

Abstract
A series of aminoacyl-triazine derivatives based upon the pan-PI3K inhibitor ZSTK474 were identified as potent and isoform-selective inhibitors of PI3Kβ. The compounds showed selectivity based upon stereochemistry with l-amino acyl derivatives preferring PI3Kβ, while their d-congeners favored PI3Kδ. The mechanistic basis of this inhibition was studied using site-directed mutants. One Asp residue, D862, was identified as a critical participant in binding to the PI3Kβ-selective inhibitors, distinguishing this class from other reported PI3Kβ-selective inhibitors. The compounds show strong inhibition of cellular Akt phosphorylation and growth of PTEN-deficient MD-MBA-468 cells.
Keywords: PI3 kinase, p110β, ZSTK474, cancer
The phosphatidylinositol 3-kinases (PI3K) are a family of lipid kinases that regulate intracellular signaling for numerous cellular events such as cell migration, growth, and survival.1 Because of their important roles in signal transduction, dysregulation in the PI3K pathway could lead to various diseases including cancer.2 Two of the key genetic alterations identified for this pathway in cancer are mutation in the PIK3CA gene, which encodes p110α (PI3Kα catalytic subunit),3 and the loss of the tumor suppressor, phosphatase and tensin homologue (PTEN).4 Recently, there has been accumulating evidence showing that PI3Kβ plays a key role in tumorigenesis driven by PTEN loss.5 It was found that down-regulation of the PIK3CB gene, which encodes p110β (PI3Kβ catalytic subunit), inhibited PI3K signaling as well as growth both in vitro and in vivo. In an animal prostate cancer model, the ablation of p110β blocked tumorigenesis and decreased Akt phosphorylation, but the same results were not observed by p110α ablation.6
The majority of inhibitors currently in clinical trials are class I pan-PI3K inhibitors.7 Because each of the PI3K isoforms has their own although overlapping physiological roles, isoform-selective PI3K inhibitors may hold some therapeutic advantages with respect to reducing off-target effects. In recent years, isoform-selective inhibitors have emerged, and compounds such as BYL719, a PI3Kα selective inhibitor, and CAL-101,8,9 a PI3Kδ-specific inhibitor, are now in phase I/II clinical trials. While these and other isoform-selective inhibitors have been reported, there is still relatively scant understanding of the mechanisms underpinning selectivity. From the structure of the adenosine triphosphate (ATP)-bound state of the enzyme (PDB: 1E8X), it is apparent that much of the inner core of the binding site is highly conserved across the class 1 isoforms.10 However, two nonconserved regions of the binding site have also been identified as capable of executing selective interactions with inhibitors (Figure 1). Region 1 from PI3Kβ 855 to 862 encompasses a loop that sits under the ribose pocket and influences the binding of PI3Kα selective inhibitor A-66 (a BYL719 analogue).11 Region 2 from PI3Kβ 772–788 corresponds to the protein kinase “P-loop”.11 This region has been shown to dictate selective inhibitor binding in a number of ways—first, a conserved methionine residue can shift to expose a cryptic binding pocket, as in the so-called “propeller-shaped” mode of binding of PI3Kδ-selective compounds like PIK-39 (a CAL-101 analogue).12 Other PI3Kδ-selective inhibitors have been shown to access a “tryptophan shelf” adjacent to a nonconserved threonine. Also, the PI3Kα-selective inhibitors PIK75 and J-32 have been shown to be sensitive to mutation at nonconserved residues of region 2.11 There are clearly other mechanisms by which inhibitors exhibit isoform selectivity. This is especially apparent in PI3Kγ-selective inhibitors that show interaction only with conserved residues of the binding site and induce little conformational change from the ATP-bound state.13
Figure 1.
