

Abstract
We describe predictions made using the Rosetta structure prediction methodology for the Eighth Critical Assessment of Techniques for Protein Structure Prediction. Aggressive sampling and all-atom refinement were carried out for nearly all targets. A combination of alignment methodologies was used to generate starting models from a range of templates, and the models were then subjected to Rosetta all atom refinement. For 50 targets with readily identified templates, the best submitted model was better than the best alignment to the best template in the Protein Data Bank for 24 domains, and improved over the best starting model for 43 domains. For 13 targets where only very distant sequence relationships to proteins of known structure were detected, models were generated using the Rosetta de novo structure prediction methodology followed by all-atom refinement; in several cases the submitted models were better than those based on the available templates. Of the 12 refinement challenges, the best submitted model improved on the starting model in 7 cases. These improvements over the starting template-based models and refinement tests demonstrate the power of Rosetta structure refinement in improving model accuracy.
1. Introduction
The CASP8 experiment provided an invaluable opportunity to stress-test our new object oriented Rosetta software suite and inspired new ideas for de novo structure prediction and comparative modeling. For all targets for which a sequence-detectable structural template existed, target-template sequence alignments were generated, and the Rosetta “rebuild-and-refine” protocol1 was used to generate low energy models. For the 13 targets for which a reliable template could not be identified, modeling was carried out using the Rosetta de novo2,3 modeling protocol. All targets were subjected to extensive high resolution refinement with the physically realistic Rosetta all-atom forcefield2.
2. Materials and Methods
All-atom refinement using a physically realistic forcefield
With a few notable exceptions4, the native conformation of a protein is likely to be its lowest free-energy state5; the goal of Rosetta structure prediction is to locate the lowest free energy structure for the target amino acid sequence. Energies are computed using the physically realistic Rosetta all-atom forcefield, which focuses largely on short-range interactions – van der Waals, hydrogen-bonding6 and desolvation7 - and dampens long-range electrostatic interactions. To capture high-resolution features of native structures and to better discriminate native-like from non-native models, all submitted models in CASP8 were subjected to Rosetta all-atom refinement2,3. Rosetta all-atom refinement employs a Monte Carlo Minimization protocol in which each attempted move consists of (1) perturbations to randomly selected backbone torsion angles, (2) discrete combinatorial optimization of sidechain rotamer conformations, and (3) quasi Newton minimization with respect to all backbone and sidechain torsion angles8.
Template detection, sequence alignment construction and ranking
To detect potential templates and construct sequence alignments, we combined several automatic methods. Accurate template ranking required using multiple detection methods with different levels of sensitivity aimed at different ranges of target-template similarity. The most sensitive methods for profile-profile (COMPASS9, PROCAIN10) and HMM-HMM comparison (HHSearch11) detect extremely distant homologs and produce estimates of statistical significance that generate the best ranking in the area of medium to high evolutionary distances. Although easily detected by such methods, close homologs may be assigned misordered rankings, mainly due to the methods’ comparison of whole families rather than single or small subset of closest sequences. Therefore, each target sequence was used as a query in several searches against the database of PDB sequences: (i) BLAST12; (ii) PSI-BLAST13 with query profiles based on homologs in the NCBI nr database, iterations 1 to 5 (iii) COMPASS14 and HHSearch on several databases of profiles/HMMs generated from PSI-BLAST alignments after iterations 1 to 8. The significant E-value cutoffs for BLAST, PSI-BLAST, and COMPASS were set to 0.001, 0.001, and 0.005, respectively; while the HHSearch probability cutoff was 0.90. In order to improve the ranking of close to medium-range homologs by subsequent PSI-BLAST iterations, we started with highly conservative E-value cutoffs for homolog inclusion (-h) at the initial iterations and relaxed these cutoffs at higher iterations. The following inclusion cutoff E-values were used for iterations 1–5: 10−40, 10−20, 10−10, 10−5, and 10−3. Significant hits identified in the above order were included in the ranked list of templates.
To further improve quality, we applied two additional alignment methods. First, each target-template pair was aligned by PROCAIN10, a method for profile comparison that incorporates additional information about predicted secondary structure and sequence motifs. Second, we used PROMALS15 to re-align the detected sequence segments based on a wider context of multiple sequences homologous to target and template. If multiple templates were available, a multiple sequence alignment of target and templates was constructed by PROMALS3D that integrates profile-profile comparison, secondary structure prediction and 3D structural information. For the targets of medium and high level of difficulty, automatic alignments were manually inspected and re-aligned, based on the analysis of functional sequence motifs, hydrophobicity patterns, and secondary structure predictions. For some targets, alignment variants were created for segments with uncertain registers. Automatic template ranking was also manually verified and modified, based on template structure quality and alignment properties (insertions/deletions, register ambiguity in secondary structure elements especially edge strands etc.). In some cases, ‘hybrid’ templates were constructed by connecting different parts of two or more template structures; and the ‘hybrid’ alignments were generated using corresponding parts in the original target-template alignments.
