 5 | yes | yes | yes | yes |
| 5 | 7 | yes | yes | yes | no |
| 6 | 10 | yes | yes | yes | yes |
| 7 | 5 | yes | yes | yes | yes |
| 8 | 7 | yes | yes | yes | no |
| 9 | 12 | yes | yes | yes | yes |
| 10 | 16 | - | - | - | - |
| 11 | 8 | yes | yes | yes | yes |
| 12 | 2 | yes | yes | yes | no |
| 13 | 2 | yes | yes | yes | yes |
| 14 | 1 | yes | yes | yes | no |
| 15 | 2 | yes | yes | yes | yes |
Figure 2.
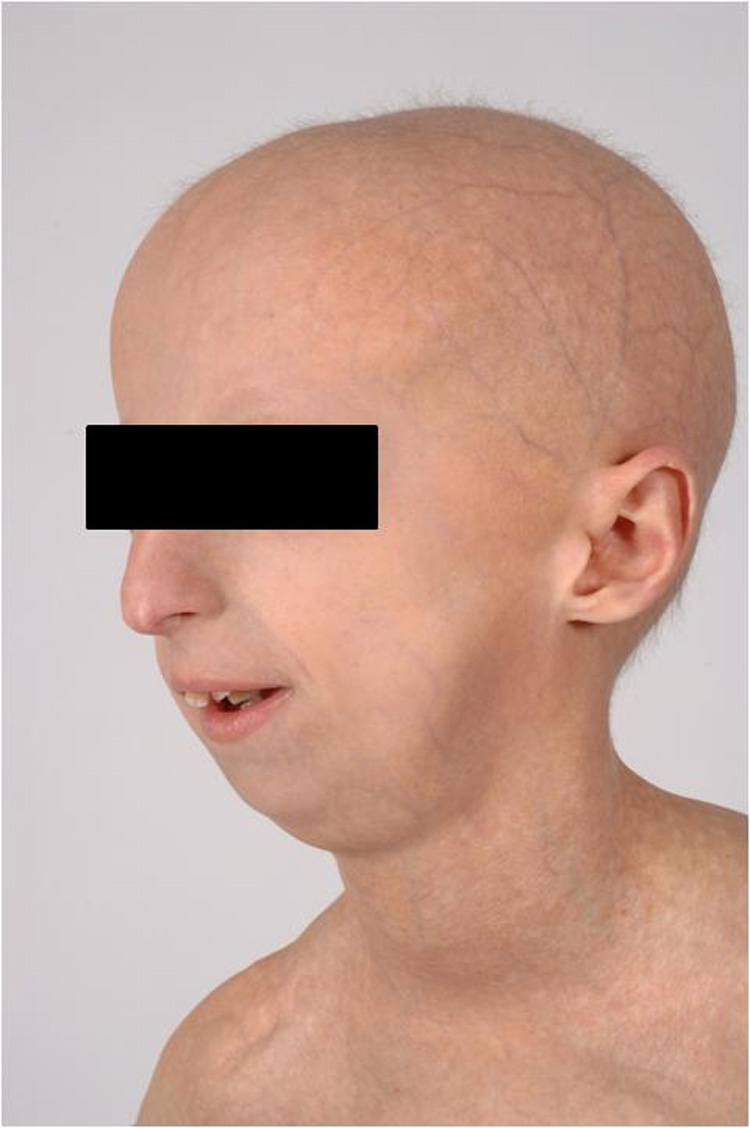
A typical Hutchinson-Gilford progeria syndrome “inverted triangle” face with a disproportionately hypoplastic midface and mandible.
Pure tone audiometry
Audiometric findings for all participants are shown in Figure 3. Conductive hearing loss ranging from slight to moderate (20–55 dB HL) was observed in the low to mid frequencies for 19 of 22 ears (86%). Air-bone gaps greater than 10 dB were present at 250 and 500 Hz in 20 of 22 ears (91%), 1000 Hz in 11 of 22 ears (50%), 2000 Hz in 5 of 22 ears (23%), and 4000 Hz in 4 of 22 ears (18%). A mixed hearing loss involving at least one frequency was observed in 2 of 22 ears.
Figure 3.