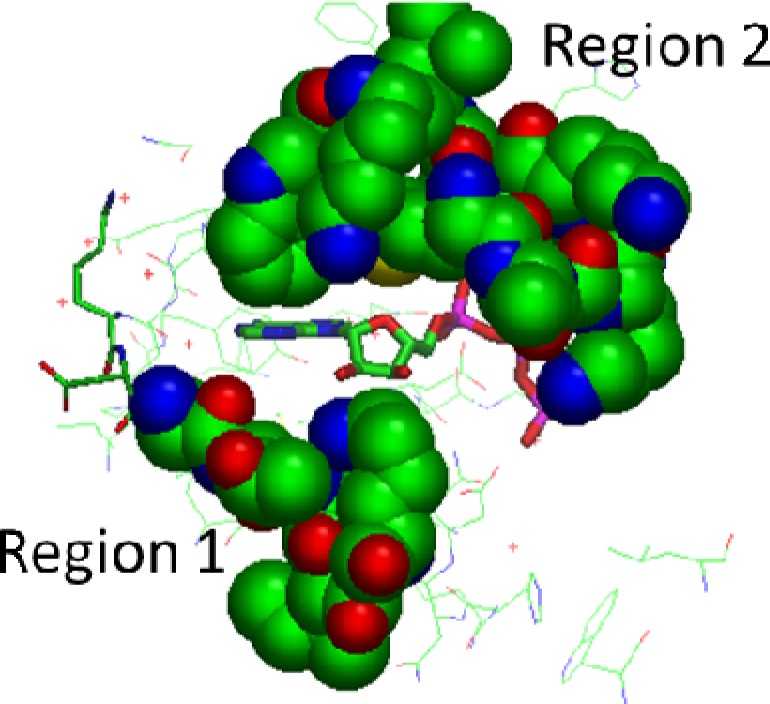
Nonconserved regions 1 and 2 of the PI3Kγ binding site (shown in space-filling representations). PDB code: 1E8X.
Until recently, the pharmacological evaluation of PI3Kβ inhibition has rested upon a series of compounds described by Thrombogenix/Kinacia.14,15 These included TGX221 and KN309 (Figure 2). Astra-Zeneca has subsequently progressed the optically pure R-enantiomer of KN309 into human trials as AZD6482 (aka KIN193).16 The molecular basis for the observed selectivity has not yet been established, although Knight et al. proposed that TGX280 (PIK108) could be accommodated in region 2, via the “propeller-shaped” mode described above. We had previously ruled out an interaction with the region 1 residues as the basis for selectivity.17
Figure 2.

Structures of PI3K inhibitors.
Recently, new PI3Kβ-selective compounds reminiscent of TGX221 have been reported. GSK2636771 has progressed to phase I/IIa clinical trials to treat advanced solid tumors with PTEN deficiency. Other analogous series have been described,18−23 and cocrystals of compounds with PI3Kγ (PDB: 4FJY, 4FJZ, and 4G11) and PI3Kδ (4AJW) support the analogy between these and the “propeller-shaped” compounds. The only reported X-ray structure of PI3Kβ in complex with the pan-PI3K inhibitor GDC9041 (PDB: 2Y3A) displays a conventional “flat” binding pose.24
Our hypothesis was that isoform-selective inhibition of PI3Kβ should be achievable by targeting the nonconserved residues of region 1. In particular, the acidic Asp862 residue at PI3Kβ is exchanged for Gln, Lys, and Asn in PI3Kα, -γ, and -δ, respectively. Adaptation of the pan-PI3K inhibitor ZSTK474 (Figure 2),25 which projects one morpholinyl ring toward this region, was considered the logical template compound.12 We have identified compounds that selectively inhibit PI3Kβ and moreover have demonstrated the role of D862 by showing that mutagenesis of that residue prevents inhibition by these compounds.
