

Abstract
Chromosome translocations are the most severe form of genome defect. Translocations represent the end product of a series of cellular mistakes and they form after cells suffer multiple DNA double strand breaks (DSBs), which evade the surveillance mechanisms that usually eliminate them. Rather than being accurately repaired, translocating DSBs are misjoined to form aberrant fusion chromosomes. Although translocations have been extensively characterized using cytological methods and their pathological relevance in cancer and numerous other diseases is well established, how translocations form in the context of the intact cell nucleus is poorly understood. A combination of imaging approaches and biochemical methods to probe genome architecture and chromatin structure suggest that the spatial organization of the genome and features of chromatin, including sequence properties, higher order chromatin structure and histone modifications, are key determinants of translocation formation.
Introduction
In 1973, Janet Rowley identified an aberrant chromosome as the cause of chronic myelogenous leukemia (CML) [1]. This momentous discovery changed the field of cancer biology forever by demonstrating that a chromosome defect could cause cancer. The now famous Philadelphia chromosome consists of a reciprocal fusion of parts of chromosome 9 and chromosome 22 and is a prototypical example of a cancer-causing chromosome translocation. The juxtaposition of the ABL gene, encoding a tyrosine kinase, on chromosome 9 and the BCR gene on chromosome 22, leads to the generation of a chimeric fusion protein with constitutive, oncogenic kinase activity [1,2]. Since the discovery of the Philadelphia chromosome, thousands of chromosome translocations have been characterized [3]. In addition to the generation of fusion proteins as in the case of BCR-ABL, translocations may lead to gene disruption or misregulation as seen in Burkitt’s lymphoma, where enhancer elements of one of three immunoglobulin loci (IGH, IGK, or IGL) become juxtaposed to the MYC gene leading to its constitutive activation [4,5]. While chromosome translocations have been particularly well characterized in blood cancers such as CML, they are similarly relevant and frequent in solid tumors as well as in non-cancerous diseases including infertility and schizophrenia [3].
The formation of a translocation requires three basic steps: 1) the occurrence of multiple DNA double strand breaks on distinct chromosomes, 2) the physical association of the broken ends, and 3) the rejoining of the broken partner chromosomes. Despite the undisputed pathological relevance of translocations, how these steps occur in vivo and in the context of the intact cell have remained largely unknown. Recent work is beginning to shed light on two key questions: What determines which chromosomes undergo translocation with each other and what determines where chromosomes break in the first place?
What determines the choice of translocation partners?
The genesis of a chromosome translocation requires that two DSBs come into physical contact to allow the illegitimate misjoining of the chromosome ends (Fig. 1). Since the physical interaction of the involved chromosomal loci is a fundamental step in the formation of translocations, their spatial arrangement is likely to directly contribute to translocation frequency [6]. Cytogenetic and biochemical evidence supports this notion.
Figure 1. The presence of breaks and the spatial arrangement of chromosomes influence translocation frequency.

Translocations cannot form in the absence of breaks. In the presence of breaks, the spatial positioning of the broken chromosomes affects translocation outcome. Proximal breaks translocate with high frequency. Distal breaks can also translocate, albeit at lower frequency.
Cytogenetic studies
Numerous cytogenetic studies have pointed to a strong correlation between spatial proximity of chromosomes or genes and their translocation frequencies by showing that proximal genome sites are more likely to form translocations than distal ones [7–12] (Fig. 1). As an example, in mouse lymphoma, translocations often involve chromosomes 12, 14, and 15, and these chromosomes are found with high frequency in a spatial cluster in normal mouse splenocytes when mapped by fluorescence in situ hybridization (FISH) [11]. Similarly, human chromosomes 4, 13 and 18, which all preferentially localize to the nuclear periphery, frequently translocate with each other, but they do not translocate with internally localized chromosomes, with which they are not in physical proximity [7]. Interestingly, translocation frequency also correlates with the degree of intermingling between chromosomes [8], strongly suggesting that the local arrangement of DSBs drives translocations. The same type of correlation applies to individual genes [9,12–14]. For example, the spatial proximity of the MYC gene, relative to its possible translocation partners IGH, IGK and IGL in Burkitts’ lymphoma, directly correlates with the observed frequency of these translocations in patients [12]. Many other similar examples exist [15–17].
