

Abstract
Microprobe X-ray absorption near edge structure (μ-XANES) measurements were used to determine directly, for the first time, the oxidation state of intracellular plutonium in individual 0.1 μm2 areas within single rat pheochromocytoma cells (PC12). The living cells were incubated in vitro for 3 hours in the presence of Pu added to the media in different oxidation states (Pu(III), Pu(IV), and Pu(VI)) and in different chemical forms. Regardless of the initial oxidation state or chemical form of Pu presented to the cells, the XANES spectra of the intracellular Pu deposits was always consistent with tetravalent Pu even though the intracellular milieu is generally reducing.
INTRODUCTION
The oxidation state of a metallic element establishes its chemical behavior. It defines a given metal ion’s coordination preferences and reactivity, which in biological systems determine the toxicity and specific utility of the metal ion as well as its bioavailability, biochemical trafficking pathways, and mechanisms for accumulation or excretion. As a consequence, organisms work to control the oxidation states of metal ions. Knowing the oxidation state speciation of metallic elements in particular environments is central to understanding and manipulating the behavior of both essential and toxic metals in biological systems.
The radioactive metal plutonium is a non-essential, highly toxic, anthropogenic element with a redox chemistry that is among the most complex in the periodic table. Pu is commonly encountered in four oxidation states (+3, +4, +5, and +6) under environmentally relevant conditions, and the standard reduction potentials that link them are similar.1 Consequently, it is possible to have a single solution containing significant amounts of Pu(III), Pu(IV), Pu(V) and Pu(VI).2 In addition, the individual redox potentials suggest that multiple Pu oxidation states are likely accessible at biologically relevant reduction potentials (Eh) and pH values.3 For example, Pu(IV), Pu(V), and Pu(VI) all are involved in the sorption of Pu on the surface of B. sphaericus bacteria,4 and soluble complexes of Pu(IV) can be rapidly reduced to Pu(III) by metal-reducing bacteria.5
Despite the facts that Pu is incorporated into living cells6 and that the oxidation state is critical to determining the chemistry and reactivity of plutonium, its redox speciation has never been directly measured in mammalian cells. Nevertheless, it is generally thought that Pu exists as Pu(IV) in vivo.7,8 This belief is founded on the general stability of tetravalent Pu in solutions near neutral pH under laboratory conditions7,9 and the indirect evidence of the biodistribution and uptake biokinetics of plutonium administered to animals in different oxidation states as compared to other elements.6,10 These studies assumed, however, that Pu reaches essentially a single, thermodynamically-controlled oxidation state in vivo, that redox cycling of Pu does not impact its biodistribution, and that naturally occurring ligands do not significantly stabilize the other oxidation states of Pu relative to Pu(IV) in vivo.
The well-established redox activity of Pu and the pH and Eh of various cellular compartments suggest that other oxidation states, particularly Pu(III), could be important to the cellular uptake and retention. In contrast to blood plasma or fluids in the lungs, the intracellular milieu encountered by Pu in mammalian cells is generally reducing due to the presence of millimolar concentrations of glutathione as well as lesser concentrations of NADPH, ascorbic acid, and uric acid.11,12 The concentrations of these cell components and the ratio of their reduced and oxidized forms vary between cell compartments, which gives different cell compartments different redox environments. Effective reducing potentials12 of approximately −316 mV are expected in the mitochondria where the glutathione and NADPH concentrations and pH all are high.13–15 In the cytosol of proliferating cells the effective reducing potential averages approximately −240 mV,11 while in the endoplasmic reticulum it is approximately −160 mV16 (under the approximation that the glutathione disulfide/glutathione couple principally determines the potential in these cellular compartments), and in the endosomes and lysosomes it is approximately −200 to −240 mV.15 In the absence of ligands that stabilize other oxidation states, such potentials are sufficient to make Pu(III) the thermodynamically stable oxidation state (Figure 1) in more acidic cell compartments such as the endosomes (pH = 5.5 – 6.5) and lysosomes (pH ~ 4.8).17,18
Figure 1.

Pourbaix diagram for 10−5 M plutonium using critically evaluated values of the relevant equilibrium constants9,45 to calculate the plutonium speciation in the absence of any added ligands. Ranges of reported Eh and pH values for the blood and various cellular compartments11–13,15–17,46 are indicated by black boxes.
To better understand the molecular-level chemistry of plutonium and how its oxidation state influences interactions with biomolecules and the associated cellular uptake and distribution, we studied mammalian cells incubated in growth media supplemented with 242Pu in one of three different oxidation states. The cellular accumulation and intracellular distribution of plutonium was quantified by synchrotron X-ray fluorescence microscopy (SXFM). In addition, the X-ray absorption near edge structure of Pu measured by X-ray fluorescence microspectroscopy (μ-XANES), a powerful technique for studying actinide speciation,19 was used to determine the oxidation state of Pu in 0.1 μm2 intracellular deposits inside individual cells. Although synchrotron-based micro-spectroscopy has been used in the past to study Pu in inorganic environmental matrices,20 this report represents its first use of μ-XANES to study the speciation of transuranic elements in individual cells. We find that whereas the initial oxidation state of the plutonium affects the amount of Pu taken in by the cells, only tetravalent Pu is present inside the cells.