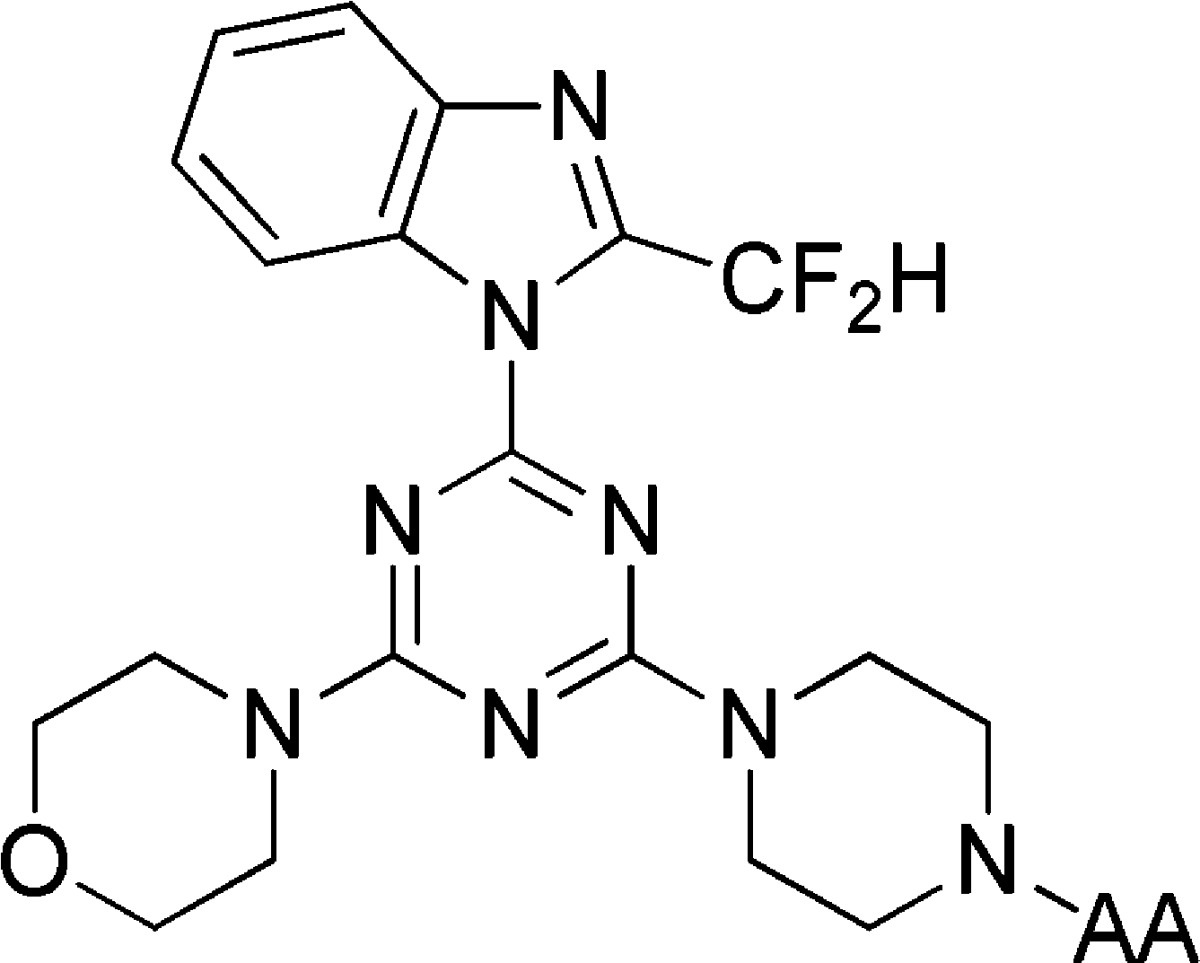
The synthesis of the substituted triazines was performed largely as previously reported (see the Supporting Information).26−28 Condensation of protected amino acid derivatives to piperazinyl analogues of ZSTK474 yielded the N-protected derivatives, which could be deprotected to give the free aminoacyl compounds 17 and 19–28. Alternatively condensation with N-acetylglycine or N,N-dimethylglycine gave the products 18 and 29, respectively.
The PI3K assay data are summarized in Table 1 and identify that the substituent has a significant influence upon isoform selectivity, consistent with our hypothesis. Compound 17 showed potent dual inhibition of PI3Kβ and PI3Kδ with very poor inhibition of the PI3Kα and PI3Kγ isoforms. Incorporation of an N-acetylglycine residue 18 resulted in pan-PI3K potency, suggesting the importance of the amino function to the observed selectivity.
Table 1. Structure and Inhibition of PI3K Isoforms by ZSTK474 Analogues.
| PI3K isoform IC50 (nM)a |
||||||
|---|---|---|---|---|---|---|
| compd | AA residue | α | β | γ | δ | β/δ selectivity ratio |
| ZSTK474 | 6 | 6 | 38 | 3 | 0.5 | |
| 17 | Gly | 1500 | 35 | 9900 | 110 | 3.0 |
| 18 | Ac-Gly | 42 | 54 | 380 | 26 | 0.48 |
| 19 | l-Ala | 3500 | 31 | 3600 | 490 | 15.7 |
| 20 | d-Ala | 3700 | 1000 | 2000 | 70 | 0.070 |
| 21 | l-Phe | 4700 | 63 | >100 μM | 2200 | 34.8 |
| 22 | d-Phe | >10 μM | >10 μM | >10 μM | 3600 | <0.36 |
| 23 | l-Ile | 6300 | 67 | >10 μM | 1000 | 15.6 |
| 24 | l-Tyr | 3300 | 220 | >10 μM | 1200 | 5.4 |
| 25 | l-Pro | 2200 | 26 | >10 μM | 390 | 15.1 |
| 26 | l-Lys | 2300 | 50 | >10 μM | 670 | 13.4 |
| 27 | β-Ala | 720 | 98 | 3900 | 58 | 0.6 |
| 28 | N-Me-l-Ala | 1500 | 260 | 8000 | 290 | 1.1 |
| 29 | N,N-diMeGly | 1600 | 2400 | >10 μM | 330 | 0.14 |
IC50 values are means of at least two duplicate experiments (Kinase-Glo). Standard errors were all within 25% of the mean. Assays were performed using 10 μM ATP.