Both the spatial arrangement of genomes and the occurrence of translocations is tissue- and cell-type specific. The comparison of translocation patterns and spatial genome organization amongst tissues further supports a role of genome organization in translocations [3,18,19]. In particular, tissue-specific translocation frequencies correlate with tissue-specific organization patterns [10,20]. In mice, for example, chromosomes 12 and 15 which frequently translocate in lymphomas are proximal in lymphocytes but not in hepatocytes, whereas chromosomes 5 and 6 which often translocate in hepatomas, are proximal in hepatocytes, but not in lymphocytes [20]. These correlations led to the proposal that tissue-specific genome organization is a major driver of chromosome translocations [18].
Genome-wide studies
While these studies provide evidence for the contribution of spatial genome organization as a determinant of the outcome of translocations, their correlative and retrospective analysis assumed that these regions form translocations, without, however, demonstrating it directly. Moreover, tumorigenic translocations are usually clonal and highly selected, and thus correlations may not accurately mirror the contribution of spatial organization to translocation frequency. Several recent studies overcame this limitation by capturing the genome-wide landscape of translocations in the absence of selection [21,22**]. These studies confirm the earlier morphological observations.
Sequencing of junctions of translocations, formed by experimentally induced single DSB at the c-myc or the Igh locus in primary B lymphocytes [21,22**], indicates that translocations occur most frequently on the same chromosome, whereas translocations with other chromosomes are much rarer [21,22**]. Considering that genome regions on the same chromosome are more proximal than loci on other chromosomes, these results highlight the notion that the relative distance of translocating partners determines formation of translocations. This interpretation is further supported by studies which used chromosome conformation capture techniques to map physical interactions on a genome-wide scale [23–25**]. In one approach, DSBs were created by integration of the ISceI restriction site in transformed pro-B cells expressing the RAG (recombination activated gene) endonuclease which cleaves the endogenous Igk locus and other sites. Translocation frequencies were then mapped and compared to the spatial arrangement of chromosomes [23**]. The most frequent translocation partners for the ISceI-induced DSBs were found within the endogenous Igk locus and within other RAG-target loci, suggesting that, as expected, the formation of DSBs is a pre-requisite for the formation of translocations. In support of this notion, the comparison of a genome-wide map of translocations involving Igh and Myc and the physical location of breaks throughout the genome in activated B lymphocytes showed that in the presence of the activation-induced cytidine deaminase AID, which triggers DSBs, the number and location of DNA breaks govern the rate of the chromosome translocations rather than the nuclear interactions [24**]. In contrast, using the same translocation capture sequence data set, a recent study arrived at the opposite conclusion, showing that the majority of AID-induced hotspots are found in domains that contact Igh at high frequency [25**]. However, when DSBs are not the limiting factor [23**], or in the absence of recurrent AID-induced DNA damage [24**], the frequency of translocations was directly related to the frequency of their pre-existing contacts. Taken together, these observations suggest that once DSBs have formed, the spatial arrangement of the chromosomal loci within the nuclear space determines their translocation frequency (Fig. 1).
Spatial proximity and translocations
Two models have been put forth for how translocations form within the nuclear 3D space [9,26,27]. The “breakage-first” model envisions that DSBs from distant locations are able to move towards each other over long distances and are then joined to form a permanent translocation. In an alternative “contact-first” model, joining of broken ends preferentially occurs between chromosomal loci that are found in close proximity before the formation of the breaks. The morphological and biochemical observations strongly support the contact- first model. Additional evidence comes from studies showing that DSBs have limited mobility within the mammalian nucleus [28–31]. Typically, in mammalian cells a DSB undergoes limited local motion with a mean squared displacement of ~1μm2/h, comparable to that of a locus on an intact chromatin fiber [28,29,32]. In contrast, similar experiments in yeast S. cerevisiae indicated increased chromosome mobility of persistent DSBs compared to intact chromosomal loci [33,34]. The observed increase was dependent on factors involved in steps of the homologous recombination (HR) repair pathway, presumably to facilitate homologous pairing during recombination [33,34]. Together with the finding that the mammalian DSB-repair protein 53BP1 promotes the end-joining of dysfunctional telomeres by increasing the local chromatin mobility [35], these studies indicate the involvement of key players of the DSB-repair pathways in controlling DSB mobility. However, even in the much smaller yeast nucleus, spatial proximity appears to play a role in determining recombination outcomes as illustrated by the fact that the MAT mating locus preferentially recombines with its most proximal potential partner rather than a distant potential partner [36].