‘Hybrid’ templates were prepared by (i) superimposing two or more templates of interest with structurally similar core using DALI16 and (ii) combining different template regions that were selected as better approximations of the target (e.g. core secondary structure elements from closest template and a loop region that was absent in the closest but was present in a more distant template and had a reasonable sequence alignment to the target). In some cases, when the ends of combined regions were outside the normal range of Cα distances in a protein chain, they were manually spliced by moving the residues of these regions to eliminate chain breaks.
All-atom energy-based selection of templates/alignments
For targets with no clear best template (i.e. several templates were detected with comparable scores and alignment coverage), all-atom energy-based selection was used to try to identify the closest template and the best alignment (Figure 1(A)). Starting models were generated by threading the sequence of the query onto the structure of the template for all the automatically generated alignments. The gaps in the unaligned regions were closed by loop-modeling and ~50 independent all atom refinement runs were carried out, initializing each Monte Carlo trajectory with a different random seed. The template and alignment most enriched in the lowest energy population of all-atom refined models was then selected and used in the subsequent modeling steps. For cases in which more than one alignment or template was enriched in the lowest energy refined models, all were used in the subsequent modeling steps.
Figure 1. Methodological improvements.

(A) Energy-based template selection for T0464 models derived from different templates and alignments. Each color represents an ensemble of all-atom refined models generated from a particular template.
(B) Distribution of GDT-TS of models generated for T0460 from the standard length fragment set (green) vs. variable fragment length set (blue).
Model generation with Rosetta
The comparative modeling targets were broadly categorized based on the sequence similarity of the closest template(s) to the target sequence into the following categories.
High sequence similarity template(s) (>50% sequence similarity)
Medium sequence similarity template(s) (20–50% sequence similarity)
Low sequence similarity template(s) (<20% sequence similarity)
The following protocols were used for the three target classes.
High sequence similarity template(s)
For these targets a conservative modeling protocol was used which does not change any atom positions in the well-aligned regions. To this end, a starting threaded model was generated using the alignment to the template with closest sequence similarity to the target sequence. Regions with insertions or deletions, and regions with relatively low sequence conservation were built using Rosetta all-atom loop modeling17. The side-chains of the template were retained for the non-loop-modeled regions. The lowest energy models with good hydrophobic burial and packing, as assessed by RosettaHoles18, were submitted.
Medium sequence similarity template(s)
A more comprehensive search of conformational space was necessary for this class of targets than for targets with high sequence similarity template(s). As described above, if multiple templates were identified with roughly equivalent sequence similarity to the query and alignment coverage, energy-based selection was performed to identify those likely to produce the best models (Figure 1(A)). Starting models based on these templates were refined using the Rosetta rebuild-and-refine protocol1. In the rebuild stage, large selected regions of the starting model, chosen in random order, were entirely rebuilt by fragment insertion followed by cyclic coordinate descent (CCD)19 loop closure. The regions chosen for rebuilding include residues surrounding gaps and insertions, loops in the starting model and sequence segments with low sequence conservation in the protein family. Each starting model was subjected to multiple independent rebuild-and-refine trajectories in which different regions of the model are stochastically selected for rebuilding. The breaking of the chain in the rebuilding step allows easier traversal of free energy barriers which would otherwise be nearly insurmountable with a continuous chain model20. Multiple rounds of the rebuild-and-refine protocol were carried out, alternating between diversification and intensification, the end result was a very low energy but diverse set of models which, in favorable cases, bracketed the global minimum. For close homology cases, only a single iteration of the rebuild-and-refine protocol was carried out.
Low sequence similarity template(s)
For targets where only distant homology was detectable, a larger number of initial target-template alignments were used, as both the template and alignment were less reliable. The iterative rebuild-and-refine protocol described above was made still more aggressive by allowing the rebuilding of secondary structure elements in addition to loops and variable regions in the rebuild step. This allowed reconfiguration of secondary structure elements, in particular the movement of helices on beta sheets; this is necessary because for such distant sequence relationships considerable shifting of secondary structural elements relative to each other frequently occurs. Iterative rebuild-and-refine has been found to be particularly effective on targets with medium-to-low sequence similarity templates21, since a single round of rebuild-and-refine is not likely to satisfactorily converge for such targets. Due to the computationally intensive nature of this protocol, it was applied primarily to domains less than 150 amino acids.
In several cases, in particular T0471, the input set of sequence alignments was modified based on the analysis of the lowest energy models, and a second round of model generation and refinement was carried out starting from the new alignments.