Compounds 19–29 were all PI3Kβ and/or PI3Kδ preferring ligands. Strikingly, the introduction of an l-alanine substituent resulted in a compound 19 with 15-fold selectivity for PI3Kβ over PI3Kδ, while the opposite stereoisomer with a d-alanine substituent 21 showed 15-fold selectivity for PI3Kδ over PI3Kβ. Pursuing the potential for l-amino acids providing for PI3Kβ selectivity in general held true. Most selective was l-Phe (21), which showed 35-fold selectivity for PI3Kβ over PI3Kδ with the l-Ile (23), l-Tyr (24), l-Pro (25), and l-Lys (26) derivatives showing 6–16-fold selectivity. Homologation to β-Ala (27) and further substitution on the amino group as in 28 and 29 dropped the preference for PI3Kβ significantly.
To test the premise that the observed PI3Kβ selectivity was due to the interaction between the primary amino group of inhibitors and the nonconserved residues of the binding site, compounds were tested against a variety of mutant forms of PI3Kβ where the nonconserved residues were exchanged for their equivalent residues in PI3Kα.
The PI3Kβ-selective compounds were very sensitive to mutation at region 1 residue D862Q (see Supplementary Figure 1 in the Supporting Information). The IC50 values were determined for 19, 21, and 23 against the WT and mutant D862Q form as well as the WT and reciprocal mutant in PI3Kα (Table 2). Note that the mutations do not hinder the catalytic properties of the mutant enzymes (Supplementary Table 1 in the Supporting Information). Each of the compounds shows a greater than 10-fold change in IC50 against the mutant form of PI3Kβ. The IC50 of 21 drops 82-fold as compared to the wild-type isoform upon introduction of the D862Q mutation. The reciprocal mutation in PI3Kα D859Q does not restore inhibitory potency of 21 at PI3Kα, although some enhancement in potency is observed for compounds 19 and 23. These data clearly distinguish the mechanism of selectivity of these compounds from that of TGX221 and TGX286, which are unaffected by the mutation D862Q in PI3Kβ.17
Table 2. Inhibition of PI3K Isoforms and in Vitro Mutants by ZSTK474 Analogues.
| PI3K isoform IC50 (nM)a |
||||||
|---|---|---|---|---|---|---|
| compd | PI3Kα WT | PI3Kα Q859D | PI3Kβ WT | PI3Kβ D862Q | fold change αWT→ αQ859D | fold change βWT→ βD862Q |
| 19 | 8200 | 2300 | 62 | 890 | ↓3.5 | ↑14 |
| 21 | 12000 | 8100 | 74 | 6000 | ↓1.4 | ↑82 |
| 23 | 9200 | 2800 | 140 | 1400 | ↓3.3 | ↑10 |
IC50 values are means of at least two duplicate experiments (Kinase-Glo). Assays were performed using 100 μM ATP. A full table including standard errors is provided as Supplementary Table 2 in the Supporting Information.
The data from these studies suggest a clear and dramatic influence of aminoacyl substitution upon PI3K binding as compared to ZSTK474. It should be noted that in all cases, the inhibitory potency is poorer against every isoform, but the loss of affinity is more modest at PI3Kβ. The presence of the acidic group D862 in PI3Kβ is effectively rescuing binding. The conformational restriction engendered by the l-α-substituent restricts the interaction to D862 alone and blocks possible interaction with E858, which is also an acidic residue in PI3Kδ (D831). This may also explain the potency of 17 and 20 at PI3Kδ.