EXPERIMENTAL
Caution! Neptunium-237 and plutonium-242 are α-emitting radionuclides that must be handled in facilities designed to safely handle transuranic elements. Many perchlorates are potentially explosive. Contamination with organic material, dehydration of the perchloric acid, and isolation of perchlorate salts were strenuously avoided in this procedure.
A solution of 242Pu(IV) (t1/2 = 3.76 × 105 years) in HNO3 was obtained from laboratory stocks. The stock solution was purified by anion exchange chromatography, selectively sorbing the Pu from 7.5 M HNO3 and eluting with 0.1 M HNO3. Oxidation of the purified Pu to the hexavalent oxidation state was accomplished by evaporating the purified, organic-free, Pu solution to incipient dryness followed by dissolution in hot perchloric acid. Cooling and dilution with water gave solutions containing Pu(VI) in 0.1 – 1 M HClO4. Solutions of PuO2(CO3)34− were prepared by addition of the Pu stock to solutions of Na2CO3. Solutions of Pu(III) were prepared by reduction of the acidic Pu(VI) solution with ascorbic acid, or by electrolytic reduction at a Pt electrode (0 V vs. Ag/AgCl). Pu(IV) solutions were prepared by electrochemical oxidation of the Pu(III) solution at a Pt electrode (+1 V vs. Ag/AgCl). The oxidation state purity of each preparation was verified by optical spectroscopy21 in 1 cm cells using a Cary-14 spectrometer modified for computer control by OLIS, Inc., and was at least 98%. Complexes of Pu(IV)-citrate and the mixed Pu(IV)-Fe(III)-transferrin complex, PuCFeNTf, were prepared from the oxidation state adjusted Pu solutions as previously described.22
A solution of 237Np obtained from laboratory stocks was adjusted to the petnavalent oxidation state and purified as previously described.23 The NpO2+ was precipitated by the addition of NaOH, washed with water, and redissolved in the minimal amount of 0.2 M HClO4. The resulting solution was diluted 15-fold with a solution of 0.01 M trisodium citrate/0.1 M NaCl and neutralized with NaOH to give a 0.005 M Np(V) stock solution.
Pheochromocytoma cells from rat adrenal glands (PC12) obtained from ATCC were grown in F12K medium supplemented with 12.5 % horse serum, 2.5% fetal calf serum, antibiotics, and antimycotics in a humidified 5% CO2 environment at 37°C. Cells were plated in serum free media at approximately 70% confluency the day before Pu exposure and serum-starved overnight. The following day, cells were exposed to 12.5 – 100 μM 242Pu in different oxidation states (Table 1) or 50 μM 237Np(V)-citrate for 3 hours at 25 °C (or at 37 °C in the case of PuCFeNTf) in complete F12K growth medium with serum (pH = 7.4, Eh = +0.36 V vs. N.H.E. at 25 °C). Cell viability in the presence of 100 μM 242Pu was checked by trypan blue exclusion after the 3 hour incubation. Viability was typically 80 – 90 % and did not differ from Pu-free controls. At the end of the incubations, the media was removed and the cells were washed three times with a 0.01 M solution of the extracellular chelating agent EGTA (ethyleneglycol bis(2-aminoethyl ether)-NN,,N ′,N′-tetraacetic acid) in 0.1 M NaCl. The cells were then suspended in 0.1 M NaCl/0.01 M EGTA and a portion was taken to determine the Pu content of the cells using liquid scintillation counting. Samples of the washed cells were prepared for SXFM measurements by depositing aliquots of the cell suspension on Formvar-coated copper electron microscopy grids and allowing them to air dry. The grids were then mounted on aluminum holders before being encapsulated between two 500 nm silicon nitride windows and sealed beneath a layer of 6 μm polypropylene or 8 μm Kapton for protection from plutonium’s α-emissions.
Table 1.
XANES parameters of the intracellular plutonium introduced to the media as various Pu(III), Pu(IV), or Pu(VI) complexes. Each line corresponds to a different intracellular Pu deposit or, in the case of Pu(III) aquo, a conventional XANES measurement on a pellet of cells.
| Number of Scans | Edge Energy (eV) a | Peak Maximum (eV) a | Normalized Peak Intensity | Peak Width (eV) b | |
|---|---|---|---|---|---|
| Oxidation State Standards | |||||
| Pu(III) | 18057 | 18063 | 1.7 | 10 | |
| Pu(IV) | 18062 | 18067 | 1.8 | 11 | |
| Pu(VI) | 18061 | 18068 | 1.3 | 13 and 14c | |
| Intracellular Deposits | |||||
| Pu added as | |||||
| Pu(III) aquod,e | 6 | 18061 | 18067 | 1.8 | 11 |
| Pu(IV) aquoe | 2 | 18062 | 18067 | 1.7 | 12 |
| Pu(IV) aquo | 2 | 18062 | 18067 | 2.0 | 11 |
| Pu(IV) aquo | 2 | 18061 | 18067 | 1.7 | 12 |
| Pu(IV) aquo | 3 | 18061 | 18067 | 1.7 | 12 |
| Pu(IV) aquo | 3 | 18061 | 18068 | 1.5 | 12 |
| Pu(IV) aquo | 1 | 18061 | 18067 | 1.8 | 12 |
| Pu(IV) citrate | 2 | 18062 | 18068 | 1.8 | 12 |
| Pu(IV) citrate | 2 | 18061 | 18067 | 1.6 | 12 |
| Pu(IV) citrate | 2 | 18061 | 18067 | 1.6 | 11 |
| Pu(IV) - PuCFeNTf | 1 | 18061 | 18068 | 1.7 | 12 |
| Pu(VI) carbonatee | 1 | 18062 | 18068 | 1.6 | 12 |
± 1 eV relative to the Zr foil standard
Width of Lorentzian peak used to fit the white line, ± 1 eV
One Lorentizian and one Gaussian peak at 18068 and 18082 eV, respectively were required to fit the overlapping white line and multiple scattering peak of the plutonyl species.