Fold recognition/free modeling targets
This category includes targets with very remote or no detectable templates. In cases in which remote sequence relationships to proteins of known structure could be detected, both template-based and free-modeling runs were carried out. Template based models were made using the aggressive rebuild-and-refine protocol described above. Free modeling was carried out using the Rosetta de novo structure prediction methodology2,3, which consists of a coarse-grained fragment-based search of conformational space followed by all-atom refinement. The initial fragment-based structure assembly step generates a large, diverse pool of models with hydrophobic cores and other protein-like features. Following all-atom refinement, final submissions were selected by clustering the lowest energy models, occasionally supplemented by visual inspection. As in CASP7, we increased the diversity of models by folding multiple sequence homologs for each target, disallowing beta hairpins, and by resampling long-range beta sheet pairings. As in previous CASP experiments, the full folding protocol was carried out on alternative domain parses, and additional sampling was carried out for parses giving rise to the lowest energy plausible structures.
We recently observed that Rosetta de novo structure generation can sample closer to the native structure if a range of fragment sizes are used. Instead of using only 3 and 9 residue fragments, we use a range of lengths. In each trajectory a single “long fragment” length is used in place of the standard 9mer insertions, and a single “short fragment” length in place of 3mer insertions. On a sixty two protein benchmark set, using 5 to 19 residue long fragments and 3 to 12 residue short fragments, the number of proteins for which at least 0.1% of the structures generated were less than 2Å rmsd to native increased from eight to thirteen. Analysis of the results suggested that longer fragments are better for helical proteins and shorter fragments for beta proteins, consistent with the larger number of residues on average in an individual helix compared to an individual strand. In CASP8, for α-helical proteins we used 5–19 residue long fragments and 3–12 residue short fragments, for α/β proteins, 5–12 residue long fragments and 3–9 residue small fragments, and for all β proteins, 4–10 residue long fragments and 3–7 residue short fragments. This resulted in improved sampling for several targets, the clearest example is T0460 (Figure 1(B)).
In several cases, manual analysis of models produced by de novo structure prediction prompted the refinement of initial target-template sequence alignments, and was followed by the next iteration of template-based modeling. The final submissions were chosen by energy and visual inspection after pooling together models from both protocols (when both were used).
Refinement CASP
We experimented with several different strategies for increasing the model quality of the CASP8 refinement challenges. For targets that were described as already close (within 2 Å RMSD) to the native structure, the structure was only subjected to gradient-based minimization of all torsion angles. To prevent large excursions from the starting model, harmonic Cα-Cα distance restraints were included that constrained residue pairs with starting distances less than 8 Å to remain within 2 Å of the starting distance. For targets that were judged to be far from the native, we performed rebuilding of targeted regions of the protein followed by relaxation of the entire structure in the Rosetta full atom forcefield as described above for low sequence-similarity template-based modeling. Selection of regions for rebuilding was done using a variety of different methods, including regions of low sequence conservation, regions with obvious packing defects, and regions identified by the assessors as being incorrect in the starting models for refinement. A preliminary version of the FoldIt interactive modeling game was used to prepare some of the refinement targets (unpublished results). The resulting models were not clearly worse or better than submissions prepared with automated algorithms, and are not further discussed here.
Robetta Server method
Comparative modeling targets
For CASP8, HHSearch11 was used as opposed to the 3D-Jury22 metaserver in previous years, for detecting fold recognition targets. Templates with HHSearch probabilities of at least 0.85 were considered CM targets, and templates with probabilities between 0.60 and 0.85 were treated astwilight-zone” and were modeled using both the de novo and comparative modeling protocols. Robetta used the highest confidence detection from BLAST12, and up to 5 of the highest confidence detections from PSI-BLAST13 or HHSearch11, to select the template for comparative modeling. After selecting templates, a parametric alignment ensemble was generated using the K*Sync alignment method23. Compared to the method used in CASP7, a more conservative approach was taken for generating the model ensemble for BLAST and PSI-BLAST targets by trimming less of the template at the regions adjoining loops to generate trimmed template variants for loop modeling. During loop modeling, an increased weight was used for the Rosetta radius of gyration (RG) score term to generate more compact loops. For modeling long loops (>17 residues), approximately ten fold more models were generated compared to the number generated for CASP7. In addition to the changes mentioned above, a number of bugs were discovered and fixed. The iterative-loosening of PSIBLAST23 E-value threshold for detecting the closest match to the target sequence was not functional in CASP7 due to a bug.
A large number of chain breaks existed in CASP7 Robetta models due to an error in the chain break filter. For CASP8, a more stringent filter was used that incrementally loosened when necessary to ensure at least 50 ensemble members, and as a result, significantly fewer models contained chain breaks.
