

Abstract
Normal cellular phenotypes that serve an oncogenic function during tumorigenesis are potential candidates for cancer targeting drugs. Within a subset of invasive primary breast carcinoma, we observed relatively abundant expression of Tetherin, a cell surface protein encoded by the Bone Marrow Stromal Cell Antigen (BST2) known to play an inhibitory role in viral release from infected immune cells of the host. Using breast cancer cell lines derived from low and intermediate histopathologic grade invasive primary tumors that maintain growth-suppressive TGFβ signaling, we demonstrate that BST2 is negatively regulated by the TGFβ axis in epithelial cells. Binding of the transcription factor AP2 to the BST2 promoter was attenuated by inhibition of the TGFβ pathway thereby increasing BST2 expression in tumor cells. In contrast, inherent TGFβ resistance characteristic of high grade breast tumors is a key factor underlying compromised BST2 regulation, and consequently its constitutive overexpression relative to non-malignant breast epithelium, and to most low and intermediate grade cancer cells. In both 2-dimensional and 3-dimensional growth conditions, BST2-silenced tumor cells displayed an enhancement in tamoxifen or staurosporine-induced apoptotic cell death together with a reduction in the S-phase fraction compared to BST2 overexpressing counterparts. In a subset of breast cancer patients treated with pro apoptotic hormonal therapy, BST2 expression correlated with a trend for poor clinical outcome, further supporting its role in conferring an anti apoptotic phenotype. Similar to the effects of gene manipulation, declining levels of endogenous BST2 induced by the phytoalexin – resveratrol, restored apoptotic function, and curbed cell proliferation. We provide evidence for a direct approach that diminishes aberrant BST2 expression in cancer cells as an early targeting strategy to assist in surmounting resistance to pro apoptotic therapies.
Introduction
Cell-intrinsic defenses against invading pathogens are a relatively new discovery [1]. To assist the host’s immune system in controlling microbial spread during productive infection, these defenses function as ‘restriction factors’. Among these factors, the bone marrow stromal antigen 2 (BST2)/tetherin is known to dramatically reduce the release of the human immunodeficiency virus, as well as a range of other viruses from infected cells [2], [3] by directly tethering virions to cells [4]. Viral infection is generally accompanied by the induction of interferon in the blood of infected individuals, which in turn initiates a cascade of feedback loops, known to involve BST2 upregulation [5]. Various response elements in the BST2 gene promoter suggest that inflammatory cytokines may be involved in regulating its expression. In blood derived human immune cells, BST2 activation and type I interferon (IFN) production appear to share the same triggers ensuring that BST2 expression, and its role in the IFN negative feedback loop are synchronized [6]. Strong evidence also suggests that a viral gene product - Vpu, counteracts the inhibitory effect of BST2 on virus release within the host [3], [7]. BST2 is a 30–36 kd protein comprised of a cytoplasmic N-terminal region, followed by a transmembrane domain, a coiled-coil extracellular domain, and a C-terminal glycosylphosphatidylinositol (GPI) anchor [8], which in the human protein may represent a second transmembrane segment rather than a GPI anchor [9]. BST2 is normally detected in dendritic cells, terminally differentiated B cells, and bone marrow stromal cells [10], and is overexpressed in some hematological cancers [11].
The role of BST2 in solid tumors is largely undetermined. Other than an association with a breast cancer xenograft metastatic to the bone [12], the regulation and function of this membrane-associated gene product in cancerous breast tissue remains unknown. In the search for functional phenotypes characteristic of clinically aggressive breast cancer, our approach is to compare these with indolent, or less aggressive breast tumors, whereby the differential of mitotic index that masks many distinctive cellular features, could be overridden. Other approaches, such as direct molecular phenotyping of tumors of varying histopathologic grade, a well-established indicator of clinical outcome and breast cancer prognostication, has served largely to confirm previously observed microscopic differences in proliferation status [13]. To provide fresh insights in this regard, we have developed and characterized a panel of 16 novel cell lines of varying histologic grade [14]–[16] that supplement routinely used breast cancer cell lines uniformly representative of high grade breast cancer. Genomic profiling of an expanded panel of breast cancer cell lines revealed BST2 among the top 100 significant genes differentially expressed in high vs. low grade tumor derived cell cultures - all in the proliferative state [16]. Striking differences in TGFβ responsiveness were a key factor underlying differential expression of other high grade associated genes [17]. Here, we demonstrate a role for the TGFβ axis in BST2 regulation, and the effects of its experimental manipulation on downstream functional endpoints of apoptosis induction, and cell proliferation. Our study identifies an underlying regulatory mechanism gone awry that activates BST2 in breast epithelial cells during tumorigenesis, and contributes to the functional hallmarks of cancer portrayed by tumor cells overexpressing this gene product. A pragmatic strategy used here for normalizing endogenous BST2 expression experimentally could be applied towards increasing the effectiveness of standard cancer therapeutics in patients.