Conventional XANES measurement on pelleted cells
XANES spectrum depicted in Figure 5b.
The intracellular distributions of the elements from P though Pu were measured simultaneously by SXFM on the Advanced Photon Source 2-ID-D hard X-ray microprobe beamline.24 The incident 18100 eV X-ray beam was focused to a 200 nm × 500 nm spot and detailed elemental maps of selected cells were obtained by raster scanning the samples in 500 nm steps with a 2 – 4 second dwell time. The fluorescence signal for each element at each spatial pixel was converted to elemental content in μg/cm2 after calibration with elemental standards as previously described.25 In the absence of a traceable concentration standard, Pu was quantified by extrapolation of the calibration curve obtained from the X-ray fluorescence signals of the multi-element thin film standards, taking into account the differing photo-electric absorption of incident photons, absorption in the Be detector window, and the fluorescence yield. The estimated absolute accuracy is better than 40% and the relative accuracy is better than 10%. The resulting elemental maps were visualized and analyzed using the program MAPS.26
After mapping the elemental distributions in the cells, μ-XANES fluorescence spectra from the Pu L3 absorption edge (18057 eV) were collected at various 200 nm × 500 nm regions of interest within the cells. Conventional X-ray absorption spectra at the Pu L3-edge also were collected on a macroscopic sample of an EGTA-washed cell pellet containing approximately 106 cells at Advanced Photon Source beamline 12-BM using fluorescent detection. This sample was encapsulated in 6 mm thick holders with Kel-F windows. In all cases, the monochromator energy at the Pu L3-edge was calibrated relative to the energy of the first inflection point of the K-edge of a Zr foil at 17998 eV. The spectra were background corrected and normalized using standard methods27 with the programs Athena28 and IFEFFIT.29
RESULTS
Although plutonium is a synthetic element with no natural biological purpose, serum-starved PC12 cells from the rat adrenal gland readily take in 242Pu from growth media initially containing Pu(III), Pu(IV), or Pu(VI) over the course of 3 hours. In parallel experiments examining the intracellular distribution of neptunium presented to the cells as pentavalent 237NpO2+, a more difficult to reduce chemical analogue of pentavalent PuO2+, we observed no measurable Np uptake in detailed SXFM scans of 4 cells, suggesting that NpO2+ is not reduced to Np(IV) over the time frame of our experiments and that uptake of pentavalent actinides by PC12 cells is limited. Consequently, experiments with Pu(V) were not pursued. In contrast, rapid low resolution (2 – 4 μm) SXFM surveys of individual cells exposed to 242Pu as Pu(III), Pu(IV), or Pu(VI) indicate that the majority of cells likely contain Pu. For cells incubated in media initially supplemented with Pu(III), the preliminary surveys indicated that 4 of 7 cells (57%) likely contained Pu. When the Pu was added to the media as Pu(IV), 18 of the 30 cells (60%) surveyed contained Pu. For the hexavalent PuO22+ system, the survey maps of 18 of 21 cells (86%) indicated Pu was present.
Detailed elemental maps of 24 of these cells were then measured with 500 nm resolution (Figures 2 and 3), from which the Pu content of the cells was quantified. They demonstrate that the Pu is truly incorporated into the cells and is not merely bound to the cell surface, revealing Pu associated with specific intracellular structures. Cell-associated Pu is found in the cytoplasm of the cells, where we also find the highest concentrations of Fe,30 and not the nucleus (Figure 3), which is marked by the cells’ highest P and Zn concentrations.31
Figure 2.

False-color SXFM images and optical micrograph of a 25 μm × 25 μm area containing an isolated PC12 cell incubated in complete F12K media supplemented with Pu(IV). Replicate XANES measurements at the Pu L3-edge depicted in Figure 5a were made on the 200 nm × 500 nm spot centered in the white circle.
Figure 3.
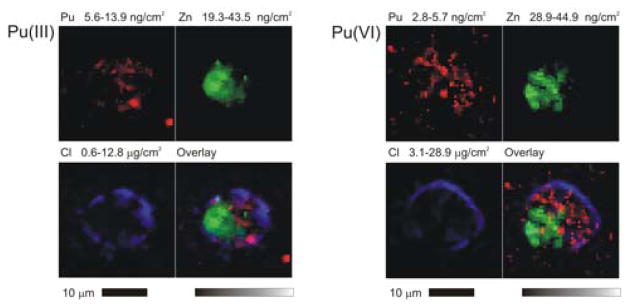
Three element colocalization maps of isolated PC12 cells incubated with Pu initially present as Pu(III) or Pu(VI) show Pu principally localizing in the cytoplasm and avoiding the nucleus, which is marked by the highest Zn concentrations. The outline of the cells appears in the Cl channel.