Free-modeling targets
Significant changes were made in our de novo structure prediction protocol for CASP8 in an effort to produce high-resolution models. By taking advantage of the computing available through Rosetta@HOME, conformational sampling was dramatically increased, and all-atom refinement was carried out on all models. As in CASP721, 4000 query-sequence models and 2000 models each for up to two homologous sequences were generated using the Rosetta fragment replacement methodology. For CASP8, up to 300,000 query-sequence models were also generated followed by all-atom refinement using the Rosetta all-atom energy function. The 4000 models for the target sequence with the lowest all-atom energies were structurally clustered with the standard query-sequence and homolog-sequence model sets which were filtered down to 2000 query-sequence models and 1000 models for each homolog to ameliorate known pathologies such as low contact-order structures. The lowest energy all-atom models from each of the 5 largest clusters were returned as the final predictions ranked based on their Rosetta all-atom energies.
Results
Manual alignments vs. Automatic alignments
To evaluate potential improvements that can be achieved by manual alignment constructionby an expert compared to current automatic methods, we compared the quality of manual and automatic sequence alignments. We applied both reference-dependent measures of alignment quality assessing the consistency with gold-standard structure alignments, and reference-independent measures assessing the closeness of structural match suggested by the sequence alignment 24. We used two types of reference-independent measures. The first measure is based on the minimum-RMSD superposition of target and template structures, guided by the residue equivalences from the evaluated alignment. Given this superposition, the quality score is calculated according to the GDT_TS formula 25, i.e. by averaging the numbers of equivalent residue pairs that are placed within the distance of 1, 2, 4, and 8 A. As the second type of reference-independent measures, we used LiveBench contact 26 scores, which do not rely on a structure superposition but assess the similarity of residue contacts suggested by the sequence alignment. In about 80% of the cases, manual alignments are better than automatic (Figure S1).
As reference-dependent measures, we calculated alignment accuracy (fraction of correctly aligned residue pairs) and coverage compared to ‘gold-standard’ structural target-template alignments by DaliLite. The average accuracy of automatic and manual alignments was approximately 50% and 60%, respectively (Figure Ss1(A), inset). Global automatic alignments were slightly longer than by manual (‘cov’ of 0.79 vs 0.71, Figure S1(A) inset) but cover approximately the same fraction of structurally alignable residue pairs (‘Qcov’ of 0.69, Figure S1(A) inset). In contrast, local automatic alignments are on average shorter than manual alignments (coverage of 0.65, data not shown) and may miss parts of the structure core.
As an example, Figure S1(B) and (C) show the structures of target T0489 and its best template (PDB ID 1j7n). The local HHsearch alignment only covers the green region whereas the manual alignment covers the green region and the orange region. The manual alignment is carefully extended over the whole structural core without compromising much of the alignment quality. While this alignment does accurately capture the relationship between template and target, it also enforces a notable structural mistake, as T0489’s N-terminal helix (Figure S1(B)) itself has a large relative displacement to the closest template (marked by arrows in Figure S1 (B) and (C)). In the target structure, this helix is almost perpendicular to the central beta-sheet, whereas in the template this helix is largely parallel to the beta-sheet. This change in helix packing is caused by the difference in the structural environment: T0489 is a single-domain protein, whereas the template has an additional N-terminal domain (not shown). The large displacement of the homologous secondary structure elements contributed to the difference between alignment quality measures, GDT-like and LiveBench contact scores. GDT-like score is more sensitive to large structural movements than the contact-based measure; thus it more heavily penalized the manual alignment of N-terminal helices.
Model Quality
In this section, we discuss our results for the targets classes described above. We exclude the four targets with high sequence similarity templates. The GDT-TS Z-scores for all targets were based on the evaluations of CASP8 predictions by the Grishin group27.
Medium sequence similarity template(s)
18 target domains fell into this category. In almost all cases, the template we used was one of the top templates identified by the assessors. Rosetta was able to improve upon the best template in 12 of the 18 targets (Figure 2(B)). A mean GDT-TS Z-score of 0.7 for predictions in this category (Figure 2(A)) shows that Rosetta was successful on these targets on a relative scale (see Figure 2(C)).
Figure 2. Model quality.

(A) Distribution of GDT-TS Z-scores of the best Rosetta model for high and medium sequence similarity template(red) low sequence similarity template(blue) fold recognition/free modeling targets(green). The dashed lines represent the mean of the distribution: 0.68 for high and medium sequence similarity template, 1.28 for low sequence similarity template and 1.5 for fold recognition/free modeling targets.
(B) Comparison of the GDT-HAs over the structurally alignable regions of the best starting model used vs. best-submitted Rosetta model.
(C) Comparison of the sequence-dependent LGA of the best template (identified by the assessors) vs. best-submitted Rosetta model.
Particularly noteworthy in this category is the atomic-level accuracy prediction of T0492 (Figure 3(A)). The first submitted model is a significant improvement over the best template (model01 GDT-HA is 74.64 and template PDB 2gcx GDT-HA is 48.57; the GDT-TS Z-score is 3.14). The core side-chains were in close agreement with the native structure (Figure 4(A)), illustrating the power of all-atom refinement.