Although cytogenetic observations, genome-wide mapping, and motion studies suggest that most translocations can be explained by the contact-first model, it is possible that distal breaks may also form translocations, but likely with reduced frequency (Fig. 1). One argument in favor of translocation formation from distal breaks is the observation of occasional long-range, apparently directed, motion of gene loci in living cells [37] and the observed ability of chromosome domains containing DSBs to move over several micrometers and cluster within the mammalian nucleus [38]. Moreover, in S. cerevisiae multiple DSBs coalesce into common repair centers [39]. While this focal assembly of repair proteins may increase their local concentration and affect the efficiency of repair [39], the spatial proximity of the involved breaks may also facilitate illegitimate misjoining. In mammalian cells, while clustering of few repair foci marked by 53BP1 has been observed in a limited number of cells [28], there is no indication that DSB clustering is the norm [28,31] and if it does happen it is likely reversible [29]. It would be interesting to assess whether congregation of repair foci in common focal centers contributes to the formation of chromosome translocations by clustering DSBs.
Why do chromosomes break where they break?
While spatial and temporal proximity of chromosomes is an essential determinant in the formation of translocations, it remains largely unclear what upstream factors predispose genomic regions to breakage and translocations in the first place. Circumstantial evidence suggests that DNA sequence features as well as chromatin properties may facilitate breakage susceptibility of genome regions (Fig. 2).
Figure 2. DNA and chromatin features in breakage susceptibility.
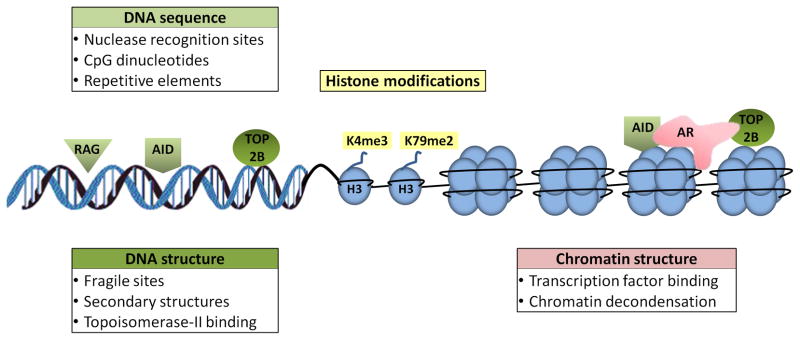
DNA sequence features (green), histone modifications (yellow), and chromatin structure (red) may facilitate breakage susceptibility and represent an important upstream event in translocation formation. (right) The combined effect of sequence and chromatin features is evident in prostate cancer, where liganded androgen receptor (AR) recruits AID and TOP2B to translocation breakage sites.
DNA sequence
DNA features that influence breakage may be sequence and/or structure related. In support of this notion, certain DNA sequences are recognized by endogenous nucleases leading to the formation of DSBs and translocations. RAG1/2 are endonucleases which create DSBs during V(D)J recombination in B- and T-cells. Translocations may form when RAG enzymes misrecognize sequences that resemble recombination signal sequences (RSS) normally found in V(D)J regions [41–43] (Fig. 2). In germinal center B cells, AID recognizes a single-stranded sequence motif during the transcription of regions involved in somatic hypermutation and class switch recombination and promotes DSBs to generate antibody diversity [44], however, misrecognition of non-Ig targets can lead to translocations [45]. AID-induced translocations were first observed in germinal center-derived B cell lymphomas, but have recently been discovered in other B cell lymphomas as well as in some solid tumors [46]. AID has also been suggested to contribute to translocations in other ways than misrecognition of RSS. In prostate cancer, AID is co-recruited with liganded androgen receptor (AR) to AR-binding DNA sequences, sensitizing them to DSB breaks and leading to the formation of translocations in the presence of genotoxic stress [47**] (Fig .2). Furthermore, several genome-wide studies have implicated off-target AID binding sites that may play a role in the formation of translocations [21,22,48–50]. These observations suggest that mis-recognition of sequences by cellular endonucleases promotes genome breakage.
What are the sequences most prone to breakage? CpG islands are one candidate. While representing only 1% of the human genome, CpG dinucleotides are present in 40–70% of bcl-2 and bcl-1 breakpoints in pro-B and pre-B lymphocytes [51], leading to the suggestion that CpGs are targeted by AID and RAG endonucleases. However, CpGs are not associated with translocations in other cell types including lymphoid-myeloid progenitors, mature B cells, and T cells [51], suggesting that if CpG islands do facilitate breakage, their presence is not sufficient to promote them and does not do so in all tissues.