The detailed elemental maps also show that measurable Pu is associated with the cells regardless of the initial form of Pu added to the media. The average Pu content per unit area of the individual cells examined was quantified from the intensity of the Pu Lα fluorescence after correction for the X-ray fluorescence background measured in representative cell-free areas near each cell. The average Pu content of the individual cells ranged between 0.6 and 43 ng/cm2, which corresponds to a total cellular content of 1 – 70 fg Pu per cell; however the amount associated with the cells varied with the initial Pu oxidation state (Figure 4). By quantifying the Pu present in the elemental maps, we find that the average uptake from growth media initially containing tetravalent Pu was 8 times greater than for media supplemented with Pu(III) or Pu(VI). The content determined by SXFM is also in agreement with the results of radioactive counting of pellets of 106 cells, which give an average cellular 242Pu content of 16 ± 5 fg per cell for incubation with 100 μM Pu(IV).
Figure 4.
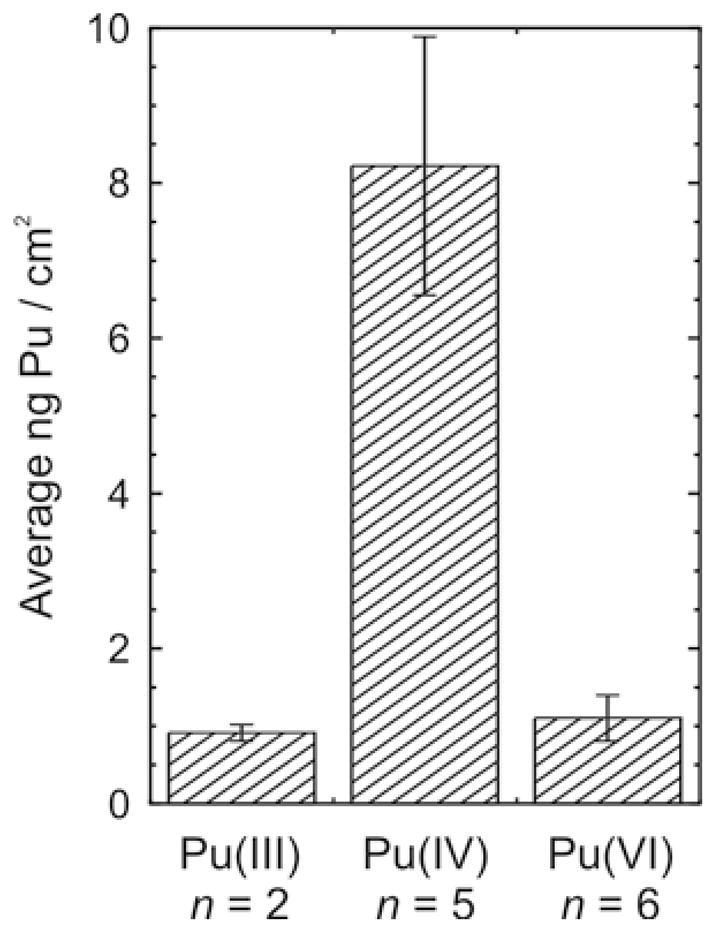
Average plutonium content of individual cells incubated for 3 hours in complete media with serum containing 100 μM Pu added as Pu(III), Pu(IV), or Pu(VI). The corresponding average total Pu contents are Pu(III), 1.7 ± 0.2 fg per cell; Pu(IV), 15.8 ± 3.2 fg per cell; and Pu(VI), 2.1 ± 0.5 fg per cell. Uncertainties are given as the standard error of the mean for measurement of n cells.
After locating intracellular Pu deposits in the elemental maps of the individual cells, μ-XANES spectra at the Pu L3-edge were collected from 11 unique 200 nm × 500 nm spots in 7 different individual cells in order to identify the oxidation state of the intracellular Pu. Because the Pu deposits in the cells exposed to Pu(III) proved to be too diffuse for effective μ-XANES measurements on individual cells, conventional XANES spectra were collected on a pellet containing approximately 106 cells that had been exposed to media initially containing Pu(III). This XANES spectrum is depicted in Figure 5. Regardless of the mode of XANES data collection, the position, intensity, and shape of the X-ray absorption white line did not change when repeated XANES scans were collected on the same area (supplemental material), indicating that X-ray induced changes in the oxidation state32 were negligible in these systems.
Figure 5.

XANES spectra of intracellular Pu demonstrate the presence of only Pu(IV) in the PC12 cells. (a) The energy, intensity, and peak shape of the normalized XANES spectra of an individual intracellular Pu deposit in a PC12 cell (——), match the reference XANES spectrum of Pu(IV) (– – –), not the reference spectra of Pu(III) (–..–) or Pu(VI) (- - - -).47 Elemental maps of the cell examined are depicted in Figure 2. (b) The XANES spectra of the intracellular Pu are all that of Pu(IV) whether the Pu is added to the media as Pu(III) (–..–, conventional XANES), Pu(IV) (——, μ-XANES), or Pu(VI) (- - - -, μ-XANES).