Figure 3. Examples of successful template-based predictions.

For each target, the native structure is shown in blue, our best-submitted model in red and best template in green. (A) T0492 (B) T0464 (C) T0429 domain 1 (D) T0487 domain 4 (E) T0407 domain 2 (F) T0457 domain 2
Figure 4. Examples of predictions with atomic-level accuracy.
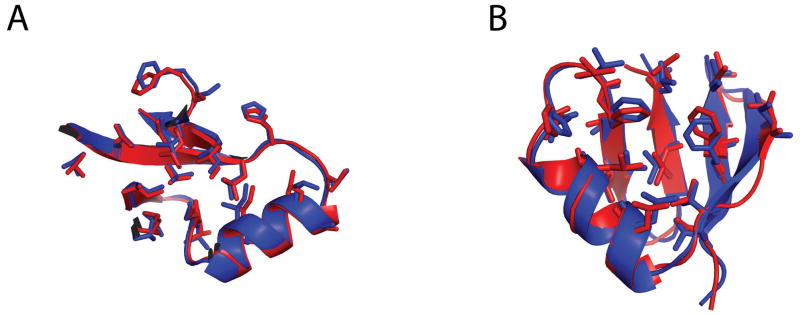
The core sidechains of our of best-submitted model (red) and native (blue) are highlighted. (A) T0492 domain 1 (B) T0513 domain 2 (predicted by the BAKER-ROBETTA server)
Domain 1 of T0429 (model04 GDT-HA is 66.79 and template PDB 2ef1 GDT-HA is 62.5; the GDT-TS Z-score is 2.31) was predicted at high resolution (Figure 3(C)). We identified an internal sequence duplication in the full-length T0429 suggesting two homologous domains. Both domains were independently refined by the iterative rebuild-and-refine protocol. The centers of the lowest energy clusters for both domains were assembled by low-resolution docking followed by refinement. Many of the loop regions improved over the template and the core sidechains were largely placed correctly.
Low sequence similarity template(s)
Of the 32 target domains in this category, 8 had a GDT-TS Z-score greater than 2.0; of these 3 had a GDT-TS Z-score greater than 3.0 (Figure 2(A)). The aggressive modeling strategy starting from a large pool of alternative target-template alignments overcame to some extent the ambiguity in the template and alignment selection. We discuss here two notable examples.
The submitted models for T0464 were quite good, with Z scores up to 3.0. On the basis of sequence similarity and alignment coverage, several suitable templates were identified for T0464. All-atom energy-based template selection as described in the methods clearly identified 2pk7 as the closest template (Figure 1(A)). Several iterations of the rebuild-and-refine method were carried out, primarily focusing sampling on the ~25 residue insertion in the target sequence. All five of our submissions were ranked as the top predictions, and subsequently our best prediction was released as a CASP refinement target to the community which was nearly 30 GDT-TS units improvement over the starting template (model05 GDT-TS is 78.77 and GDT-TS Z-score is 2.88) (Figure 3(B)). We further improved the model accuracy in the refinement experiment (starting model GDT-TS: 77 and refined model GDT-TS: 81) by all-atom loop-modeling around the two segments identified by the assessors as being in error.
T0487 is the largest structured protein ever evaluated in CASP (685 residues). The overall strategy for predicting the structure of this protein involved refining each domain separately to test different alignment variants, assembling the best individually refined domains onto the full-length 2f8s template, and refining the complete model again. Out of the five domains comprising this target, we did reasonably well on two (T0487 domain 2 model01 GDT-TS is 50.37 and GDT-TS Z-score is 1.28; T0487 domain 5 model01 GDT-TS is 59.63 and GDT-TS Z-score is 1.73) and very well on T0487 domain 4 (model01 GDT-TS is 79.21 and GDT-TS Z-score is 4.50). Two template sequences (1yvu/2f8s and 1u04) that correspond to the full-length target were identified with BLAST, while additional template sequences related to individual domains were identified with PSI-BLAST (1w9h includes domain 1 and 5, and 1r4k, 1si2, and 1r6z include domain 3). A structure based alignment of identified templates displayed sequence regions with an inconsistent hydrophobicity profile. Because the hydrophobicity patterns of a template with lower resolution (2f8s) agreed better with the PROMALS target family alignment than the template with the best resolution (1u04), the lower resolution template (2f8s) was chosen.