Alu repeats which constitute an estimated 11% of the human genome have been proposed to serve as recombination hotspots for translocations by virtue of non-allelic homologous recombination [52]. However, in an engineered system to quantify translocations, the introduction of identical or divergent Alu repeats adjacent to induced DSB sites did not alter the frequency of translocations [53], suggesting that the presence of homology per se is not a driver of translocation frequency [53,54]. In support, the presence of Alu elements at sequenced translocation junctions in patient cases has been sparse and anecdotal, further suggesting that Alu elements are not universal markers of breakpoints [55].
Common fragile sites (CFS) have also been linked to translocations [56]. CFSs are cytologically defined regions of chromosomes containing gaps and constrictions in metaphase under partial replication stress, and these regions have been shown to be prone to breakage [57]. A recent large scale analysis of 746 cancer cell lines revealed extensive co-incidence of fragile sites with regions of cancer-causing homozygous deletions strongly supporting their role in tumorigenesis [58]. A potential link between fragile sites and translocation formation comes from the observation that exposure of thyroid cells to chemicals that induce fragile sites promotes RET/PTC translocations [59]. Although no single mechanism appears to account for the emergence of CFSs, some common sequence features have been identified. CFSs are enriched in strings of AT-dinucleotide repeats that give these regions high DNA helix flexibility and the ability to form stable non-B DNA secondary structures, which may inhibit DNA replication [60]. Indeed, translocations have been postulated to form at AT palindromic sequences through a mechanism involving cruciform DNA structures that may be prone to breakage [61], and computational analysis of five translocation genes (CBFB, HMGA1, LAMA4, MLL, and AFF4) revealed significantly higher AT content than control regions [62].
More direct evidence for DNA secondary structures in breakage and translocations was the discovery that the major breakpoint region of bcl-2 adopts a stable non-B DNA structure that is targeted by RAG in a sequence-independent manner [63**]. By containing stable regions of single-strandedness, this DNA structure promotes RAG-mediated cleavage of the bcl-2 locus and formation of the t(14;18) translocation in follicular lymphoma [63]. This secondary structure may be a “G-quadruplex,” a four-stranded DNA structure that can spontaneously form in G-rich sequences [64]. Further support that non-B DNA structures contribute to genomic instability and translocations comes from mouse studies in which the integration of sequences that form triplex H-DNA or left-handed Z-DNA increased chromosome breakage, deletion, and translocation events [65].
Topological features of DNA may also contribute to breakage susceptibility. Topoisomerase II (TOP2) generates a transient DSB to regulate under- and overwinding of DNA, for example in mitotic chromosomes and in replication, but also during transcription [66,67 ]. The normally beneficial function of TOP2 may at times, however, have detrimental effects. The TOP2 beta isoform has been shown to associate with androgen receptor upon transcriptional activation and to trigger DSBs at TMPRSS2 and ERG breakpoints in prostate cancer [68**] (Fig. 2). Interestingly, cancer patients who are treated with TOP2 poisons such as epipodophyllotoxins, which potentiate the DNA cleavage activity of TOP2, form therapy-related leukemias harboring characteristic balanced chromosome translocations most often involving the MLL gene and a partner gene [66,69].
Chromatin structure and histone modifications
Circumstantial evidence suggests that various aspects of chromatin may play a role in chromosome breakage susceptibility and translocations. Genome-wide mapping of translocating regions after a single DSB is introduced at the c-myc or Igh locus in primary B lymphocytes found that DSBs occur primarily in transcriptionally active regions [21,22**]. Along the same lines, two studies have documented breakpoints in, or near, transcriptionally active genome regions [47,70**]. In anaplastic large cell lymphoma, several genes in the vicinity of translocation breakpoints are highly expressed before translocations occur [70]. Similarly, liganded androgen receptor, a potent transcriptional activator, binds near the breakpoints of TMPRSS2, ERG, and ETV, which are involved in translocations in prostate cancer and under genotoxic stress, induces translocations [47**]. One interpretation of these observations is that chromatin remodeling and binding of transcription factors may predispose genomic regions to breakage and translocations [71].