The edge energies of the resulting XANES spectra were determined from the first inflection point of each spectrum. The normalized XANES spectra were simulated as previously described33 with a combination of an arc tangent centered at the edge energy, representing the edge step; a Lorentzian function, representing the white line peak; and, if needed, one or two Gaussian peaks, which account for the first EXAFS oscillation and also multiple scattering from the “-yl” oxygens for samples containing the linear dioxo PuO2m+ (m = 1 or 2) moiety. The energy and line shape of the XANES spectra given in Figure 5 and Table 1 are similar for all of the samples studied regardless of the initial oxidation state or chemical form of the Pu. When these spectra are compared to the XANES of Pu standards in known oxidation states (Figure 5a), the spectra of the intracellular Pu clearly match those of the Pu(IV) standard in every case. This is completely consistent with the parametric analysis of the XANES. As summarized in Table 1, the absorption edge energies and the position of the peak maxima both are consistent with the intracellular Pu being present as either Pu(IV) or Pu(VI). However, the normalized peak intensities reported in Table 1 are substantially higher than those reported for plutonyl(VI) species34 or for our Pu(VI) standard, and the XANES spectra of all our samples lack the multiple scattering shoulder from O=Pu=O approximately 15 eV above the peak maximum that is a characteristic feature of actinyl(V) and actinyl(VI) XANES.35 This is true even when Pu is deposited in the cells from media containing PuO22+ (Figure 5b). Therefore, we find that all detectable intracellular Pu is present as Pu(IV).
The generally strong optical absorbance of the PuO22+ complexes also made it possible to examine the oxidation state stability of the hexavalent PuO22+ complexes after addition to the media. When 100 μM PuO2(CO3)34− was added to the complete serum-containing F12K media, Pu(VI) absorption bands were observed at 808 nm and 850 nm. The 808 nm band arises from PuO2(CO3)34−,36 the Pu(VI) complex added to the media, while the 850 nm band is consistent with the presence of another PuO22+ complex. (PuO2)2(OH)4 is reported to possess a sharp absorption band at 850 nm,37 but the peak may be due to PuO22+ complexation by other ligands in the media. These two peaks persist over the entire course of the incubation, slowly decreasing in intensity to ca. 80% of the initial value in 3 hours (supplemental material). The Pu(VI) complexes are not stable indefinitely, however. All absorption bands attributable to PuO22+ complexes in the growth media disappear within 24 hours.
DISCUSSION
Regardless of the initial oxidation state, all of the intracellular Pu examined was found to be Pu(IV) even though the reported Eh and pH ranges of the endosomes and lysosomes, which are the eventual Pu deposition sites in liver cells,38 would favor the formation of Pu(III) in aqueous solution. One explanation for this observation could be perturbation of the expected oxidation state of Pu by the ionizing radiation from the Pu or the synchrotron.
The relatively long half life of 242Pu, the small amount and high-isotopic purity of the Pu used in these experiments (99.96 atom% 242Pu, 0.035 atom% 239Pu, < 4 × 10−4 atom% 241Pu), and the limited time frame of these experiments make radiolytically induced changes in the Pu oxidation state speciation39 irrelevant during the Pu uptake experiments and the subsequent X-ray measurements in these systems. The measured range of intracellular 242Pu contents represent truly low levels of radioactivity, corresponding to only 0.01 – 0.9 disintegrations per day per cell, and the 242Pu concentration in the media never exceeded 100 μM. Such low levels of radioactivity are incapable of significantly perturbing the Pu oxidation state over the course of our experiments. The very low radioactivity of our samples has an additional advantage, as it also minimizes radiation effects on the cells themselves, allowing examination of the chemical behavior of Pu in these systems without substantial radiological perturbation of their intrinsic behavior.
External ionizing radiation from the X-rays used to examine the cells also has the potential to perturb the oxidation states of Pu, and radiation induced redox of actinides from intense beams of synchrotron X-rays has been observed in some instances.32 We observe no evidence of this in our samples. The XANES spectra recorded from repeated scans of the same area do not change, and the individual near edge spectra always were only consistent with Pu(IV). This was true for both focused μ-XANES scans of individual intracellular spots and conventional XANES measurements on a collection of pelletized cells. Consequently, neither the intrinsic radioactivity of the Pu nor the X-rays from the synchrotron appear to affect the oxidation state in our samples.
Because the XANES measurements find only Pu(IV) associated with cells, and because substantially more Pu is taken into the cells when it is presented as Pu(IV), it is tempting to postulate that only tetravalent Pu is efficiently taken in by PC12 cells and that reduction of Pu(VI) or oxidation of Pu(III) must occur in the media or at the cell surface4 before uptake. This would be entirely consistent with the body of in vivo biokinetic studies of Pu uptake.6 In the absence of ligands that stabilize Pu(III) or Pu(VI) relative to Pu(IV), the Eh and pH of the media used for our in vitro studies imply that Pu(IV) will be the stable oxidation state in the growth media. Moreover, the 8-fold lower uptake of Pu from media initially containing Pu(III) or Pu(VI) (Figure 4) is consistent with this hypothesis, as is rate of Pu(VI) reduction implied by the spectrophotometric measurements. Although we could not monitor it directly, it is also possible that a substantial fraction of the original Pu(III) remains in the media at the end of the incubation since Pu(III) is able to persist for 2 hours or more in neutral solutions under certain conditions.40 Unfortunately, the effect of such redox reactions prior to Pu uptake by the cells cannot easily be decoupled from other mechanisms for retarding uptake, such as complexation by ligands in the growth media that render the Pu(III) or Pu(VI) less bioavailable than Pu(IV). Given the limited molecular-level knowledge of the biological chemistry of Pu and the presence of serum in the cell medium, we cannot exclude this possibility. However, if complexation of Pu(III) or Pu(VI) were to be an important mechanism in regulating Pu uptake in this system, it would require especially abundant ligands because the plutonium concentration in the growth media of the Pu(III) and Pu(VI) experiments was 100 μM.