Materials and Methods
Primary Breast Carcinoma Cell Culture
The development of low, intermediate, and high grade immortalized primary breast cancer cell lines has been previously described [14]–[15]. All tumor cell lines were routinely propagated in optimized MCDB170 medium supplemented with 2% fetal bovine serum [18]. Treatments included exposure to 4 ng/ml TGFβ1 (R&D Systems, Minneapolis, MN, USA), 20 µM TGFβ inhibitor - SB-431542 hydrate (Sigma, St. Louis, MO, USA), 5 µM tamoxifen (Sigma) or 20–100 µM resveratrol (Sigma).
Immunolocalization
Archived blocks of formalin fixed primary breast cancer samples collected with the written informed consent of the donor were obtained from the Department of Pathology, and used with the approval of the California Pacific Medical Center IRB committee. Each sample was graded and reviewed by participating pathologists (ZM, JLB). Sections of tumor tissue arrays and individual tumor blocks were deparaffinized and used for immunoperoxidase localization of a rabbit anti human BST2 antibody (FabGennix, Frisco, TX, USA). Membrane/cytoplasmic staining of BST2 in greater than 30% tumor cells were scored as a positive result.
For immunocytochemistry on cell cultures, paraformaldehyde-fixed tumor cells were permeablized with Triton X-100, incubated with rabbit anti BST2 followed by Alexa Fluor® 488 anti rabbit (Life Technologies, Grand Island, NY, USA), counterstained with propidium iodide (PI), and analyzed by confocal microscopy. Images from all cell lines were acquired at a constant voltage gain setting of the detector channel used. Data acquisition for no antibody control, and anti BST2 stained samples was performed under identical conditions.
For Western blotting, whole cell lysates were resolved by SDS-PAGE on 12% gels, transferred to PVDF membranes and probed with rabbit anti BST2, or mouse anti tubulin (Abcam, Cambridge, MA, USA) followed by peroxidase-labeled secondary antibody (Sigma), and chemiluminescent detection.
Quantitative Real Time PCR (QPCR)
RNA isolated with the RNeasy kit (Qiagen Valencia, CA, USA) was used to synthesize cDNA for relative transcript quantitation. BST2, AP2, and STAT3 sequence specific primers were employed in QPCR assays by SYBR Green analysis. ACTB control was included in each PCR reaction. Relative expression of BST2 in tumor cell lines was normalized against mean ΔCt values of short-term primary cultures of nonmalignant human breast epithelial cells (n = 3). Relative expression of target genes was calculated as previously described [17].
Chromatin Immunoprecipitation (ChIP) Assay
ChIP reagents were used as per manufacturer’s instructions (Upstate Biotechnology, Lake Placid, NY, USA) with minor modifications. Tumor cells fixed with 1% paraformaldehyde were sonicated, followed by immunoprecipitation of DNA-AP2 complexes with anti AP2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Normal rabbit IgG served as a negative control. After de-crosslinking and purification, DNA was subjected to PCR with AccuPrime™ Taq DNA Polymerase (Life Technologies) and oligonucleotides for putative AP2 binding regions in the BST2 promoter. Input controls were included in PCR reactions. PCR products were resolved on 2% agarose gels and visualized by ethidium bromide staining. Oligonucleotide sequences used for PCR amplification of ChIP DNA are as follows:
BST2 Promoter B Forward:ACAGTTGGCTGGCACCCAGTT
BST2 Promoter B Reverse:GAGGGTGCTGGAATCTTCTACGG
RNA Interference
For BST2 silencing, tumor cells were transfected with siCONTROL non–targeting siRNA or a set of 4 human BST2 specific siRNAs (Thermo Scientific) using Lipofectamine 2000 (Life Technologies).