Histone modifications modulate transcription, replication, DSB repair, and recombination, making them potential candidates in DSB susceptibility and translocation mechanisms [72]. H3K4me3 has been implicated in both RAG and AID-mediated DSB mechanisms. The RAG2 plant homeodomain finger binds to H3K4me3 at the Ig locus in V(D)J recombination, and mutation of this domain greatly diminishes the efficiency of recombination [73–75]. Furthermore, H3K4me3 stimulates RAG activity at sites other than its natural recognition site, especially at cryptic sites [76]. In T-cells, H3K4me3 peaks at cryptic RAG binding sites in certain translocation breakpoints and it has been proposed that this binding promotes translocations in T-cell leukemias [76]. Similarly, genome-wide analysis of AID-induced cleavage sites identified four non-immunoglobulin genes that accumulate high rates of mutations and participate in translocations [48]. Like the natural immunoglobulin target genes cleaved by AID [77], three of these four non-immunoglobulin genes featured enrichment of H3K4me3 at their breakage sites, as well as clusters of repeat DNA sequences. Genome-wide changes in H4K20 monomethylation in mice led to defective DSB repair, Ig class-switch recombination and to translocations involving the IgH locus [78]. H3K79 methylation has also been implicated in DNA recombination and possibly translocation formation. In prostate cancer cells, liganded androgen receptor induces enrichment of H3K79me2 at breakpoint regions, and overexpression of the H3K79 methyltranferase DOT1L increases the frequency of translocations in the presence of androgen and genotoxic stress [47**]. Genome-wide mapping of a set of histone modifications in a prostate cancer cell line has indicated possible enrichment of active chromatin marks, H3K4me3, H3K36me3, and acetylated H3, over the TMPRSS2-ERG translocation region [79]. In addition, the regions corresponding to prostate breakpoints other than TMPRSS2-ERG showed an entirely different pattern in that they were depleted of these active marks and instead enriched in the repressive mark H3K27me3, indicating that the relationship between histone marks and breakage susceptibility is complex [79]. Nevertheless, the sum of these observations points to the possibility of a combined effect of DNA sequence features, chromatin structure and histone modifications in chromosome breakage susceptibility.
Conclusions
Chromosome translocations are one of the most severe forms of genomic damage and their pathological relevance is undeniable. Yet, 40 years after the discovery of the first cancer-causing translocation, our understanding of their genesis is still rudimentary. A major reason for the lack of mechanistic insight into translocation formation has been the absence of sharp experimental tools to probe them. The recent application of chromosome conformation and deep-sequencing methods complements the traditionally used imaging approaches and extends these studies to a global, genome-wide level. But many questions remain; none maybe more pressing than what the temporal aspects of translocations are. How long does it take for a translocation to form? How rapidly do two unrepaired DSBs find each other? How do they move within the nuclear space? These questions will require the visualization of translocation formation in living cells. Such approaches, which are becoming technically feasible, will also allow probing of individual steps in translocation formation and to experimentally manipulate factors that affect translocation frequency, particularly chromatin structure and histone modifications.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 2.Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature. 1985;315:758–761. doi: 10.1038/315758a0. [DOI] [PubMed] [Google Scholar]
- 3.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 4.Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A. 1982;79:7837–7841. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misteli T. Higher-order genome organization in human disease. Cold Spring Harb Perspect Biol. 2010;2:a000794. doi: 10.1101/cshperspect.a000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bickmore WA, Teague P. Influences of chromosome size, gene density and nuclear position on the frequency of constitutional translocations in the human population. Chromosome Res. 2002;10:707–715. doi: 10.1023/a:1021589031769. [DOI] [PubMed] [Google Scholar]
- 8.Branco MR, Pombo A. Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol. 2006;4:e138. doi: 10.1371/journal.pbio.0040138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meaburn KJ, Misteli T, Soutoglou E. Spatial genome organization in the formation of chromosomal translocations. Semin Cancer Biol. 2007;17:80–90. doi: 10.1016/j.semcancer.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290:138–141. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- 11.Parada LA, McQueen PG, Munson PJ, Misteli T. Conservation of relative chromosome positioning in normal and cancer cells. Curr Biol. 2002;12:1692–1697. doi: 10.1016/s0960-9822(02)01166-1. [DOI] [PubMed] [Google Scholar]