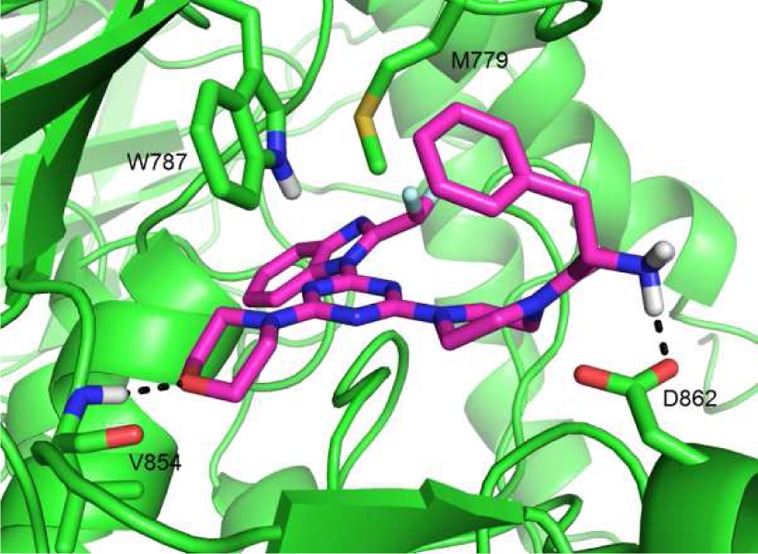
Docking simulations support this model. In the wild-type enzyme, 21 adopted the anticipated pose closely overlaying the reported pose of ZSTK474 in PI3Kδ12 and making a specific interaction of the free amine with D862 (Figure 3). In the D862Q mutant form, not only is this specific interaction lost, but the compound can no longer adopt this pose, suggesting that the aspartyl residue acts to compensate for other unfavorable interactions associated with the inclusion of a phenylalanine substituent.
Figure 3.
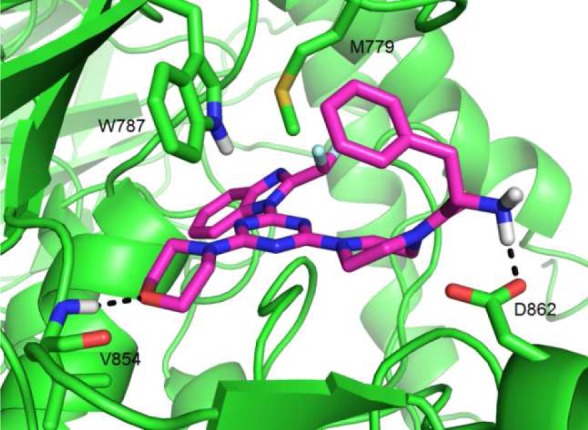
Docking solution for compound 21 into a model of PI3Kβ derived from crystal structure PDB: 2Y3A.
Finally, we assessed whether these compounds exhibited cell-based activity that might make them worthy of further pharmacological study and comparison to other recently reported PI3Kβ inhibitors.19 The PTEN-deficient cell line MDA-MB-468 was used to measure inhibition of Akt phosphorylation and inhibition of cell growth. Dose-dependent inhibition of both activities was observed with each of the inhibitors tested. Compound 21 showed strong cellular inhibition of Akt phosphorylation relative to the direct enzyme assay with IC50 values <10 nM (Supplementary Figure 2 in the Supporting Information). Compound 21 showed comparable potency to ZSTK474 in cell growth despite both lower potency at PI3Kβ and poor potency at the other isoforms (Table 3). While improved cell permeability relative to ZSTK474 may provide one explanation for this efficacy, other factors may be contributing such as cellular metabolism or activity at other PI3K pathway enzymes. In a screen (KinomeScan) of 21 versus another 96 kinases, only the class II PI3K PI3KC2β showed binding affinity comparable to that seen for PI3Kβ. The results generally mirror that seen for ZSTK474 itself29 (Supplementary Table 3 in the Supporting Information).
Table 3. Inhibition of Cell Growth in Breast Cancer Cell Line by ZSTK474 Analogues.
| MDA-MB-468 growth EC50 (μM) | |
|---|---|
| ZSTK474 | 3.2 ± 1.4 |
| 17 | 15 ± 5.3 |
| 19 | 13 ± 1.2 |
| 21 | 4.6 ± 1.1 |
| 23 | 11 ± 1.1 |
In summary, on the basis of the observed X-ray structure of ZSTK474 in PI3Kδ, a series of l-aminoacylpiperazine-substituted analogues have been prepared that exhibit excellent potency and selectivity for PI3Kβ isoforms. The configuration of the amino acids is pivotal to the selectivity, underpinning a well-defined interaction with the nonconserved binding site residue D862. The inhibitors show an alternate mechanistic basis for selectivity in comparison to other recently reported selective inhibitors. The compounds show inhibition of PI3Kβ-dependent function in PTEN-deficient cancer cells and effectively inhibit growth of the cell line. The compounds provide a basis for the further study of PI3Kβ function in a number of disease contexts.