The fourth domain of T0487 (res. 177–265) forms an SH3-like barrel known as a PAZ domain. Of the identified templates, the individual PAZ domain of human eIF2c1 bound to a 3′ siRNA-like deoxynucleotide overhang (1si2) represented the closest non-NMR template sequence28. A PROMALS3D multiple alignment of this domain to all templates was adjusted manually to preserve hydrophobicity patterns and conserved residues in the 3′ overhang-binding site. Two templates were chosen for initial refinement: 1) the individual PAZ domain template 1si2 and 2) a hybrid of this template substituting the first 19 residues from the full-length template 2f8s. Various alignment variants were tested by model refinement, with the lowest energy model corresponding to one alignment variant with the hybrid template. After assembly of this alignment variant into the full-length template 2f8s, subsequent refinement produced the excellent model shown in Figure 3(D).
Fold recognition/free modeling targets
This category contains 13 target domains that were predicted by free modeling. Where remote templates could be detected, both template-based and free-modeling protocols were carried out. Of the 13 targets, 9 had a GDT-TS Z-score greater than 2.0, of these 5 had a GDT-TS Z-score greater than 3.0 (Figure 2(A)). Some of the major successes in this category included T0405 domain 1, T0407 domain 2, T0460, T0467, T0468 (closely related to T0467), T0482 and T0496 domain 1 (one of the two ‘new fold’ targets in CASP8).
Target T0476 (model01 GDT-TS is 50.00 and GDT-TS Z-score is 3.31) was predicted using a combination of template-based and free modeling methods (Figure 5(E)). We surmised that the four cysteines in the target sequence (residues 4, 7, 47 and 50) might be involved in coordinating a metal ion and hence will be in close spatial proximity in the native structure. We filtered the threaded models based on several alternative alignments to distant templates for models that satisfied this spatial requirement. The N-terminal part of the sequence was based on 2q5h and the C-terminal part was modeled de novo in the context of the rest of the protein. When compared to the native structure, the template-based part agreed well while the de novo part deviated significantly.
Figure 5. Examples of successful predictions in the fold-recognition/free-modeling category.

In each panel, the native structure is on the left and our best-submitted model on the right. (A) T0405 (B) T0460 (C) T0467 (D) T0468 (E) T0476 (F) T0482 (G) T0496 domain 1 (H) T0513 (prediction made by BAKER-ROBETTA server)
T0460 is a successful prediction resulting from the improved fragment assembly protocol developed after CASP7 (Figure 5(B))(model03 GDT-TS is 59.27 and GDT-TS Z-score is 4.85). This protocol uses a broad range of fragment sizes in place of the constant 9-mer and 3-mer sets used in previous experiments. For different fragment lengths, different types of fragments are found, and making models with these different sets considerably increases model diversity. This increased diversity can be extremely valuable at the all-atom refinement stage, since low resolution models that are within 2–3Å of the native structure can frequently be distinguished based on their very low energies after all-atom refinement. In each independent trajectory, two randomly selected fragment lengths were used to avoid any one fragment set from overly dominating the set of produced models. For T0460 using a selection from the range of 5–12-mers instead of 9-mers, and then 3–9mers instead of 3-mers, increases the average GDT-TS of the best 1% models by GDT-TS from 53.92 to 61.66.
For the all-beta T0468, the Rosetta de novo method was modified to disfavor the formation of local strand pairings connected by a hairpin (Figure 5(D)). As noted previously30,31, the Rosetta de novo method forms low contact order strand pairings more frequently than observed in native protein structures. To encourage formation of long-range of strand pairings, stochastically selected low contact order strands were energetically penalized. Models not in the top 25% by contact order and top 20% by energy were automatically rejected. Our model 3 (model03 GDT-TS is 59.48 and GDT-TS Z-score is 2.42) was the best submitted prediction for this target.
Refinement targets
The CASP8 refinement challenges provided an excellent test of Rosetta’s all-atom energy function and refinement methods, independent of alignment and template identification. Our refinement methods improved the GDT-HA of the starting model provided by the organizers in 7 of 12 cases. Three challenges in which the refined model was better than the starting model are shown in Figure 6. For TR432, the all-atom loop-modeling followed by constrained minimization yielded the best model submitted for this target (starting model GDT-HA 77.9 and our best model GDT-HA 80.2) (Figure 6(A)). For TR488, our models 1 to 3 were the most accurate models submitted (Figure 6(B); GDT-HA of 81.1, 82.1, and 78.2, compared to 75.3 for the starting model); for the first two structures, we explicitly remodeled the ligand-binding loop using the inverse kinematic loop modeling protocol32,33 incorporated into the latest object-oriented version of Rosetta. Finally, as discussed above, the Rosetta all-atom force field was able to identify energetically favorable localized conformation changes from the starting structure for TR464 (Figure 6(C) and (D); starting model GDT-HA is 53.4 and submitted model01 GDT-HA is 58.2), which itself was a model that had undergone intense Rosetta refinement from a starting template.
Figure 6. Examples of successful predictions in the refinement category.

