

Abstract
The majority of patients with non-HIV-related collapsing focal segmental glomerular sclerosis (FSGS) have idiopathic disease. Only a few genetic forms associated with rare syndromes have been described in families. Here we report two families with multiple members who have collapsing FSGS with no clear associated secondary etiology. Genetic analysis revealed a defect in the TRPC6 gene in one family, but excluded all known common inherited podocyte defects in the other family. The course and response to treatment differed dramatically among members of the same family.
Keywords: focal glomerulosclerosis, collapsing, glomerulopathy, hereditary glomerulonephropathy
Introduction
Collapsing glomerulopathy (CG) is considered to be an aggressive variant of focal segmental glomerulosclerosis (FSGS). While it may be associated with infections such as HIV disease, autoimmune diseases, and certain medications, CG comprises from 9% to 24% of patients with idiopathic FSGS [20]. A number of rare genetic syndromes have been associated with collapsing FSGS, including action myoclonus renal failure, mandibuloacral dysplasia or WT1 gene-associated syndromes [1, 18]. We report here two families with multiple members who had collapsing FSGS and include genetic analysis of commonly known inherited genetic podocyte defects. The course and response to therapy differed dramatically among members of the same family.
Case histories
First family
A 37-year-old Hispanic woman underwent evaluation as a potential kidney donor for her brother who was on hemodialysis. She was found to have a normal physical examination, a normal serum creatinine, and a negative urinalysis. Three months later and prior to kidney donation, she developed the sudden onset of edema and shortness of breath.
The patient denied taking any medications. She was monogamous with only one sexual partner, had no blood transfusions, and no history of intravenous drug abuse or any other risk factors for HIV infection. She had a history of preeclampsia during her first pregnancy and a second normal pregnancy thereafter. Family history included the brother with ESRD due to collapsing FSGS and a sister who also had recently developed FSGS (Figure 1).
Figure 1.
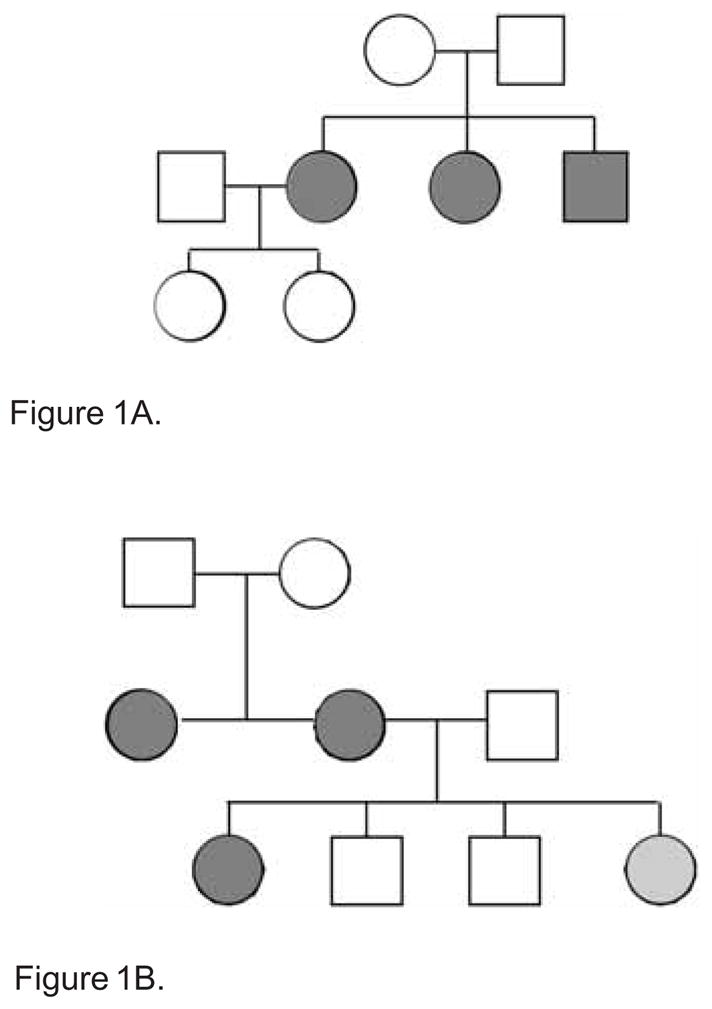
Figure 1A. Family history of the first family. Example: ○ = female; □ = male;
 = FSGS
= FSGS
Figure 1B. Family history of the second family. Example: ○ = female; □ = male;
= FSGS;
 = trace proteinuria
= trace proteinuria
Physical examination revealed a blood pressure of 166/94 mmHg and both periorbital and ankle edema. Laboratory evaluation showed a urinalysis with 4+ proteinuria but no hematuria. 24-h urine protein excretion was 12 g/d. Serum creatinine was 0.7 mg/dl, serum albumin 1.8 g/dl, and total cholesterol 400 mg/dl. Serology for hepatitis B and C, CMV and antinuclear antibody was negative, and serum complement was within the normal range. HIV testing was negative.