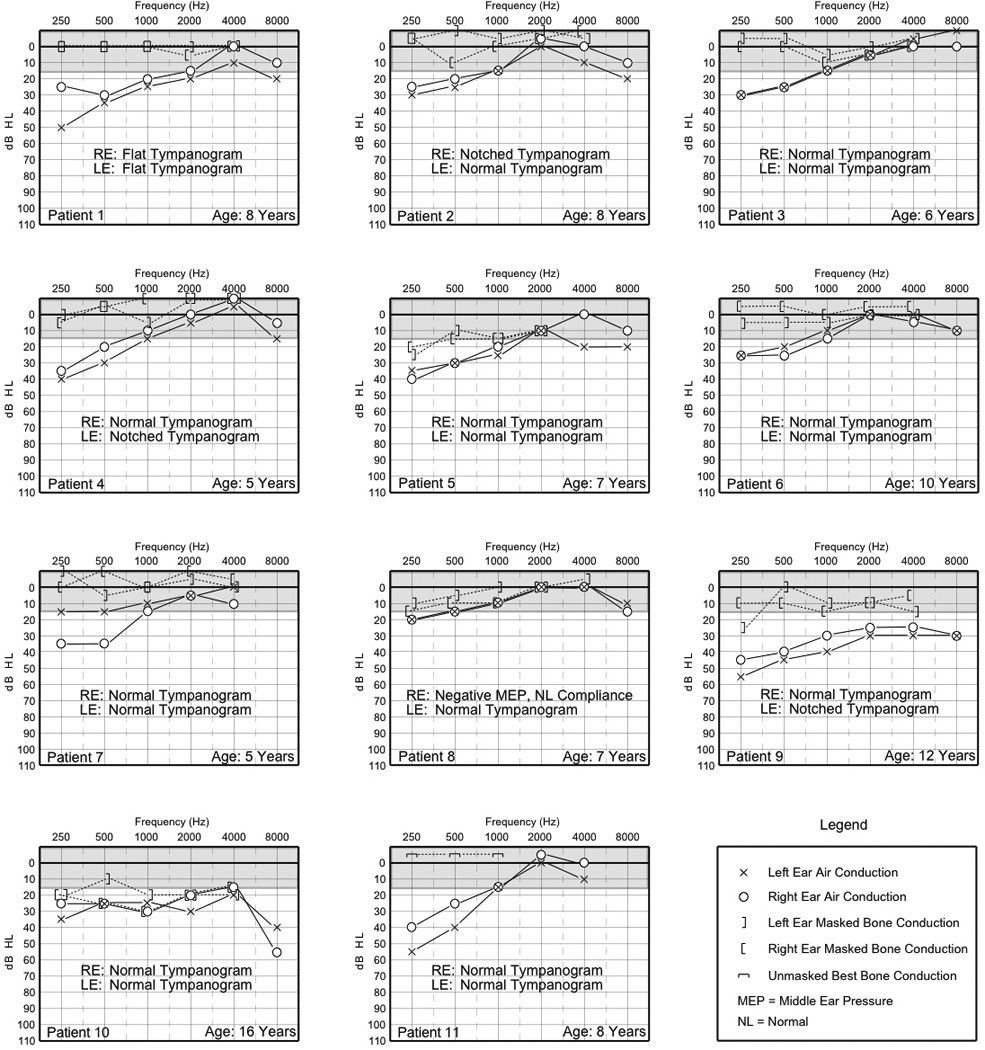
Individual audiograms and tympanometry of 11 patients. RE=right ear, LE=left ear.
Sensorineural hearing loss was observed in both ears of a single patient (patient 10) who was the oldest in this group (age 16 years). His sensorineural hearing loss ranged from a slight to mild degree for nearly the entire frequency range from 250 to 4000 Hz before sloping to a moderate hearing loss at 8000 Hz in his right ear. There was a low frequency conductive hearing loss and a slight to mild sensorineural hearing loss for 1000 to 8000 Hz in the left ear. No high frequency hearing loss of sensorineural origin was observed in any of the remaining 10 patients undergoing comprehensive audiologic evaluations.
Tympanometry, Acoustic Reflexes and Otoacoustic Emissions
Normal peak compensated static compliance and middle ear pressure (type A) were documented in 15 of 22 ears (68%). Of these, 14 ears (93%) exhibited pure-tone thresholds with a significant air-bone gap for at least one frequency other than 250 Hz. Three ears (14%) had notched (“W-shaped”) 226-Hz tympanograms. There was reduced static compliance with normal middle ear pressure (Type As) for a single ear (4.5%). Flat tympanograms (Type B) occurred in both ears of a single patient (2 of 22 ears, 9%) and negative middle ear pressure (Type C) was documented in a single ear (4.5%). Both the patient with bilateral flat tympanograms and the patient with negative middle ear pressure had evidence of middle ear fluid on physical exam.