Glossary
Abbreviations
- ATP
adenosine triphosphate
- Fmo
fluorenylmethyloxycarbonyl
- PI3K
phosphoinositide 3-kinase
- PTEN
phosphatase and tensin homologue
Supporting Information Available
Full experimental details relating to the synthesis of compounds and methods for biochemical and cellular assays. This material is available free of charge via the Internet at http://pubs.acs.org.
J.-A.P. and M.S.M. are recipients of Australian Postgraduate Award (APA) Scholarships. M.S.M. was the recipient of a top-up scholarship from the CRC for Cancer Therapeutics. This work was funded through the National Institutes of Health Grants CA43460 and CA62924, the Virginia and D.K. Ludwig Fund for Cancer Research (USA), the Cancer Council Victoria No. 436708, and a National Health and Medical Research Council Grant No. 545943 (Australia).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Vanhaesebroeck B.; Stephens L.; Hawkins P. PI3K signalling: the path to discovery and understanding. Nat. Rev. Mol. Cell. Biol. 2012, 13, 195–203. [DOI] [PubMed] [Google Scholar]
- Courtney K. D.; Corcoran R. B.; Engelman J. A. The PI3K Pathway As Drug Target in Human Cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y.; Wang Z. H.; Bardelli A.; Silliman N.; Ptak J.; Szabo S.; Yan H.; Gazdar A.; Powell D. M.; Riggins G. J.; Willson J. K. V.; Markowitz S.; Kinzler K. W.; Vogelstein B.; Velculescu V. E. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554–554. [DOI] [PubMed] [Google Scholar]
- Li J.; Yen C.; Liaw D.; Podsypanina K.; Bose S.; Wang S. I.; Puc J.; Miliaresis C.; Rodgers L.; McCombie R.; Bigner S. H.; Giovanella B. C.; Ittmann M.; Tycko B.; Hibshoosh H.; Wigler M. H.; Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- Wee S.; Wiederschain D.; Maira S. M.; Loo A.; Miller C.; deBeaumont R.; Stegmeier F.; Yao Y. M.; Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S.; Liu Z.; Zhang S.; Liu P.; Zhang L.; Lee S. H.; Zhang J.; Signoretti S.; Loda M.; Roberts T. M.; Zhao J. J. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth S. J.; Silva F. A.; Cecil A. R. L.; Tomassi C. D.; Hill T. J.; Raynaud F. I.; Clarke P. A.; Workman P. Progress in the Preclinical Discovery and Clinical Development of Class I and Dual Class I/IV Phosphoinositide 3-Kinase (PI3K) Inhibitors. Curr. Med. Chem. 2011, 18, 2686–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo J. J.; Furman M.; Winer E. S. CAL-101: A phosphatidylinositol-3-kinase p110-delta inhibitor for the treatment of lymphoid malignancies. Expert Opin. Invest. Drugs 2012, 21, 15–22. [DOI] [PubMed] [Google Scholar]
- Meadows S. A.; Vega F.; Kashishian A.; Johnson D.; Diehl V.; Miller L. L.; Younes A.; Lannutti B. J. PI3Kdelta inhibitor, GS-1101 (CAL-101), attenuates pathway signaling, induces apoptosis, and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma. Blood 2012, 119, 1897–1900. [DOI] [PubMed] [Google Scholar]
- Walker E. H.; Perisic O.; Ried C.; Stephens L.; Williams R. L. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 1999, 402, 313–320. [DOI] [PubMed] [Google Scholar]