A renal biopsy (Table 1) contained 30 glomeruli, 50% of which showed focal segmental and mostly global collapse of the glomerular capillary tufts with features of wrinkling and retraction of basement membrane with partial or complete occlusion of the capillary lumina. There was no mesangial or endothelial proliferation. Patchy interstitial edema and inflammation along with focal tubular atrophy were observed. There were areas of mild tubular dilatation, a few containing hyaline casts. Immunofluorescence was negative, and on electron microscopy the glomerular basement membranes (GBM) were wrinkled and collapsed with capillary luminal narrowing or occlusion with diffuse foot process effacement. There were no electron dense deposits or tubuloreticular inclusions. The histopathologic diagnosis was collapsing focal segmental and global glomerulosclerosis. Sequencing-based genetic analysis for defects in the NPHS2, TRPC6, and ACTN4 genes revealed no defects.
Table 1.
| Affected family members | Biopsy | Treatment | Response | |
|---|---|---|---|---|
| First family | ||||
| Proband (1) (37 years old, Hispanic woman) | L.M. |
|
|
Complete remission |
| IF | Negative | |||
| E.M. |
|
|||
| Sister (2) (40 years old) | L.M. |
|
|
ESRD + Tx (normally functioning renal graft) |
| IF | 1+ IgM staining in the mesangium | |||
| E.M. |
|
|||
| Brother (3) (38 years old) | L.M. |
|
None | ESRD + Tx (normally functioning renal graft) |
| Second Family | ||||
| Proband (4) (21 years old, white woman) | L.M. |
|
|
ESRD + Tx (normally functioning renal graft) |
| IF | IgM and C3 mesangial staining | |||
| E.M. |
|
|||
| Maternal aunt (5) (34 years old) | L.M. | 18 glomeruli: 2 globally sclerotic; several with areas of focal tuft collapse; minimal mesangial proliferation |
|
ESRD + Tx CKD |
| IF | IgM and C3 mesangial staining | |||
| Mother (6) (38 years old) | L.M. | 72 glomeruli: 9 globally sclerotic, 3 with segmental glomerulosclerosis and several with mesangial sclerosis |
|
CKD Stage 1 |
| IF | IgM mesangial staining | |||
| E.M. | 1 glomerulus with collapse of the tuft and others with partial tuft collapse | |||
The patient was started on diuretics, an HMG CoA reductase inhibitor, and cyclosporine 100 mg twice daily. Two months later the patient had less edema and declining proteinuria. One year after the initiation of cyclosporine, she was edema free and in partial remission (urine protein 2.4 g/d and serum creatinine 1.3 mg/dl). At 18 months she had improved further with a urine protein 0.625 g/d and with serum creatinine 1.3 mg/dl. Cyclosporine was discontinued. During the 9 years of follow-up, the patient has been treated with losartan, amlodipine and atorvastatin and remains in remission with urine dipstick revealing trace to 1+ protein, 24-h urine protein < 1 g/d and normal serum creatinine and serum albumin. Urinalyses performed on her two asymptomatic female children (ages 7 and 10 years old) revealed no proteinuria or hematuria.