Cell Proliferation and Apoptosis
Growth potential was measured as colony counts 7-days after plating single cell suspensions in 3% Matrigel (BD Biosciences, San Jose, CA, USA). To determine S-phase distribution, cells were pulse labeled with 10 µM BrdU for 1 hr, and stained with anti BrdU (Santa Cruz Biotechnology), and FITC-conjugated secondary antibody (Life Technologies). Proliferating cells represented by FITC-stained nuclei were counted as a percentage of all nuclei over 10 microscopic fields. Similarly, percent apoptosis were measured by immunolocalization of anti cleaved caspase 3 (Cell Signaling Technology, Danvers, MA, USA) in untreated vs. tamoxifen, or staurosporine treated tumor cell populations.
Cell Cycle Analysis
Cells exposed to 10 µM BrdU for 1 hr were fixed with 70% ethanol, incubated with anti BrdU and FITC conjugated secondary antibody, counterstained with PI, and analyzed by FACScan (BD Biosciences) using CellQuest software (BD Biosciences).
Clinical Outcome Analysis
A publicly available primary breast cancer microarray dataset - GSE4922 [19] was used for evaluating clinical outcome of BST2 overexpressing patient subsets. The Kaplan-Meier estimates were used to plot survival curves, and the p-value from a log-rank test used to determine the statistical significance of hazard ratios. Disease-free survival was defined as the time interval from surgery until the recurrence or last date of follow up. All survival statistics were performed in the R package.
Results
Differential Expression of BST2 in Primary Breast Cancer
Immunoperoxidase localization of anti BST2 on 3 tissue arrays comprised of 234 primary breast tumor cores demonstrated strong membrane staining in the majority of intermediate and high grade tumors (grades 2 and 3, respectively), while minimal to no immunostaining was observed in low grade (grade 1) tumors (Table 1). BST2 immunolocalization in tissue arrays was verified in full-sized sections of tumor tissue blocks in several cases (Figure 1A–C). BST2 positive primary tumors with a concurrent DCIS component were found to display strong staining in both pre invasive and invasive tumor cells (Figure 1C).
Table 1. BST2 immunolocalization in primary invasive breast cancer.
| Histological grade | Number of cases | Positive tumors (%) | Negative tumors (%) |
| 1 | 30 | 8 (27) | 22 (73) |
| 2 | 120 | 103 (86) | 17 (14) |
| 3 | 134 | 126 (94) | 8 (6) |
The association between BST2 positive status and histological grade is significant (p<0.001).
Figure 1. BST2 overexpression is associated with high histological grade of primary breast cancer.
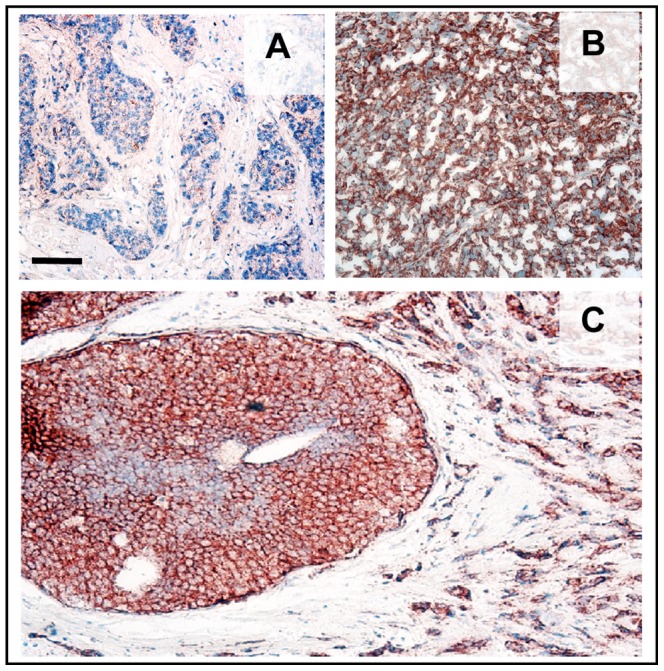
A. Low grade invasive primary breast tumor showing weak or no BST2 expressing cancer cells. Blue – hematoxylin counterstain. B. High grade invasive primary breast tumor displaying strong BST2 localization in cancer cells (brown). C. Homogeneous BST2 immunostaining in cells of coexisting ductal carcinoma in situ (DCIS), and invasive tumor. Bar –50 µm.