For each target, the native structure is shown in blue, our best-submitted model in red and starting model in green (A) TR432 residues 32–47 (B) TR488 residues 11–18 (C) TR464 residues 19–27 (D) TR464 residues 39–44
Robetta server results
In general, Robetta’s performance compared to other servers improved as the target’s difficulty increased. The average GDT-TS Z-scores of the best Robetta models with high, medium and low sequence similarity templates, and for fold recognition and free modeling targets were 0.20, 0.23, 0.66 and 1.42 respectively. For T0416 domain 2(server-only GDT-TS Z-score is 3.0) and T0464(server-only GDT-TS Z-score is 2.41), increasing the sampling for modeling long loops may have been a factor in their successful predictions. T0416 domain 2 is a domain insertion and was modeled as a long loop in the context of a BLAST template, and T0464 contained a 33 residue insertion. T0462(server-only Z-score is 3.61) consisted of two domains modeled with different templates that were successfully assembled by Robetta’s domain assembly method34. Among all predictors Robetta’s best model for T0449 was the top performer with a GDT-TS Z-score of 1.70 and 3.10 among human/server and server only predictors, respectively.
Among the top performing de novo Robetta models, the server only target, T0513 domain 2, particularly stood out (Figure 5(H)). For this target, increased sampling and all-atom minimization using Rosetta@HOME, and ranking based on all-atom energy led to a successful high-resolution prediction for model01 which had a GDT-TS score of 70.65(server-only GDT-TS Z-score is 2.72). As shown in Figure 4(B), model01 had an RMSD to the native structure of 0.84 Å over 39 residues with accurate placement of core side-chains. This was an outstanding result considering it was from a fully automated method and is comparable to the best human predictions among all de novo targets.
What went wrong
The goal of protein structure prediction is to produce high accuracy models for every protein sequence. Our CASP8 predictions that did not reach atomic accuracy illustrate that the considerable amount of methods development still required to achieve this goal. As we have observed previously, the primary barrier to more accurate structure prediction is conformational sampling: for most of the cases where we failed to produce a model with atomic level accuracy, the Rosetta refined crystal structure has lower energy than any model we generated during CASP8. Developing more effective conformational sampling algorithms and protocols is a critical area for current research in protein structure prediction.
In cases where we did produce good models we generally failed to rank these as the first of our five submissions. This issue is also largely due to inadequate conformational sampling. As noted earlier, the Rosetta all atom energy decreases rapidly as conformations approach within 2Å rmsd of the native state and the native jigsaw-puzzle like sidechain packing starts to be achieved. However, if no structures generated are within 2Å rmsd of the native structure, even the most accurate of the models will have incorrectly modeled regions that can considerably increase the overall energy. Thus, amongst a population of models greater than 2–3Å from the native structure, the lowest energy models are not necessarily more accurate than other models, and hence ranking based on energy will often fail to identify the best model. In such cases the most effective strategy has been to cluster the lowest energy models and submit the cluster centers, but there is no rigorous way to rank these predictions.
Conclusions
The performance of Rosetta in CASP8 was quite good, with models considerably improved over the best template for 24 of the 71 domains. For CASP8, we used a completely rewritten object-oriented version of Rosetta which was recently publicly released. The modularity of Rosetta3 made it straightforward to try out many new ideas and approaches. Our methods evolved considerably during CASP8 and different strategies were used for different targets. Since CASP8, we have set up a comprehensive benchmark of comparative modeling challenges derived from previous CASPs and we are testing each of the protocols used in CASP together with alternative approaches for generating alignment ensembles. Our goal is to have an almost completely automated and consistent protocol ready for CASP9 that can be applied to any protein sequence.
Acknowledgments
We thank the participants of Rosetta@HOME distributed computing network, the Blue Gene supercomputer at the Argonne Leadership Computing Facility and the Ranger supercomputer at the Texas Advanced Computing Center for providing the computing resources for this work. This work was supported by NIH GM76222 (DB), GM67165 (NVG) and Welch foundation grant I1505 (NVG). We thank Patrick Barth, Ian Davis, Andrew Leaver-Fay, Hua Cheng and Yong Wang for help with predictions.