Her older sister, a 40-year-old Hispanic woman, was also evaluated as a potential kidney donor for her brother and was also found to have neither hypertension nor proteinuria, and a normal serum creatinine. She had no history of medication use, drug abuse, transfusions, promiscuity, or other risk factors for HIV infection. Two months after her evaluation, she developed periorbital and pedal edema and weight gain. 24-h urine showed proteinuria 11 g/day, creatinine of 2 mg/dl, and total cholesterol 463 mg/dl. Serology for hepatitis B and C, HIV, and antinuclear antibody was negative. Serum complement was within the normal range.
A renal biopsy (Table 1) contained 12 glomeruli, 3 of which showed segmental to global collapse of the capillary walls with a distinctive retraction and absence of increased matrix or cell proliferation. Prominent hyperplasia of visceral epithelial cells was observed. There was focal tubular atrophy and mild interstitial fibrosis. Immunofluorescence revealed only 1+ IgM staining in the mesangium. Electron microscopy showed extensive wrinkling of the basement membranes, with total foot process effacement but no electron dense deposits or TRIs.
The patient was treated elsewhere with oral prednisone 60 mg daily, diuretics, an ACE inhibitor, and an HMG CoA reductase inhibitor. Over the next 3 months her kidney function declined to Stage 5 of CKD (GFR < 15 ml/min/1.73 m2) and dialysis was started. She remained on dialysis for 5 years until receiving a deceased donor renal transplant. She is maintained on tacrolimus, mycophenolate mofetil, and low-dose prednisone immunosuppression. Current serum creatinine is 0.96 mg/dl, and urinalysis is negative for proteinuria.
The brother, a 38-year-old Hispanic man, originally underwent evaluation elsewhere for nephrotic syndrome accompanied by Stage 5 CKD. Biopsy at that time (Table 1) contained 13 glomeruli, 11 of which were globally sclerotic. One demonstrated segmental sclerosis and the remaining glomerulus showed collapse of capillary loops and increased mesangial matrix. The interstitium was expanded by fibrous connective tissue and scattered lymphocytic infiltration with atrophy and focal dilatation of the tubules. He progressed rapidly to ESRD and was started on hemodialysis. After both sisters were excluded as potential donors due to their own kidney disease, he received a living unrelated renal transplant from his wife. This failed due to chronic allograft nephropathy but without recurrence of the FSGS. After 2 more years of hemodialysis, he subsequently received a deceased donor renal transplant. At 6 years following the transplant, he is maintained on a regimen of cyclosporine, leflunimide, and low-dose prednisone. His serum creatinine is 0.7 mg/dl, and he has no proteinuria or signs of recurrent disease.
Second family
A 21-year-old white woman developed ankle edema and nephrotic-range proteinuria during the 5th month of pregnancy. Her history included no medication use and no drug abuse. Her family history included a mother with collapsing FSGS discovered at the age of 38 during evaluation as a potential kidney donor for her own sister, and the maternal aunt with ESRD due to collapsing FSGS. The patient’s sister had a urinalysis showing only trace proteinuria; two brothers ages 21 and 19 years old had normal urinalyses.