In our previous expression profiling study of 14 novel primary breast cancer cell lines of varying histological grade (grade 1: n = 3; grade 2: n = 7; and grade 3: n = 4) developed in our laboratory, and 51 breast epithelial cell lines established by others [16], BST2 was among the most highly expressed genes in grade 3 tumor lines identified by Significance Analysis of Microarrays (SAM). Here, we have established functional phenotypes resulting from differential BST2 expression by using breast cancer cell lines of varying grade. Grade associated variation in BST2 transcript levels was first confirmed by QPCR analysis of breast cancer cell lines normalized to that of non-malignant primary breast epithelial cultures derived from reduction mammoplasty tissue (Figure 2A). Using a subset of 3 grade 3, and 5 grade 1 & 2 cell lines, differences in transcript levels were further confirmed by protein quantitation (Figure 2B), and by immunofluorescence microscopy (Figure 2C). Grade 3 tumor cell lines expressed abundant BST2 protein, whereas considerably lower or no protein expression was observed in grade 1 & 2 cell lines. Consistent with its localization in other cell types in various human tissues, BST2 immunostaining of unfixed live breast cancer cells displayed significant accumulation at the cell surface (Figure 2D).
Figure 2. Differential BST2 expression in primary breast cancer of varying histological grade is maintained in tumor-derived cell lines.

A . QPCR based BST2 transcript levels in 17 breast cancer cell lines normalized to expression in non-malignant breast epithelium. B. Western blot analysis of BST2 protein (25–35 kd) in breast cancer cells. Tubulin used as a loading control. C. Microscopic images of BST2 immunostaining (green) in fixed, permeabilized breast cancer cells. Nuclei counterstained with propidium iodide (red). Bar –50 µm. D. BST2 immunolocalized at the cell membrane in live, unfixed breast cancer cells. Bar –25 µm.
Negative Regulation of BST2 Expression by TGFβ
Exposure of grade 1 & 2 tumor cell lines to TGFβ for 24 hr resulted in further suppression of BST2 transcripts whereas no change was observed in grade 3 cell lines by QPCR analysis (Figure 3A), suggesting transcriptional regulation of BST2 in TGFβ responsive tumor cells. BST2 reduction was accompanied by a significant (p<0.01) increase in expression of AP2, a transcription factor positively regulated by TGFβ. Additionally, expression of STAT3, a gene negatively regulated by TGFβ, was reduced under these conditions (Figure 3B). Transcriptional alterations of above mentioned test genes were not induced by TGFβ exposure of grade 3 cell lines. In contrast to these effects, treatment of 2 independent grade 1 tumor cell lines with the TGFβ inhibitor - SB431542, induced up to 2-fold increase in BST2 (p<0.01) (Figure 3C).
Figure 3. Regulation of BST2 expression by TGFβ in breast cancer cells.