- **12.Roix JJ, McQueen PG, Munson PJ, Parada LA, Misteli T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat Genet. 2003;34:287–291. doi: 10.1038/ng1177. This is one of the first studies to demonstrate that the higher-order genome organization contributes to the formation of chromosome translocations by showing that the physical proximity of MYC gene to its translocation partners correlates with their observed clinical incidence. [DOI] [PubMed] [Google Scholar]
- 13.Neves H, Ramos C, da Silva MG, Parreira A, Parreira L. The nuclear topography of ABL, BCR, PML, and RARalpha genes: evidence for gene proximity in specific phases of the cell cycle and stages of hematopoietic differentiation. Blood. 1999;93:1197–1207. [PubMed] [Google Scholar]
- 14.Soutoglou E, Misteli T. On the contribution of spatial genome organization to cancerous chromosome translocations. J Natl Cancer Inst Monogr. 2008:16–19. doi: 10.1093/jncimonographs/lgn017. [DOI] [PubMed] [Google Scholar]
- 15.Lukasova E, Kozubek S, Kozubek M, Kjeronska J, Ryznar L, Horakova J, Krahulcova E, Horneck G. Localisation and distance between ABL and BCR genes in interphase nuclei of bone marrow cells of control donors and patients with chronic myeloid leukaemia. Hum Genet. 1997;100:525–535. doi: 10.1007/s004390050547. [DOI] [PubMed] [Google Scholar]
- 16.Roccato E, Bressan P, Sabatella G, Rumio C, Vizzotto L, Pierotti MA, Greco A. Proximity of TPR and NTRK1 rearranging loci in human thyrocytes. Cancer Res. 2005;65:2572–2576. doi: 10.1158/0008-5472.CAN-04-4294. [DOI] [PubMed] [Google Scholar]
- 17.Nikiforov YE, Koshoffer A, Nikiforova M, Stringer J, Fagin JA. Chromosomal breakpoint positions suggest a direct role for radiation in inducing illegitimate recombination between the ELE1 and RET genes in radiation-induced thyroid carcinomas. Oncogene. 1999;18:6330–6334. doi: 10.1038/sj.onc.1203019. [DOI] [PubMed] [Google Scholar]
- 18.Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 19.Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2:292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- 20.Parada LA, McQueen PG, Misteli T. Tissue-specific spatial organization of genomes. Genome Biol. 2004;5:R44. doi: 10.1186/gb-2004-5-7-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **21.Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. In this study, together with [22], the authors developed genome-wide, sequencing-based, translocation capture techniques to identify translocation partners of single DSBs located at the c-myc or IgH locus in activated B lymphocytes. They demonstrated that most of the translocations occurred in cis with induced DSB and involved transcribed chromosome regions. Experiments in the presence or absence of AID suggested AID-dependent rearrangements which can be found in B lymphomas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **22.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. see reference [21] [DOI] [PMC free article] [PubMed] [Google Scholar]
- **23.Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. In this study, DSBs were induced in various known chromosome locations and translocations were isolated in ATM deficient, G1-arrested, pro-B cells. The translocation genome-wide map was then compared to the spatial arrangement of chromosomes mapped by a conformation capture technique (Hi-C). The authors demonstrate that intrachromosomal and interchromosomal translocations correlate with their relative spatial proximity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **24.Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484:69–74. doi: 10.1038/nature10909. This study demonstrates that in absence of AID, the frequency of Igh or Myc translocations with their partners is directly correlated with their contact frequency, while in presence of AID, the presence of recurrent DNA damage is the determining factor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **25.Rocha PP, Micsinai M, Kim JR, Hewitt SL, Souza PP, Trimarchi T, Strino F, Parisi F, Kluger Y, Skok JA. Close Proximity to Igh Is a Contributing Factor to AID-Mediated Translocations. Mol Cell. 2012;47:873–885. doi: 10.1016/j.molcel.2012.06.036. Using the genome-wide translocation map data combined with 4C data on the interactions of Igh, they found a positive correlation between Igh translocation frequency and Igh interaction frequency in presence of AID, contrary to what is reported in [24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savage JR. A brief survey of aberration origin theories. Mutat Res. 1998;404:139–147. doi: 10.1016/s0027-5107(98)00107-9. [DOI] [PubMed] [Google Scholar]