Postpartum proteinuria persisted at 16g/d; her serum creatinine was 0.6 mg/dl. The patient was serologically negative for hepatitis B, hepatitis C, HIV, ANA, and cytoplasmic and perinuclear ANCA. Apercutaneous kidney biopsy showed collapsing FSGS (Table 1) with 14 of 34 glomeruli having segmental solidification and/or collapse of the tufts and mild hyperplasia of the overlying podocytes. Patchy interstitial fibrosis and tubular atrophy of 20 – 30% of the parenchyma was present. IF showed only IgM and C3 mesangial staining. By electron microscopy, GBM showed focal wrinkling and folding, and podocytes showed extensive foot process effacement, hypertrophy, and focal detachment from the underlying GBM. There were no electron dense deposits or TRI’s.
The nephrotic syndrome did not respond to 6 months of prednisone and subsequently she was treated with cyclosporine. However, she progressed to ESRD 10 months after the biopsy. After 4 months of hemodialysis she received a deceased donor renal transplant. She is maintained on tacrolimus and mycophenolate mofetil with a serum creatinine of 1.0 – 1.3 mg/dl and no evidence of recurrent disease (urinalysis without proteinuria).
The patient’s maternal aunt had proteinuria for 7 years. At a time when her serum creatinine was 1.3 mg/dl and her proteinuria was 2 g/d, she had a kidney biopsy (Table 1), which showed minimal mesangial proliferation on light microscopy with 2 globally sclerotic glomeruli out of 18 total. Several glomeruli showed areas of focal tuft collapse. IgM and C3 were present in the mesangium. She developed nephrotic syndrome with 15 g proteinuria per day, hypertension and progressive renal failure with a serum creatinine rising to 4.5 mg/dl despite a trial of prednisone therapy. Repeat renal biopsy showed 20 – 30 glomeruli with 80% showing glomerulosclerosis, 10% showing segmentally sclerosed glomeruli, and 10% showing “minimal change”. She progressed to ESRD and received a prophylactic one haplotype match renal transplant from her brother 10 years ago. She is now maintained on cyclosporine, prednisone, and mycophenolate mofetil with a serum creatinine ranging from 1.7 to 2.2 mg/dl and urinalysis showing no proteinuria.
The patient’s mother was found to have dipstick-positive (2+) proteinuria at the time of a planned kidney donation to her own sister 10 years ago. A biopsy at that time (Table 1) revealed 72 glomeruli with 9 globally sclerotic. Three showed segmental glomerulosclerosis and several showed mesangial sclerosis. IF was positive for mesangial IgM staining. Electron microscopy revealed one glomerulus with collapse of the tuft and two others with partial tuft collapse. ACE inhibition therapy reduced her proteinuria. Nine years after biopsy her serum creatinine is 0.9 mg/dl and urine protein to creatinine ratio is 0.9.
Sequence analysis of the exons and splice sites of ACTN4 and NPHS2 did not reveal any likely disease-causing defects. In TRPC6, an arginine to cystine amino acid substitution at residue 895 was detected in the proband and her mother, the two affected individuals from whom DNA samples were available. This R895C substitution is identical to one that was previously described in a family from Mexico that confers markedly increased current amplitude on the encoded TRPC6 channel [19].
Discussion
Most cases of CG not associated with HIV infection are idiopathic. However, both reactive and genetic forms have been described. The exact pathogenetic mechanisms leading to the development of CG have not yet been fully elucidated. In murine models CG may occur secondary to a number of different initiators, including HIV-1 gene products, oxidative stress, and alterations in podocyte functions [2]. How these seemingly different insults lead to the same pattern of CG injury remains to be clarified. In the clinical setting, based on the observation that many associated disorders involve a perturbation of the immune system, a potential role for immune activation has been hypothesized [14]. The higher prevalence of the idiopathic CG in blacks, although not as pronounced as in HIV-associated nephropathy (HIVAN), does suggest some genetic susceptibility [2, 22]. Furthermore, a genetic predisposition is supported by the correlation between a certain MHC haplotype and CG in the one reported family with multiple members with collapsing FSGS. This Mexican family had five nephrotic siblings, all of whom had the collapsing pattern of FSGS and the same MHC haplotype. However, the same MHC haplotype was also present in unaffected members of this family [3]. Thus, these data support the hypothesis that a common genetic trait may predispose to the development of CG, but that environmental factors may also play a pivotal role.