- Zheng Z. H.; Amran S. I.; Zhu J. X.; Schmidt-Kittler O.; Kinzler K. W.; Vogelstein B.; Shepherd P. R.; Thompson P. E.; Jennings I. G. Definition of the binding mode of a new class of phosphoinositide 3-kinase alpha-selective inhibitors using in vitro mutagenesis of non-conserved amino acids and kinetic analysis. Biochem. J. 2012, 444, 529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt A.; Miller S.; Williams O.; Le D. D.; Houseman B. T.; Pacold J. I.; Gorrec F.; Hon W. C.; Liu Y.; Rommel C.; Gaillard P.; Ruckle T.; Schwarz M. K.; Shokat K. M.; Shaw J. P.; Williams R. L. The p110 delta structure: mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M.; Ruckle T.; Ji H.; Ardissone V.; Rintelen F.; Shaw J.; Ferrandi C.; Chabert C.; Gillieron C.; Francon B.; Martin T.; Gretener D.; Perrin D.; Leroy D.; Vitte P. A.; Hirsch E.; Wymann M. P.; Cirillo R.; Schwarz M. K.; Rommel C. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [DOI] [PubMed] [Google Scholar]
- Jackson S. P.; Schoenwaelder S. M.; Goncalves I.; Nesbitt W. S.; Yap C. L.; Wright C. E.; Kenche V.; Anderson K. E.; Dopheide S. M.; Yuan Y.; Sturgeon S. A.; Prabaharan H.; Thompson P. E.; Smith G. D.; Shepherd P. R.; Daniele N.; Kulkarni S.; Abbott B.; Saylik D.; Jones C.; Lu L.; Giuliano S.; Hughan S. C.; Angus J. A.; Robertson A. D.; Salem H. H. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat. Med. 2005, 11, 507–514. [DOI] [PubMed] [Google Scholar]
- Knight Z. A.; Gonzalez B.; Feldman M. E.; Zunder E. R.; Goldenberg D. D.; Williams O.; Loewith R.; Stokoe D.; Balla A.; Toth B.; Balla T.; Weiss W. A.; Williams R. L.; Shokat K. M. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006, 125, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander S.; Kull B.; Bjorkman J.; Ulvinge J. C.; Oakes N.; Emanuelsson B.; Andersson M.; Skarby T.; Inghardt T.; Fjellstrom O.; Gustafsson D. Human target validation of phosphoinositide 3-kinase (PI3K)beta; effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kbeta inhibitor. J. Thromb. Haemostasis 2012, 10, 2127–2136. [DOI] [PubMed] [Google Scholar]
- Frazzetto M.; Suphioglu C.; Zhu J.; Schmidt-Kittler O.; Jennings I. G.; Cranmer S. L.; Jackson S. P.; Kinzler K. W.; Vogelstein B.; Thompson P. E. Dissecting isoform selectivity of PI3K inhibitors: The role of non-conserved residues in the catalytic pocket. Biochem. J. 2008, 414, 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.; Schulz M. J.; Xie R.; Zeng J.; Luengo J. I.; Squire M. D.; Tedesco R.; Qu J.; Erhard K.; Mack J. F.; Raha K.; Plant R.; Rominger C. M.; Ariazi J. L.; Sherk C. S.; Schaber M. D.; McSurdy-Freed J.; Spengler M. D.; Davis C. B.; Hardwicke M. A.; Rivero R. A. Rational Design, Synthesis, and SAR of a Novel Thiazolopyrimidinone Series of Selective PI3K-beta Inhibitors. ACS Med. Chem. Lett. 2012, 3, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.; Erhard K.; Hardwicke M. A.; Luengo J. I.; Mack J. F.; McSurdy-Freed J.; Plant R.; Raha K.; Rominger C. M.; Sanchez R. M.; Schaber M. D.; Schulz M. J.; Spengler M. D.; Tedesco R.; Xie R.; Zeng J. J.; Rivero R. A. Synthesis and structure-activity relationships of imidazo[1,2-a]pyrimidin-5(1H)-ones as a novel series of beta isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2230–2234. [DOI] [PubMed] [Google Scholar]
- Sanchez R. M.; Erhard K.; Hardwicke M. A.; Lin H.; McSurdy-Freed J.; Plant R.; Raha K.; Rominger C. M.; Schaber M. D.; Spengler M. D.; Moore M. L.; Yu H. Y.; Luengo J. I.; Tedesco R.; Rivero R. A. Synthesis and structure-activity relationships of 1,2,4-triazolo[1,5-a]pyrimidin-7(3H)-ones as novel series of potent beta isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3198–3202. [DOI] [PubMed] [Google Scholar]