Five patients successfully underwent acoustic reflex testing. Of note, most of the patients were unable to tolerate this testing due to ear canal discomfort from the probe. Three of five patients (60%) had absent acoustic reflexes, all of whom exhibited conductive hearing loss on pure-tone audiometry.
Otoacoustic emission testing was performed in nine patients, eight of whom had DPOAE for the mid- to high frequencies. In the remaining case, excessive patient noise precluded observation of otoacoustic emissions.
Discussion
There are many known otolaryngologic manifestations of HGPS. Unusual facial features include decreased subcutaneous fat with significant wrinkling of the skin, prominent eyes (due to loss of periorbital fat), thin nasal bridge with a flattened and broad nasal tip, circumoral cyanosis and prominent ears with absent ear lobes.1, 2, 10 Patients have significant alopecia, including absence of eyebrows and eyelashes, and prominent scalp veins.1 Affected patients also undergo osteolysis of all bones formed by membranous ossification; this includes the facial bones and skull. Osteolysis of the mandible leads to shortening of the horizontal and ascending rami and progressive retrognathia; osteolysis of the maxilla leads to an ogival (high arched) palate.1, 10 The subsequent small size of the mandible and maxilla results in several dental abnormalities including crowding of the teeth, double rows of teeth, delayed tooth eruption and hypodontia.1, 2, 10
Our physical exam findings mirror those that have been previously described. In addition to the characteristically small or absent lobule, we noted that these patients also have a shortened ear canal and stiff auricular cartilages. The outer one third of the EAC canal consists of an incomplete cylinder of elastic cartilage surrounded by dense fibrous tissue and subcutaneous fat. The inner, bony canal is composed of a complete cylinder of bone.11 There are also significant differences in the skin of the cartilaginous and bony EAC. The skin of the bony canal is very thin and there are few hairs and sebaceous glands. This is in contrast to the skin of the cartilaginous canal, which is thick and has numerous hairs as well as sebaceous and ceruminous apocrine glands.11, 12 In our patients, the outer one third of the canal was consistently hypoplastic. We know that the apocrine glands found in the ear canal are similar to those found in the breast.13 Since patients with HGPS are known to have hypoplastic breast tissue, it is not surprising that the apocrine glands of the external auditory canal could also be hypoplastic; this might lead to the accumulation of a dry, hard wax present in most patients. The thin skin in the remaining bony portion of the canal can lead to discomfort during otoscopic examination and probe placement for audiologic measures in patients with HGPS. Lack of subcutaneous fat in the scalp of HGPS patients can also make bone conduction testing painful and challenging to perform. It is important for health care providers to check regularly for cerumen impaction and provide extra care when examining the ears and performing audiologic tests on individuals with HGPS.
Because HGPS is a rare genetic disorder, its associated audiologic features have not been well documented in the literature. A case published by Nelson in 1962 and later in 1965 describes a patient with a progressive, moderate, bilateral mixed hearing loss. Nelson states that, although the patient’s initial diagnosis was “senile deafness,” a presbycusic pattern of hearing loss was not present.14, 15 In 1981, Baker et al. reported a case of a 9-year-old girl with HGPS who was briefly mentioned to have a conductive hearing loss.16 In 1993, Hall and Denneny reported a case of a five-year-old girl with HGPS in whom a mild-to-moderate low-to-mid frequency conductive hearing loss was documented. Tympanometry (Type A) was normal bilaterally, but acoustic reflexes were absent.16 The patient subsequently underwent middle ear explorations, during which fibrous adhesions were noted on the stapes footplate and between the incudostapedial joint and the tympanic membrane. After these fibrous adhesions were lysed, the patient showed significant closure of the air-bone gap in the right ear and improvement only at 2000 Hz in the left ear. This patient also received auditory brainstem response (ABR) testing at the time of surgery that supported the presence of a conductive hearing loss, but also revealed delayed interpeak latencies in waves I to V bilaterally; the authors hypothesized that may be due to the early central nervous system effects of cerebral vascular disease.17
Our results extend the observations of Merideth et al. and represent the first and largest cohort describing the otologic and audiologic phenotype of HGPS. The presence of a low-frequency conductive hearing loss in nearly all of our patients most likely represents the effect of a middle ear abnormality. This pattern of hearing loss in the presence of mostly normal 226-Hz tympanometry is suggestive of a stiffening pathology such as otosclerosis. The presence of a notched W-shaped 226-Hz tympanogram for three ears is typical of that observed in a mass controlled middle ear system resulting from pathologies such as tympanosclerosis or excessive scar-tissue formation.18
The presence of otoacoustic emissions is consistent with normal peripheral hearing sensitivity in the mid- to high frequencies and adequate backward transmission through the middle ear system. This was the case despite low frequency conductive hearing loss that notably occurred in conjunction with essentially normal tympanometry.