Certain rare syndromes (e.g. action myoclonus renal failure, mandibuloacral dysplasia or WT1 gene associated syndromes) have also been linked to genetic forms of CG [1, 18]. Furthermore, some genes susceptible to CG may encode for proteins involved in mitochondrial function. Primary coenzyme Q10 deficiency secondary to defects in the COQ2 gene has been described as a cause of CG [10]. The fact that the biphosphonate medication pamidronate may cause CG and is also a mitochondrial toxin may imply that in these cases mitochondrial damage is the crucial pathogenetic factor in the CG.
Mutations of the gene encoding α-actinin-4 that causes autosomal dominant FSGS inheritance [13] have also been associated with the development of CG in one of two affected family members [7]. Cytoskeletal derangements of the podocytes caused by ACTN4 mutations may contribute to the development of CG, a theory supported by a recent study that reported the development of CG in ACTN4-deficient mice [12]. Mutations in genes like NPHS2, which encodes podocin, TRPC6, which encodes the canonical transient receptor potential 6 ion channel, CD2AP which encodes a protein found at the slit diaphragm of the glomerulus and PLCE1 encoding phospholipase C epsilon have all also been incriminated in genetic forms of FSGS, but not of the collapsing type [18]. In our patients genetic testing in the first family did not reveal any defects in ACTN4, NPHS2, or TRPC6, in the proband. Testing for WT1 and mitochondrial dysfunction forms of CG, etc., was not performed due to lack of routine availability of these genetic tests and because the familial pedigrees did not fit the genetic syndrome of these tests. Moreover, the clinical response to therapy with a complete remission of the nephrotic syndrome makes an inherited defect in a structural component of the podocyte unlikely. In the second family, an arginine to cystine amino acid substitution at residue 895 was detected in the proband and her mother, the two affected individuals from whom DNA samples were available. Of interest, this substitution is the same as one previously observed in a family with hereditary FSGS from Mexico [19]. A genetic defect in TRPC6 must now be added to the genetic forms of the disease associated with the collapsing form of FSGS.
Other etiologies of CG were excluded in our patients. Infections are the most common cause of secondary or reactive forms of CG with HIV and HIV-associated nephropathy (HIVAN) being the predominant cause. Our patients had no risk factors for HIV infection (specifically no blood transfusions, intravenous drug abuse, history of multiple sexual partners or other common risks for HIV infection), had an absence of TRI’s (one of the hallmarks of HIV associated nephropathy) on their renal biopsies, and were tested serologically negative for HIV infection at time of diagnosis of FSGS. Moreover, they had no manifestations of this disease despite long-term follow-up. Indeed, they were negative for a variety of other infectious agents as well and had no symptoms or signs suggesting an on-going infection. Moreover, our patients, even within families, lived in different households making the possibility of simultaneous viral infection unlikely. Exposure to certain medications like bisphosphonates, mainly pamidronate [15] and in one case interferon-α [21] has been linked with the development of CG. In our cases there was no history of drug exposure prior to the onset of renal disease. Rarely, autoimmune diseases like SLE and adult Still’s disease as well as malignancies (mostly multiple myeloma and lymphomas) have been associated with CG [2]. In the solitary Mexican family with familial CG several members had features suggestive of an autoimmune disease. ANA were negative in all of our patients, no symptoms suggestive of autoimmune diseases were present and no one developed any collagen-vascular disease or malignancy during follow up. Furthermore, 2 of our patients had undergone the extensive work-up as potential kidney donors for their siblings and had been found healthy.