While the actual mechanisms of Pu uptake are of practical importance and the initial oxidation state of the Pu introduced to the media affects the amount of Pu taken up by the cells, we detect only Pu(IV) inside the cells. This is true for the 11 individual deposits studied by μ-XANES and also for the average of the millions of intracellular deposits probed together in the conventional XANES experiment with the pelletized cells. The exclusive presence of Pu(IV) inside the cells implies that the Pu is sequestered in a form that insulates it from the average pH or Eh encountered in the endosomes/lysosomes, or that the Pu complexes formed inside the cell, for example Pu-ferritin complexes,41 substantially stabilize Pu(IV) relative to Pu(III).
Along similar lines, Durbin and coworkers have used conventional XANES spectroscopy to examine the oxidation state speciation of the transuranium element neptunium in bulk samples of mouse femora after in vivo exposure to Np(V) injected as NpO2Cl.42 They found the Np to be principally present as Np(IV), with an upper limit of 20% Np(V) content, and suggested that Np(IV) was stabilized by formation of strong citrate or phosphate complexes in the bone. Unlike Pu in the lysosomes, however, the trivalent oxidation state of Np(III) is not expected to be accessible at the bone surfaces because the Eh and pH (7.3 ± 0.2)43 of the bone fluid is only sufficient to drive reduction of Np(V) to Np(IV) even in the absence of citrate or phosphate ligands.44
As direct measurements of the oxidation state of Pu in mammalian cells, our experiments augment the existing body of biokinetic data for plutonium distribution, which demonstrate that Pu(IV) dominates the rate determining step(s) in the uptake and distribution of Pu in mammals.6 Our study highlights, for the first time, what form of Pu is present intracellularly in a model cell line after the processes that are readily probed by metal uptake and partitioning kinetics have run their course. It demonstrates that the chemistry of Pu(IV) not only determines the rate of uptake and the organs or cells where Pu accumulates, but that Pu(IV) also dominates the intracellular chemistry, even though Pu(III) could, in theory, exist in the cell compartments that are the major route of entry for most materials taken up by the cells. Such information is essential to understanding how synthetic elements such as Pu interact with individual molecules in living systems and how biological mechanisms to control the oxidation states of essential and toxic metal ions operate.
Supplementary Material
Acknowledgments
The authors thank Dr. L. Soderholm for invaluable assistance in the earlier stages of this work, as well as the staff of APS XOR beamlines 2-ID and 12-BM and the Actinide Facility for the support and infrastructure that made these experiments possible. The work at Argonne National Laboratory and use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CHI11357, and by the University of Chicago and the Department of Energy under section H.35 of this contract awarded to UChicago Argonne LLC, operator of Argonne National Laboratory.
Footnotes
Supporting Information Available: XANES spectra and time dependence of Pu(VI) optical absorption. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Silva RJ, Nitsche H. Environmental Chemistry. In: Hoffman DC, editor. Advances in Plutonium Chemistry, 1967-200. American Nuclear Society; La Grange Park, Illinois: 2002. pp. 89–117. [Google Scholar]
- 2.Cleveland JM. The Chemistry of Plutonium. Gordon and Breach; New York: 1970. p. 653. [Google Scholar]
- 3.Allard B, Kipatsi H, Liljenzin JO. J Inorg Nucl Chem. 1980;42:1015–1027. [Google Scholar]
- 4.Panak PJ, Nitsche H. Radiochim Acta. 2001;89:499–504. [Google Scholar]
- 5.(a) Francis AJ, Dodge CJ, Gillow JB. Environ Sci Technol. 2008;42:2355–2360. doi: 10.1021/es072016w. [DOI] [PubMed] [Google Scholar]; (b) Boukhalfa H, Icopini GA, Reilley SD, Neu MP. Appl Environ Microbiol. 2007;73:5897–5903. doi: 10.1128/AEM.00747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durbin PW. Actinides in Animals and Man. In: Morss LR, Edelstein NM, Fuger J, editors. The Chemistry of the Actinide and Transactinide Elements. Vol. 5. Springer; Dordrecht: 2006. pp. 3339–3440. [Google Scholar]
- 7.Taylor DM. Chemical and Physical Properties of Plutonium. In: Hodge HC, Stannard JN, Hursh JB, editors. Uranium, Plutonium, Transplutonic Elements. Vol. 36. Springer-Verlag; New York: 1973. pp. 323–347. [Google Scholar]