- 27.Savage JR. Cancer. Proximity matters. Science. 2000;290:62–63. doi: 10.1126/science.290.5489.62. [DOI] [PubMed] [Google Scholar]
- 28.Jakob B, Splinter J, Durante M, Taucher-Scholz G. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc Natl Acad Sci U S A. 2009;106:3172–3177. doi: 10.1073/pnas.0810987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JH. In situ visualization of DNA double-strand break repair in human fibroblasts. Science. 1998;280:590–592. doi: 10.1126/science.280.5363.590. [DOI] [PubMed] [Google Scholar]
- 31.Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol. 2007;9:675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chubb JR, Boyle S, Perry P, Bickmore WA. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr Biol. 2002;12:439–445. doi: 10.1016/s0960-9822(02)00695-4. [DOI] [PubMed] [Google Scholar]
- 33.Dion V, Kalck V, Horigome C, Towbin BD, Gasser SM. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol. 2012;14:502–509. doi: 10.1038/ncb2465. [DOI] [PubMed] [Google Scholar]
- 34.Mine-Hattab J, Rothstein R. Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol. 2012;14:510–517. doi: 10.1038/ncb2472. [DOI] [PubMed] [Google Scholar]
- 35.Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bressan DA, Vazquez J, Haber JE. Mating type-dependent constraints on the mobility of the left arm of yeast chromosome III. J Cell Biol. 2004;164:361–371. doi: 10.1083/jcb.200311063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chuang CH, Carpenter AE, Fuchsova B, Johnson T, de Lanerolle P, Belmont AS. Long-range directional movement of an interphase chromosome site. Curr Biol. 2006;16:825–831. doi: 10.1016/j.cub.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 38.Aten JA, Stap J, Krawczyk PM, van Oven CH, Hoebe RA, Essers J, Kanaar R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 2004;303:92–95. doi: 10.1126/science.1088845. [DOI] [PubMed] [Google Scholar]
- 39.Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- 40.Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol. 2011;11:251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- 41.Marculescu R, Le T, Simon P, Jaeger U, Nadel B. V(D)J-mediated translocations in lymphoid neoplasms: a functional assessment of genomic instability by cryptic sites. J Exp Med. 2002;195:85–98. doi: 10.1084/jem.20011578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raghavan SC, Kirsch IR, Lieber MR. Analysis of the V(D)J recombination efficiency at lymphoid chromosomal translocation breakpoints. J Biol Chem. 2001;276:29126–29133. doi: 10.1074/jbc.M103797200. [DOI] [PubMed] [Google Scholar]
- 43.Numata M, Saito S, Nagata K. RAG-dependent recombination at cryptic RSSs within TEL-AML1 t(12;21)(p13;q22) chromosomal translocation region. Biochem Biophys Res Commun. 2010;402:718–724. doi: 10.1016/j.bbrc.2010.10.092. [DOI] [PubMed] [Google Scholar]
- 44.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, Bolland DJ, Chen HT, Corcoran AE, Nussenzweig A, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–1038. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okazaki IM, Kotani A, Honjo T. Role of AID in tumorigenesis. Adv Immunol. 2007;94:245–273. doi: 10.1016/S0065-2776(06)94008-5. [DOI] [PubMed] [Google Scholar]
- **47.Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, Zhang J, Rose DW, Fu XD, Glass CK, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. This paper demonstrates that transcription factor binding underlies translocations in prostate cancer and that AID may have off-target effects in solid tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kato L, Begum NA, Burroughs AM, Doi T, Kawai J, Daub CO, Kawaguchi T, Matsuda F, Hayashizaki Y, Honjo T. Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes. Proc Natl Acad Sci U S A. 2012;109:2479–2484. doi: 10.1073/pnas.1120791109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Mol Cell. 2011;41:232–242. doi: 10.1016/j.molcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 51.Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kolomietz E, Meyn MS, Pandita A, Squire JA. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer. 2002;35:97–112. doi: 10.1002/gcc.10111. [DOI] [PubMed] [Google Scholar]