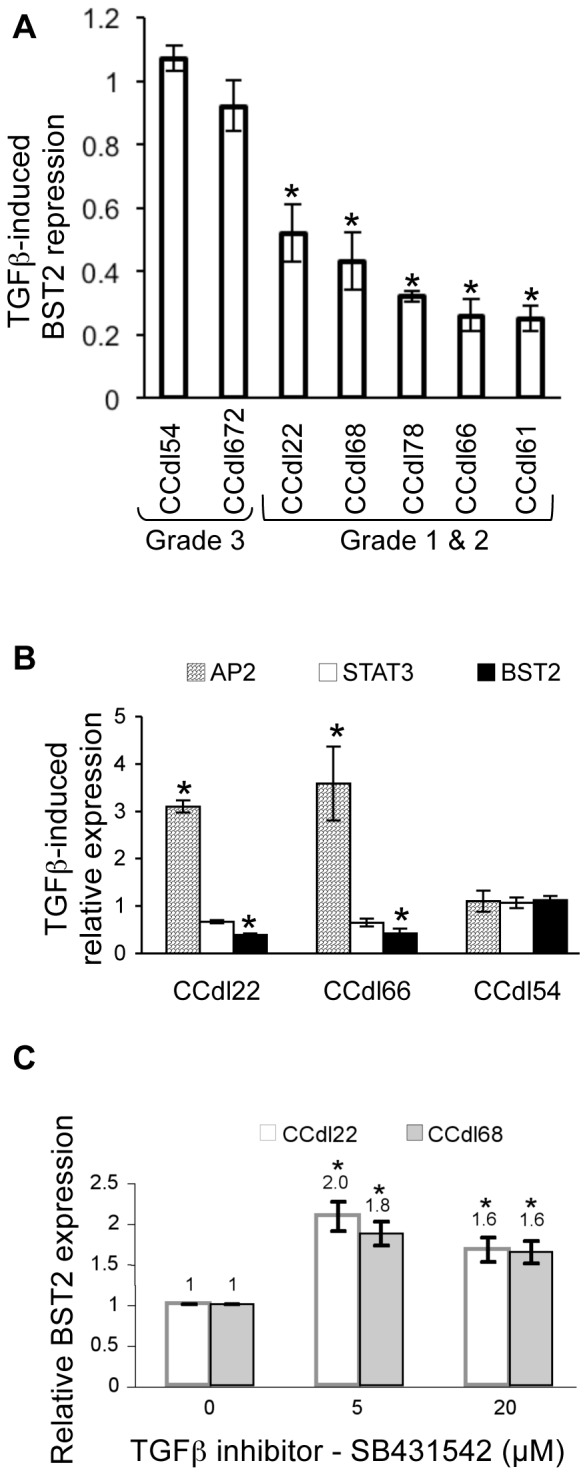
A. Reduction in baseline BST2 expression detected in 5 grade 1 & 2 cell lines after 24 hr TGFβ (4 ng/ml) treatment. Each QPCR reaction was carried out in triplicate, and values normalized to ACTB expression and to vehicle-only controls. Asterisks represent statistically significant differences (p<0.01) between untreated and TGFβ-treated samples. B. TGFβ-induced shift in AP2 and STAT3 transcript levels under the same treatment conditions employed in panel A. BST2 reduction in grade 1 (CCdl22) & 2 (CCdl66) cell lines is accompanied by a decline in STAT3 levels, while AP2 transcripts increase significantly. No significant changes were observed in grade 3 (CCdl54) cells. Each assay was done in triplicate. Asterisks indicate a significant difference (p<0.01) between untreated and TGFβ-treated samples. C. Inhibition of the TGFβ pathway with 24 hr SB-431542 treatment increased BST2 expression in grade 1 cell lines (CCdl22, CCdl68). Data acquired in triplicate shows significant differences (p<0.01) between test and vehicle-only cell samples, indicated by asterisks.
Site-specific Binding of AP2 to the BST2 Promoter
Towards a direct analysis of BST2 regulation, we first evaluated the 240 bp region of the human BST2 promoter and exon 1 by Vector NTI Suite software and detected the presence of AP2 and STAT binding sites within this region (shown diagrammatically in Figure 4A). Since QPCR analysis showed more than 3-fold induction of AP2 expression by TGFβ within responsive low grade tumor cells, whereas only marginal to no change was detected in STAT3 (Figure 3B), and STAT1 transcript levels (data not shown), thus further analysis of AP2 binding was prioritized. ChIP assays were carried out using anti AP2, and promoter-specific oligonucleotides. Strong AP2 binding to the BST2 promoter was observed in 5/5 grade 1 & 2 tumor cell lines whereas no binding was detected in 2/2 grade 3 lines, and in MCF7 cells (Figure 4B, top panel). QPCR analysis of ChIP-derived DNA from all test samples confirmed gel based findings of AP2 recruitment to the BST2 promoter (Figure 4B, bottom panel). Finally, a direct role for TGFβ in mediating AP2 binding to the BST2 promoter was confirmed in 3 independent primary breast tumor lines treated with the TGFβ inhibitor - SB 431542 prior to ChIP analysis (Figure 4C). Baseline TGFβ levels (approximately 1 ng/ml in 2% serum supplemented growth media used here) sufficed to induce AP2 binding to the BST2 promoter in 3/3 TGFβ responsive low/intermediate grade breast cancer cell lines. While TGFβ induction of AP2 transcripts was observed by direct QPCR measurements (Figure 3C), this effect was not detectable by ChIP likely due to differences in assay sensitivity. However, AP2 binding to the BST2 promoter was effectively disrupted in the presence of the TGFβ inhibitor in all test cell lines (Figure 4C). Taken together, these observations suggest a model wherein AP2 binding to the BST2 promoter triggers gene suppression and serves as the basis of differential BST2 expression in low vs. high grade tumors (Figure 4D).