References
- 1.Qian B, Raman S, Das R, Bradley P, McCoy AJ, Read RJ, Baker D. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450(7167):259–264. doi: 10.1038/nature06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 3.Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309(5742):1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 4.Baker D, Sohl JL, Agard DA. A protein-folding reaction under kinetic control. Nature. 1992;356(6366):263–265. doi: 10.1038/356263a0. [DOI] [PubMed] [Google Scholar]
- 5.Anfinsen CB, Haber E, Sela M, White FH., Jr The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc Natl Acad Sci U S A. 1961;47:1309–1314. doi: 10.1073/pnas.47.9.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morozov AV, Kortemme T, Tsemekhman K, Baker D. Close agreement between the orientation dependence of hydrogen bonds observed in protein structures and quantum mechanical calculations. Proc Natl Acad Sci U S A. 2004;101(18):6946–6951. doi: 10.1073/pnas.0307578101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35(2):133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 8.Das R, Baker D. Macromolecular modeling with rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 9.Sadreyev R, Grishin N. COMPASS: a tool for comparison of multiple protein alignments with assessment of statistical significance. J Mol Biol. 2003;326(1):317–336. doi: 10.1016/s0022-2836(02)01371-2. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Sadreyev RI, Grishin NV. PROCAIN: protein profile comparison with assisting information. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21(7):951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 12.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 13.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadreyev RI, Tang M, Kim BH, Grishin NV. COMPASS server for remote homology inference. Nucleic Acids Res. 2007;35(Web Server issue):W653–658. doi: 10.1093/nar/gkm293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pei J, Grishin NV. PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics. 2007;23(7):802–808. doi: 10.1093/bioinformatics/btm017. [DOI] [PubMed] [Google Scholar]
- 16.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v. 3. Bioinformatics. 2008;24(23):2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Schueler-Furman O, Andre I, London N, Fleishman SJ, Bradley P, Qian B, Baker D. RosettaDock in CAPRI rounds 6–12. Proteins. 2007;69(4):758–763. doi: 10.1002/prot.21684. [DOI] [PubMed] [Google Scholar]
- 18.Sheffler W, Baker D. RosettaHoles: rapid assessment of protein core packing for structure prediction, refinement, design, and validation. Protein Sci. 2009;18(1):229–239. doi: 10.1002/pro.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canutescu AA, Dunbrack RL., Jr Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003;12(5):963–972. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradley P, Baker D. Improved beta-protein structure prediction by multilevel optimization of nonlocal strand pairings and local backbone conformation. Proteins. 2006;65(4):922–929. doi: 10.1002/prot.21133. [DOI] [PubMed] [Google Scholar]
- 21.Das R, Qian B, Raman S, Vernon R, Thompson J, Bradley P, Khare S, Tyka MD, Bhat D, Chivian D, Kim DE, Sheffler WH, Malmstrom L, Wollacott AM, Wang C, Andre I, Baker D. Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home. Proteins. 2007;69 (Suppl 8):118–128. doi: 10.1002/prot.21636. [DOI] [PubMed] [Google Scholar]
- 22.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19(8):1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 23.Chivian D, Baker D. Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Res. 2006;34(17):e112. doi: 10.1093/nar/gkl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pei J, Grishin NV. MUMMALS: multiple sequence alignment improved by using hidden Markov models with local structural information. Nucleic Acids Res. 2006;34(16):4364–4374. doi: 10.1093/nar/gkl514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zemla A, Venclovas C, Moult J, Fidelis K. Processing and analysis of CASP3 protein structure predictions. Proteins-Structure Function and Genetics. 1999:22–29. doi: 10.1002/(sici)1097-0134(1999)37:3+<22::aid-prot5>3.3.co;2-n. [DOI] [PubMed] [Google Scholar]
- 26.Rychlewski L, Fischer D, Elofsson A. LiveBench-6: Large-scale automated evaluation of protein structure prediction servers. Proteins-Structure Function and Genetics. 2003;53(6):542–547. doi: 10.1002/prot.10535. [DOI] [PubMed] [Google Scholar]
- 27.Shi S, Pei J, Sadreyev R, Kinch LN, Majumdar I, Tong J, Cheng H, Kim BH, Grishin NV. Analysis of CASP8 targets, predictions and assessment methods. Database: The Journal of Biological Database and Curation. 2009 doi: 10.1093/database/bap003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma JB, Ye K, Patel DJ. Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature. 2004;429(6989):318–322. doi: 10.1038/nature02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4(1):1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 30.Bonneau R, Ruczinski I, Tsai J, Baker D. Contact order and ab initio protein structure prediction. Protein Sci. 2002;11(8):1937–1944. doi: 10.1110/ps.3790102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradley P, Malmstrom L, Qian B, Schonbrun J, Chivian D, Kim DE, Meiler J, Misura KM, Baker D. Free modeling with Rosetta in CASP6. Proteins. 2005;61 (Suppl 7):128–134. doi: 10.1002/prot.20729. [DOI] [PubMed] [Google Scholar]
- 32.Mandell D, Coustias EA, Kortemme T. Sub-Angstrom Accuracy in Protein Loop Reconstruction by Robotics-Inspired Conformational Sampling. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coustias EA, Seok C, Wester MJ, Dill KA. Resultants and Loop Closure. International Journal of Quantum Chemistry. 2006;106:176–189. [Google Scholar]
- 34.Wollacott AM, Zanghellini A, Murphy P, Baker D. Prediction of structures of multidomain proteins from structures of the individual domains. Protein Sci. 2007;16(2):165–175. doi: 10.1110/ps.062270707. [DOI] [PMC free article] [PubMed] [Google Scholar]