- 53.Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 54.Weinstock DM, Elliott B, Jasin M. A model of oncogenic rearrangements: differences between chromosomal translocation mechanisms and simple double-strand break repair. Blood. 2006;107:777–780. doi: 10.1182/blood-2005-06-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aplan PD. Chromosomal translocations involving the MLL gene: molecular mechanisms. DNA Repair (Amst) 2006;5:1265–1272. doi: 10.1016/j.dnarep.2006.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arlt MF, Durkin SG, Ragland RL, Glover TW. Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst) 2006;5:1126–1135. doi: 10.1016/j.dnarep.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 57.Ozeri-Galai E, Bester AC, Kerem B. The complex basis underlying common fragile site instability in cancer. Trends Genet. 2012;28:295–302. doi: 10.1016/j.tig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 58.Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gandhi M, Dillon LW, Pramanik S, Nikiforov YE, Wang YH. DNA breaks at fragile sites generate oncogenic RET/PTC rearrangements in human thyroid cells. Oncogene. 2010;29:2272–2280. doi: 10.1038/onc.2009.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inagaki H, Ohye T, Kogo H, Kato T, Bolor H, Taniguchi M, Shaikh TH, Emanuel BS, Kurahashi H. Chromosomal instability mediated by non-B DNA: cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res. 2009;19:191–198. doi: 10.1101/gr.079244.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurahashi H, Inagaki H, Ohye T, Kogo H, Tsutsumi M, Kato T, Tong M, Emanuel BS. The constitutional t(11;22): implications for a novel mechanism responsible for gross chromosomal rearrangements. Clin Genet. 2010;78:299–309. doi: 10.1111/j.1399-0004.2010.01445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burrow AA, Williams LE, Pierce LC, Wang YH. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009;10:59. doi: 10.1186/1471-2164-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **63.Raghavan SC, Swanson PC, Wu X, Hsieh CL, Lieber MR. A non-B-DNA structure at the Bcl-2 major breakpoint region is cleaved by the RAG complex. Nature. 2004;428:88–93. doi: 10.1038/nature02355. The authors propose that RAG-mediated cleavage of stable secondary DNA structures predisposes to translocations in follicular lymphoma. [DOI] [PubMed] [Google Scholar]
- 64.Katapadi VK, Nambiar M, Raghavan SC. Potential G-quadruplex formation at breakpoint regions of chromosomal translocations in cancer may explain their fragility. Genomics. 2012;100:72–80. doi: 10.1016/j.ygeno.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 65.Wang G, Carbajal S, Vijg J, DiGiovanni J, Vasquez KM. DNA structure-induced genomic instability in vivo. J Natl Cancer Inst. 2008;100:1815–1817. doi: 10.1093/jnci/djn385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Felix CA, Kolaris CP, Osheroff N. Topoisomerase II and the etiology of chromosomal translocations. DNA Repair (Amst) 2006;5:1093–1108. doi: 10.1016/j.dnarep.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 67.Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- **68.Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, Isaacs WB, Bova GS, Liu W, Xu J, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–675. doi: 10.1038/ng.613. This paper demonstrated a link between transcription machinery and topoisomerase-IIB induced cleavage as a basis for prostate cancer translocations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mays AN, Osheroff N, Xiao Y, Wiemels JL, Felix CA, Byl JA, Saravanamuttu K, Peniket A, Corser R, Chang C, et al. Evidence for direct involvement of epirubicin in the formation of chromosomal translocations in t(15;17) therapy-related acute promyelocytic leukemia. Blood. 2010;115:326–330. doi: 10.1182/blood-2009-07-235051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mathas S, Kreher S, Meaburn KJ, Johrens K, Lamprecht B, Assaf C, Sterry W, Kadin ME, Daibata M, Joos S, et al. Gene deregulation and spatial genome reorganization near breakpoints prior to formation of translocations in anaplastic large cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:5831–5836. doi: 10.1073/pnas.0900912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mathas S, Misteli T. The dangers of transcription. Cell. 2009;139:1047–1049. doi: 10.1016/j.cell.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rando OJ. Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr Opin Genet Dev. 2012;22:148–155. doi: 10.1016/j.gde.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu Y, Subrahmanyam R, Chakraborty T, Sen R, Desiderio S. A plant homeodomain in RAG-2 that binds Hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity. 2007;27:561–571. doi: 10.1016/j.immuni.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramon-Maiques S, Kuo AJ, Carney D, Matthews AG, Oettinger MA, Gozani O, Yang W. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc Natl Acad Sci U S A. 2007;104:18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shimazaki N, Tsai AG, Lieber MR. H3K4me3 stimulates the V(D)J RAG complex for both nicking and hairpinning in trans in addition to tethering in cis: implications for translocations. Mol Cell. 2009;34:535–544. doi: 10.1016/j.molcel.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci U S A. 2010;107:22190–22195. doi: 10.1073/pnas.1016923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22:2048–2061. doi: 10.1101/gad.476008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]