The therapy of collapsing FSGS remains a dilemma. Treatment recommendations are based on expert opinions and retrospective data [2, 20]. Drug regimens that are used to treat FSGS including steroids, cyclosporine and other immunosuppressive agents have been suggested. However, collapsing FSGS often responds poorly to treatment and progression to ESRD is frequent and more rapid than in other patterns of FSGS. A course of oral steroids for 6 months is a recommended first line therapy [20]. On the other hand, some studies suggest that earlier aggressive treatment may achieve higher remission rates [8, 14]. Therefore, administration of cyclosporine or MMF or a combination of both may lead to better outcomes [20]. Calcineurin inhibitors in particular have proven efficacious in the treatment of steroid-resistant classic FSGS [6] and may be a reasonable and promising first-line therapy in patients with CG and massive proteinuria. Of interest, in our first case, the administration of cyclosporine early in the course of the disease led to complete remission, while her sister with similar clinical and histologic features at onset progressed to renal failure during steroid therapy. The second family with a known genetic defect in TRPC6 was resistant to therapy.
In conclusion, familial CG is a rare entity with an often progressive clinical course and poor response to treatment. Interestingly, CG and noncollapsing forms of FSGS may both be caused by mutations in the same genes. All patients with familial FSGS should undergo testing for genetic defects associated with the syndrome. While some patients may have known genetic defects and be resistant to therapy, the absence of such defects should still lead to potentially aggressive treatment to reverse the disorder as shown in our first case. Future recognition of the responsible hereditary and environmental factors leading to this lesion as well as better understanding of the pathophysiologic mechanisms underlying the development of collapsing FSGS may provide us with more potent therapeutic strategies.
References
- 1.Albaqumi M, Barisoni L. Current views on collapsing glomerulopathy. J Am Soc Nephrol. 2008;19:1276–1281. doi: 10.1681/ASN.2007080926. [DOI] [PubMed] [Google Scholar]
- 2.Albaqumi M, Soos TJ, Barisoni L, Nelson PJ. Collapsing glomerulopathy. J Am Soc Nephrol. 2006;17:2854–2863. doi: 10.1681/ASN.2006030225. [DOI] [PubMed] [Google Scholar]
- 3.Avila-Casado MC, Vargas-Alarcon G, Soto ME, Hernandez G, Reyes PA, Herrera-Acosta J. Familial collapsing glomerulopathy: Clinical, pathological and immunogenetic factors. Kidney Int. 2003;63:233–239. doi: 10.1046/j.1523-1755.2003.00713.x. [DOI] [PubMed] [Google Scholar]
- 4.Barisoni L, Schnaper HW, Kopp JB. A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin J Am Soc Nephrol. 2007;2:529–542. doi: 10.2215/CJN.04121206. [DOI] [PubMed] [Google Scholar]
- 5.Barisoni L, Diomedi-Casamei F, Santorelli FM, Caridi G, Thomas DB, Emma F, Piemonte F, Ghiggeri GM. Collapsing glomerulopathy associated with inherited mitochondrial injury. Kidney Int. 2008;74:237–243. doi: 10.1038/sj.ki.5002767. [DOI] [PubMed] [Google Scholar]
- 6.Cattran DC, Appel GB, Hebert LA, Hunsicker LG, Pohl MA, Hoy WE, Maxwell DR, Kunis CL. A randomized trial of cyclosporine in patients with steroid-resistant focal segmental glomerulosclerosis. North America Nephrotic Syndrome Study Group. Kidney Int. 1999;56:2220–2226. doi: 10.1046/j.1523-1755.1999.00778.x. [DOI] [PubMed] [Google Scholar]