Our audiometric findings are similar to those in the case report by Hall and Denneny, where abnormal fibrous adhesions involving the stapes footplate and the incudostapedial joint were found. This could explain the increased stiffness and/or increased mass of the middle ear system. Patients with HGPS have joint contractures and decreased joint mobility2, so perhaps the same process that leads to lack of joint mobility in the extremities also causes stiffness of the ossicular chain joints, tendons and ligaments. Moreover, lack of normal lamin A has been shown to cause accelerated osteoclastogenesis and impaired osteoblastogenesis leading to premature osteoporosis in HGPS patients.19 Clayton et al. reported a clinical association between osteoporosis and otosclerosis, suggesting these processes may be related.20
Our cross sectional data also attempt to address whether there is a progressive pattern of hearing loss in patients with HGPS. Although comprehensive audiometric data could not be obtained on any patient younger than 5 years old, a clear progression of the conductive hearing loss was not seen with age. High-frequency sensorineural hearing loss was observed only in one of our patients (patient 10), who was the oldest in this cohort. This raises the possibility that a presbycusic pattern of hearing loss may occur. With advanced age, the pathogenesis involved in HGPS may also affect the peripheral and central auditory system and lead to hearing loss similar to neural and sensory presbycusis. A further longitudinal study of HGPS subjects would be necessary to delineate this.
Given the largely normal otoacoustic emissions in the mid- to high frequencies and the tendency of many newborn and school-based hearing screenings to focus on the mid- and high frequencies, the hearing loss in HGPS may be missed by routine screening. Therefore it is important for children with HGPS to undergo regular, formal audiologic testing. Although the degree of hearing loss in most of our patients is mild, there is evidence that even minimal hearing loss can have an adverse impact on social, behavioral and academic performance in pediatric patients.21, 22, 23 Auditory communication accommodations such as preferential seating in the classroom and FM auditory listening devices may be helpful.24
Twenty-nine percent of our patients (4 of 14) reported a history of recurrent otitis media. The literature suggests a 20–30% rate of recurrent otitis media (OM) in the general population in the first several years of life, with rates decreasing after three years of age.25 Our patients with recurrent otitis media ranged in age from 7 to 12 years, slightly older than the typical group with recurrent OM. One explanation may be that the stiffening of elastic cartilage in the pinna also occurs in the elastic cartilage of the eustachian tube, preventing its normal function of equalizing middle ear pressure. In addition, the osteolysis of facial bones leading to a hypoplastic maxilla and high arched palate may result in eustachian-tube dysfunction and predispose patients with HGPS to recurrent otitis media with effusion. Hence, HGPS patients should be monitored on a regular basis for the presence of middle ear disease.
Conclusion
It is important for providers caring for HGPS patients to be aware of the expected manifestations of this disease. Our study has demonstrated that abnormal lamin A leads to specific otologic and audiologic abnormalities; HGPS patients should be monitored and managed accordingly.
Acknowledgements
This work was supported by the intramural research programs of the National Human Genome Research Institute and the National Institute on Deafness and Other Communication Disorders.
The authors thank Kelly King, AuD and Carter van Waes, MD PhD for critical review of this manuscript.
Footnotes
Financial disclosure: none
Conflict of interest: none
Part of this data was presented at the Triological Society Annual Meeting, Las Vegas, NV, April 30th-May 1st, 2010.