- 8.Durbin PW. Health Phys. 1975;29:495–510. doi: 10.1097/00004032-197510000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Lemire RJ, Fuger J, Nitsche H, Rand MH, Potter P, Rydberg J, Spahiu K, Sullivan JC, Ullman WJ, Vitorge P, Wanner H. Chemical Themodynamics of Neptunium and Plutonium. Vol. 4 Elsevier; New York: 2001. [Google Scholar]
- 10.(a) Carritt J, Fryxell R, Kleinschmidt J, Kleinschmidt R, Langham W, San Pietro A, Schaffer R, Schnap B. J Biol Chem. 1947;171:273–283. [Google Scholar]; (b) Scott KG, Axelrod DJ, Fisher H, Crowley JF, Hamilton JG. J Biol Chem. 1948;176:283–293. [PubMed] [Google Scholar]; (c) Larsen RP, Oldham RD, Bhattacharyya MH, Moretti ES, Austin DJ. Radiation Res. 1981;87:37–49. [PubMed] [Google Scholar]; (d) Sullivan MF, Ryan JL, Gorham LS, McFadden KM. Radiation Res. 1979;80:116–121. [PubMed] [Google Scholar]
- 11.Schafer FQ, Buettner GR. Free Radical Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 12.Martinovich GG, Cherenkevich SN, Sauer H. Eur Biophys J. 2005;34:937–942. doi: 10.1007/s00249-005-0470-3. [DOI] [PubMed] [Google Scholar]
- 13.Berry MN, Phillips JW. Biochem Soc Trans. 2000;28:131–135. doi: 10.1042/bst0280131. [DOI] [PubMed] [Google Scholar]
- 14.Moyle J, Mitchell P. Biochem J. 1973;132:571–585. doi: 10.1042/bj1320571. [DOI] [PMC free article] [PubMed] [Google Scholar]; Costantini P, Chernyak B, Petronilli VPB. J Biol Chem. 1996;271:6746–6751. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 15.Austin CD, Wen X, Gazzard L, Nelson C, Scheller RH, Scales SJ. Proc Nat Acad Sci. 2005;102:17987–17992. doi: 10.1073/pnas.0509035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang C, Sinskey AJ, Lodish HF. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 17.Ohkuma S, Poole B. Proc Nat Acad Sci. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demaurex N. Physiology. 2002;17:1–5. doi: 10.1152/physiologyonline.2002.17.1.1. [DOI] [PubMed] [Google Scholar]
- 19.Denecke M, Somogyi A, Janssens K, Simon R, Dardenne K, Noseck U. Microsc Microanal. 2007;13:165–172. doi: 10.1017/S1431927607070316. [DOI] [PubMed] [Google Scholar]
- 20.(a) Kaplan DI, Powell BA, Duff MC, Demirkanli DI, Denham M, Fjeld RA, Molz FJ. Environ Sci Technol. 2007;41:7417–7423. doi: 10.1021/es0706302. [DOI] [PubMed] [Google Scholar]; (b) Duff MC, Newville M, Hunter DB, Bertsch PM, Sutton SR, Triay IR, Vaniman DT, Eng P, Rivers ML. J Synchrotron Rad. 1999;6:350–352. doi: 10.1107/S0909049598016811. [DOI] [PubMed] [Google Scholar]; (c) Bielewski M, Eriksson M, Himbert J, Betti M, Belloni F, Falkenberg G. J Radioanal Nucl Chem. 2009;282:355–359. [Google Scholar]; (d) Powell BA, Duff MC, Kaplan DI, Fjeld RA, Newville M, Hunter DB, Bertsch PM, Coates JT, Eng P, Rivers ML, Serkiz SM, Sutton SR, Triay IR, Vaniman DT. Environ Sci Technol. 2006;40:3508–3514. doi: 10.1021/es052353+. [DOI] [PubMed] [Google Scholar]
- 21.Keller C. The Chemistry of the Transuranium Elements. Verlag Chemie; Weinheim: 1971. pp. 426–430. [Google Scholar]
- 22.Jensen MP, Gorman-Lewis D, Aryal BP, Paunesku T, Vogt S, Rickert PG, Seifert S, Lai B, Woloschak GE, Soderholm L. Nature Chem Biol. doi: 10.1038/nchembio.594. in press http://dx.doi.org/10.1038/nchembio.594. [DOI] [PMC free article] [PubMed]
- 23.Jensen MP, Nash KL. Radiochimica Acta. 2001;89:557–564. [Google Scholar]
- 24.Cai Z, Lai B, Xiao Y, Xu S. J Phys IV. 2003;104:17–20. [Google Scholar]
- 25.McRae R, Lai B, Vogt S, Fahrni CJ. J Struct Biol. 2006;155:22–29. doi: 10.1016/j.jsb.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Vogt S. J Phys IV France. 2003;104:635–638. [Google Scholar]
- 27.Prins R, Koningsberger DE. X-ray Absorption: Principles, Applications, Techniques for EXAFS, SEXAFS, and XANES. Wiley-Interscience; New York: 1988. [Google Scholar]
- 28.Ravel B, Newville M. J Synchrotron Rad. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- 29.Newville M. J Synchrot Radiat. 2001;8:322–324. doi: 10.1107/s0909049500016964. [DOI] [PubMed] [Google Scholar]