- 7.Choi HJ, Lee BH, Cho HY, Moon KC, Ha IS, Nagata M, Choi Y, Cheong HI. Familial focal segmental glomerulosclerosis associated with an ACTN4 mutation and paternal germline mosaicism. Am J Kidney Dis. 2008;51:834–838. doi: 10.1053/j.ajkd.2008.01.018. [DOI] [PubMed] [Google Scholar]
- 8.Chun MJ, Korbet SM, Schwartz MM, Lewis EJ. Focal segmental glomerulosclerosis in nephrotic adults: presentation, prognosis, and response to therapy of the histologic variants. J Am Soc Nephrol. 2004;15:2169–2177. doi: 10.1097/01.ASN.0000135051.62500.97. [DOI] [PubMed] [Google Scholar]
- 9.D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004;43:368–382. doi: 10.1053/j.ajkd.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 10.Diomedi-Casamei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer L, Barisoni L, Pastore A, Muda AO, Valente ML, Bertini E, Emma F. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 11.Gupta SK, Eustace JA, Winston JA, Boydstun II, Ahuja TS, Rodriguez RA, Tashima KT, Roland M, Franceschini N, Palella FJ, Lennox JL, Klotman PE, Nachman SA, Hall SD, Szczech LA. Guidelines for the management of chronic kidney disease in HIV-infected patients: recommendations of the HIV medicine association of the infectious diseases society of America. Clin Infect Dis. 2005;40:1559–1585. doi: 10.1086/430257. [DOI] [PubMed] [Google Scholar]
- 12.Henderson JM, Al-Waheeb S, Weins A, Dandapani SV, Pollak MR. Mice with altered alpha-actinin-4 expression have distinct morphologic patterns of glomerular disease. Kidney Int. 2008;73:741–750. doi: 10.1038/sj.ki.5002751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodríguez-Pérez JC, Allen PG, Beggs AH, Pollak MR. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 14.Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int. 1999;56:2203–2213. doi: 10.1046/j.1523-1755.1999.00769.x. [DOI] [PubMed] [Google Scholar]
- 15.Markowitz GS, Appel GB, Fine PL, Fenves AZ, Loon NR, Jagannath S, Kuhn JA, Dratch AD, D’Agati VD. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J Am Soc Nephrol. 2001;12:1164–1172. doi: 10.1681/ASN.V1261164. [DOI] [PubMed] [Google Scholar]
- 16.Moore TL, Bandlamudi R, Alam SM, Nesher G. Parvovirus infection mimicking systemic lupus eythematosus in a pediatric population. Semin Arthritis Rheum. 1999;28:314–318. doi: 10.1016/s0049-0172(99)80015-8. [DOI] [PubMed] [Google Scholar]
- 17.Moudgil A, Nast CC, Bagga A, Wei L, Nurmamet A, Cohen AH, Jordan SC, Toyoda M. Association of parvovirus B19 infection with idiopathic collapsing glomerulopathy. Kidney Int. 2001;59:2126–2133. doi: 10.1046/j.1523-1755.2001.00727.x. [DOI] [PubMed] [Google Scholar]
- 18.Pollak MR. Focal segmental glomerulosclerosis: recent advances. Curr Opin Nephrol Hypertens. 2008;17:138–142. doi: 10.1097/MNH.0b013e3282f5dbe4. [DOI] [PubMed] [Google Scholar]
- 19.Reiser J, Polu KR, Möller CC, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwimmer JA, Markowitz GS, Valeri A, Appel GB. Collapsing glomerulopathy. Semin Nephrol. 2003;23:209–218. doi: 10.1053/snep.2003.50019. [DOI] [PubMed] [Google Scholar]
- 21.Stein DF, Ahmed A, Sunkhara V, Khalbuss W. Collapsing focal segmental glomerulosclerosis with recovery of renal function: an uncommon complication of interferon therapy for hepatitis C. Dig Dis Sci. 2001;46:530–535. doi: 10.1023/a:1005638913487. [DOI] [PubMed] [Google Scholar]
- 22.Thomas DB, Franceschini N, Hogan SL, Ten Holder S, Jennette CE, Falk RJ, Jennette JC. Clinical and pathologic characteristics of focal segmental glomerulosclerosis pathologic variants. Kidney Int. 2006;69:920–926. doi: 10.1038/sj.ki.5000160. [DOI] [PubMed] [Google Scholar]