References
- 1.Hennekam RCM. Hutchinson-Gilford Progeria Syndrome: Review of the Phenotype. Am J Med Genet Part A. 2006;140A:2603–2624. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- 2.Merideth MA, Gordon LB, Clauss S, et al. Phenotype and Course of Hutchinson-Gilford Progeria Syndrome. The New England Journal of Medicine. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olive M, Harten I, Mitchell R, et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Atheroscler Thromb Vasc Biol. 2010;30(11):2301–2309. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eriksson M, Brown WT, Gordon L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–297. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldman RD, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBusk RL. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr. 1972;80:697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 7.Clark J. Uses and abuses of hearing loss classification. ASHA. 1981;23:493–500. [PubMed] [Google Scholar]
- 8.Margolis RH, Heller JW. Screening tympanometry: criteria for medical referral. Audiology. 1987;26:197–208. doi: 10.3109/00206098709081549. [DOI] [PubMed] [Google Scholar]
- 9.Jerger JF. Clinical experience with impedance audiometry. Archives of Otolaryngology. 1970;92:311–324. doi: 10.1001/archotol.1970.04310040005002. [DOI] [PubMed] [Google Scholar]
- 10.Gordon L, McCarten K, Giobbie-Hurder A. Disease Progession in Hutchinson-Gilford Progeria Syndrome: Impact on Growth and Development. Pediatriacs. 2007;120:824–833. doi: 10.1542/peds.2007-1357. [DOI] [PubMed] [Google Scholar]
- 11.Kelly K, Mohs D. The External Auditory Canal: Anatomy and Physiology. Otolaryng. Clinics of North America. 1996;25(2):725–740. [PubMed] [Google Scholar]
- 12.Cummings C, Haughy B, Thomas JR. Cummings Otolaryngology: Head and Neck Surgery. 4th edition 2005. [Google Scholar]
- 13.Toyoda Y, Sakuri A, Mitani Y, et al. Earwax, osmidrosis and breast cancer: why does one SNP (538G>A) in the human ABC transporter ABCC11 gene determine earwax type? FASEB J. 2009;23:2001–2013. doi: 10.1096/fj.09-129098. [DOI] [PubMed] [Google Scholar]
- 14.Nelson M. Progeria: Audiologic aspects. Archives of Pediatrics. 1962;79:87–90. [PubMed] [Google Scholar]
- 15.Nelson M. Progeria: Audiologic aspects. Final Report of a Case. Annals of Otology, Rhinology and Laryngology. 1965;74:376–385. doi: 10.1177/000348946507400208. [DOI] [PubMed] [Google Scholar]
- 16.Baker P, Baba N, Boesel C. Cardiovascular Abnormalities in Progeria. Arch Pathol Med. 1981;105:384–386. [PubMed] [Google Scholar]
- 17.Hall JW, Denneny JC. Audiologic and Otolaryngologic Findings in Progeria: Case Report. J Am Acad Audiol. 1993;4:116–121. [PubMed] [Google Scholar]
- 18.Shanks J, Lilly D, Margolis R, Wiley T, Wilson R. Tympanometry: ASHA working group on aural acoustic-immittance measurements committee on audiologic evalaluation. Journal of Speech and Hearing Disorders. 1988:354–377. [PubMed] [Google Scholar]
- 19.Rauner M, Sipos W, Goettsch C. Inhibition of Lamin A/C Attenuates Osteoblast Differentiation and Enhances RANKL-Dependent Osteoclastogenesis. J Bone and Mineral Research. 2009;24(1):78–85. doi: 10.1359/jbmr.080902. [DOI] [PubMed] [Google Scholar]
- 20.Clayton A, Mikulec A, Mikulec K. Association of osteoporosis and otosclerosis in women. J Laryngology Otology. 2004;118(8):617–621. doi: 10.1258/0022215041917790. [DOI] [PubMed] [Google Scholar]
- 21.Penn T, Grantham DW, Gravel S. Simulated Conductive Hearing Loss in Children. J Am Acad Audiol. 2004;15:300–310. doi: 10.3766/jaaa.15.4.4. [DOI] [PubMed] [Google Scholar]
- 22.Crandell C. Speech Recognition in Noise by Children with Minimal Sensorineural Hearing Loss. Ear & Hearing. 1993;14(3):210–216. doi: 10.1097/00003446-199306000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Bess FH, Dodd-Murphy J, Parker RA. Children with minimal sensorineural hearing loss: prevalence, educational performance and functional status. Ear Hear. 1998;19:339–354. doi: 10.1097/00003446-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Medel L. Speech Perception Benefits from Sound Field FM Amplification. Am J Audiol. 2003;12:114–124. doi: 10.1044/1059-0889(2003/019). [DOI] [PubMed] [Google Scholar]
- 25.Pichichero M. Recurrent and Persistent Otitis Media. Pediatr Inf Dis J. 2000;19:911–916. doi: 10.1097/00006454-200009000-00034. [DOI] [PubMed] [Google Scholar]