- Certal V.; Halley F.; Virone-Oddos A.; Delorme C.; Karlsson A.; Rak A.; Thompson F.; Filoche-Romme B.; El-Ahmad Y.; Carry J. C.; Abecassis P. Y.; Lejeune P.; Vincent L.; Bonnevaux H.; Nicolas J. P.; Bertrand T.; Marquette J. P.; Michot N.; Benard T.; Below P.; Vade I.; Chatreaux F.; Lebourg G.; Pilorge F.; Angouillant-Boniface O.; Louboutin A.; Lengauer C.; Schio L. Discovery and optimization of new benzimidazole- and benzoxazole-pyrimidone selective PI3Kbeta inhibitors for the treatment of phosphatase and TENsin homologue (PTEN)-deficient cancers. J. Med. Chem. 2012, 55, 4788–4805. [DOI] [PubMed] [Google Scholar]
- Certal V.; Halley F.; Virone-Oddos A.; Thompson F.; Filoche-Rommé B.; El-Ahmad Y.; Carry J.-C.; Delorme C.; Karlsson A.; Abecassis P.-Y.; Vincent L.; Bonnevaux H.; Nicolas J.-P.; Morales R.; Michot N.; Vade I.; Louboutin A.; Perron S.; Doerflinger G.; Tric B.; Monget S.; Lengauer C.; Schio L. Preparation and optimization of new 4-(morpholin-4-yl)-(6-oxo-1,6-dihydropyrimidin-2-yl)amide derivatives as PI3Kβ inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 6381–6384. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Lopez de Turiso F.; Shin Y.; Brown M.; Cardozo M.; Chen Y.; Fong D.; Hao X.; He X.; Henne K.; Hu Y.-L.; Johnson M. G.; Kohn T.; Lohman J.; McBride H. J.; McGee L. R.; Medina J. C.; Metz D.; Miner K.; Mohn D.; Pattaropong V.; Seganish J.; Simard J. L.; Wannberg S.; Whittington D. A.; Yu G.; Cushing T. D. Discovery and in Vivo Evaluation of Dual PI3Kβ/δ Inhibitors. J. Med. Chem. 2012, 55, 7667–7685. [DOI] [PubMed] [Google Scholar]
- Zhang X. X.; Vadas O.; Perisic O.; Anderson K. E.; Clark J.; Hawkins P. T.; Stephens L. R.; Williams R. L. Structure of Lipid Kinase p110 beta/p85 beta Elucidates an Unusual SH2-Domain-Mediated Inhibitory Mechanism. Mol. Cell 2011, 41, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaguchi S.; Fukui Y.; Koshimizu I.; Yoshimi H.; Matsuno T.; Gouda H.; Hirono S.; Yamazaki K.; Yamori T. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J. Natl. Cancer Inst. 2006, 98, 545–556. [DOI] [PubMed] [Google Scholar]
- Matsuno T.; Kato M.; Tsuchida Y.; Takahashi M.; Yaguchi S.; Terada S. Synthesis and aromatase-inhibitory activity of imidazolyl-1,3,5-triazine derivatives. Chem. Pharm. Bull. 1997, 45, 291–296. [DOI] [PubMed] [Google Scholar]
- Matsuno T.; Kato M.; Sasahara H.; Watanabe T.; Inaba M.; Takahashi M.; Yaguchi S.; Yoshioka K.; Sakato M.; Kawashima S. Synthesis and antitumor activity of benzimidazolyl-1,3,5-triazine and benzimidazolylpyrimidine derivatives. Chem. Pharm. Bull. 2000, 48, 1778–1781. [DOI] [PubMed] [Google Scholar]
- Rewcastle G. W.; Gamage S. A.; Flanagan J. U.; Frederick R.; Denny W. A.; Baguley B. C.; Kestell P.; Singh R.; Kendall J. D.; Marshall E. S.; Lill C. L.; Lee W. J.; Kolekar S.; Buchanan C. M.; Jamieson S. M. F.; Shepherd P. R. Synthesis and Biological Evaluation of Novel Analogues of the Pan Class I Phosphatidylinositol 3-Kinase (PI3K) Inhibitor 2-(Difluoromethyl)-1-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]-1H-benzimidazole (ZSTK474). J. Med. Chem. 2011, 54, 7105–7126. [DOI] [PubMed] [Google Scholar]
- Kong D. X.; Dan S. G.; Yamazaki K.; Yamori T. Inhibition profiles of phosphatidylinositol 3-kinase inhibitors against PI3K superfamily and human cancer cell line panel JFCR39. Eur. J. Cancer 2010, 46, 1111–1121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.