- 30.Aryal BP, Gorman-Lewis D, Paunesku T, Wilson RE, Vogt S, Lai B, Woloschak GE, Jensen MP. Int J Radiat Biol. doi: 10.3109/09553002.2011.584941. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Dillon CT, Lay PA, Kennedy BJ, Stampfl APJ, Cai ZH, Ilinski P, Rodrigues W, Legnini DG, Lai B, Maser J. J Biol Inorg Chem. 2002;7:640–645. doi: 10.1007/s00775-002-0343-5. [DOI] [PubMed] [Google Scholar]; (b) Paunesku T, Vogt S, Lai B, Maser J, Stojicevic N, Thurn KT, Osipo C, Liu H, Legnini D, Wang Z, Lee C, Woloschak GE. Nano Lett. 2007;7:596–601. doi: 10.1021/nl0624723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Skanthakumar S, Gorman-Lewis D, Locock A, Chiang M-H, Jensen MP, Burns PC, Fein J, Jonah CD, Attenkofer K, Soderholm L. Changing Np redox speciation in the synchrotron beam. In: Soderholm L, Joyce J, Nicol MF, Shuh D, Tobin JG, editors. Proceedings of the Materials Research Society, Volume 802: Actinides Basic Science, Applications, and Technology. Vol. 802. Materials Research Society; Warrendale, PA: 2004. pp. 151–156. [Google Scholar]; (b) Wilk PA, Shaughnessy DA, Wilson RE, Nitsche H. Environ Sci Technol. 2005;39:2608–2615. doi: 10.1021/es040080x. [DOI] [PubMed] [Google Scholar]; (c) Wilson RE. PhD Dissertation. University of California; Berkeley: 2005. [Google Scholar]; (d) Hu Y-J, Kestrel Schwaiger L, Booth CH, Kukkadapu RK, Cristiano E, Kaplan D, Nitsche H. Radiochim Acta. 2010;98:655–663. [Google Scholar]
- 33.Jensen MP, Bond AH. J Am Chem Soc. 2002;124:9870–9877. doi: 10.1021/ja0178620. [DOI] [PubMed] [Google Scholar]
- 34.Conradson SD, Abney KD, Begg BD, Brady ED, Clark DL, Den Auwer C, Ding M, Dorhout PK, Espinosa-Faller FJ, Gordon PL, Haire RG, Hess NJ, Hess RF, Keogh DW, Lander GH, Lupinetti AJ, Morales LA, Neu MP, Palmer PD, Paviet-Hartmann P, Reilly SD, Runde WH, Tait CD, Veirs DK, Wastin F. Inorg Chem. 2004;43:116–131. doi: 10.1021/ic0346477. [DOI] [PubMed] [Google Scholar]
- 35.(a) Hudson EA, Rehr JJ, Bucher JJ. Phys Rev B. 1995;52:13815–13826. doi: 10.1103/physrevb.52.13815. [DOI] [PubMed] [Google Scholar]; (b) Den Auwer C, Simoni E, Conradson S, Madic C. Eur J Inorg Chem. 2003:3843–3859. [Google Scholar]
- 36.Varlashkin PG, Begun GM, Peterson JR. Radiochim Acta. 1984;35:211–218. [Google Scholar]
- 37.Reilley SD, Neu MP. Inorg Chem. 2006;45:1839–1846. doi: 10.1021/ic051760j. [DOI] [PubMed] [Google Scholar]
- 38.(a) Seidel A, Krüger E, Weiner M, Hotz G, Balani M, Thies WG. Radiat Res. 1985;104:191–199. [PubMed] [Google Scholar]; (b) Boocock G, Danpure CJ, Popplewell DS, Taylor DM. Radiat Res. 1970;42:381–396. [PubMed] [Google Scholar]; (c) Rahman YE, Lindenbaum A. Radiat Res. 1964;21:575–583. [PubMed] [Google Scholar]
- 39.Newton T. The Kinetics of the Oxidation-Reduction Reactions of Uranium, Neptunium, Plutonium, and Americium in Aqueous Solutions. National Technical Information Center; Springfield, VA: 1975. [Google Scholar]; Vladimirova MV. J Radioanal Nucl Chem. 1990;143:445–454. [Google Scholar]
- 40.Seeger PA, Rokop SE, Palmer PD, Henderson SJ, Hobart DE, Trewhella J. J Am Chem Soc. 1997;119:5118–5125. [Google Scholar]
- 41.(a) Den Auwer C, Llorens I, Moisy P, Vudaud C, Goudard F, Barbot C, Solari PL, Funke H. Radiochim Acta. 2005;93:699–703. [Google Scholar]; (b) Bruenger FW, Stover BJ, Stevens W. Health Phys. 1971;21:679–687. doi: 10.1097/00004032-197111000-00009. [DOI] [PubMed] [Google Scholar]
- 42.Durbin PW, Kullgren B, Xu J, Raymond KN, Allen PG, Bucher JJ, Edelstein NM, Shuh DK. Health Phys. 1998;75:34–50. doi: 10.1097/00004032-199807000-00007. [DOI] [PubMed] [Google Scholar]
- 43.Neuman M, Neuman W. Calcified Tissue International. 1980;31:135–145. doi: 10.1007/BF02407174. [DOI] [PubMed] [Google Scholar]
- 44.Vitorge P, Capdevila H, Maillard S, Faure MH, Vercouter T. J Nucl Sci Technol. 2002;(Suppl 3):713–716. [Google Scholar]
- 45.Guillaumont R, Fanghänel T, Fuger J, Grenthe I, Neck V, Palmer DA, Rand MH. Update on the Chemical Themodynamics of Uranium, Neptunium, Plutonium, Americium, and Technetium. Vol. 5 Elsevier; New York: 2003. [Google Scholar]
- 46.(a) Tapper H, Sundler R. Biochem J. 1990;272:407–414. doi: 10.1042/bj2720407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kneen M, Farinas J, Li YX, Verkman AS. Biophys J. 1998;74:1591–1599. doi: 10.1016/S0006-3495(98)77870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ziegler E. In: The Redox Potential of the Blood In Vivo and In Vitro. Thomas Charles C., editor. Springfield; Illinois: 1965. [Google Scholar]; (d) Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY. Proc Nat Acad Sci. 1998;95:6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiang MH, Soderholm L, Antonio MR. Eur J Inorg Chem. 2003:2929–2936. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.