Figure 4. Differential AP2 binding to the BST2 promoter in primary breast cancer cell lines of varying histologic grade.

A. Schematic of the BST2 promoter region spanning 239 bp upstream of the transcriptional start site, including 111 bp of exon 1. Numbering is relative to the translation start site, highlighted in green (+1). Potential cis-regulatory elements shown are either on the plus (+) or minus (–) DNA strands. Regulatory binding sequences: AP2 (blue), STAT1 (red) and STAT3 (purple). B. Top panel - Chromatin immunoprecipitation (ChIP) performed with anti AP2, or control IgG in 8 breast cancer cell lines. Significant AP2 recruitment to the BST2 promoter observed only in grade 1 (CCdl22, CCdl68, CCdl67) & grade 2 (CCdl66, CCdl61) cell lines. Bottom panel - DNA from ChIP samples in top panel analyzed by QPCR. Primers encompassing putative AP2 binding sites from −221 to +6 were used. Melt curves were analyzed to ensure amplification of a single product. Each reaction was performed in triplicate. The plot represents relative binding efficiency determined by 2-ΔΔCT, where ΔCT is the difference between input CT and immunoprecipitated CT; ΔΔCT is the difference between AP2-immunoprecipitated ΔCT and IgG-immunoprecipitated ΔCT. Asterisks represent statistical significance (p<0.01). C . Inhibition of AP2 binding to the BST2 promoter in TGFβ-responsive primary breast cancer cell lines. Prior to processing for ChIP, cells were treated with vehicle, 4 ng/ml TGFβ, or 20 uM TGFβ inhibitor - SB-431542. Note striking reduction in AP2 binding in the presence of SB-431542 in 3 independent test cell lines. D. Hypothetical representation of BST2 transcriptional regulation by the TGFβ axis. Intact TGFβ regulation mediated by AP2 binding to the BST2 promoter enables maintenance of low baseline expression in grade 1 & 2 breast cancer cells.
Induction of Apoptosis and Inhibition of Cell Proliferation Promoted by Loss of BST2 Expression
To evaluate its functional role in breast cancer, BST2 overexpressing cells were transfected with BST2 siRNA and evaluated as 3-dimensional Matrigel cultures. Reduction in BST2 expression was confirmed both by QPCR and immunofluorescence (Figure 5A). As measured by immunofluorescence localization of anti cleaved caspase 3, a decline in BST2 levels detectably enhanced apoptotic cell death induced by 2 independent pro apoptotic drugs - 4-hydoxy tamoxifen or staurosporine (Figure 5B).
Figure 5. Functional consequences of endogenous BST2 overexpression in high grade primary breast cancer cells.

A. Top panel – Reduction of BST2 expression after transient knockdown by BST2 siRNA transfection of grade 3 tumor cells (CCdl54). Values normalized to ACTB expression. Bottom panel – BST2 immunolocalization (green) on day-6 post knockdown, showing sustained reduction in BST2 siRNA transfected cells plated in Matrigel; nuclei counterstained with PI (red). Bar - 50 µm. B . BST2 knockdown enhances drug-mediated apoptotic cell death in 6-day old tumor cultures plated in Matrigel, and treated with 5 µM tamoxifen, or 5 µM staurosporine for 24 hrs. Top panel – immunolocalization of anti cleaved caspase 3 (green); nuclei counterstained with PI (red). Bar - 50 µm. Bottom panel – Data plotted as percent apoptotic cells in 2 independent BST2 overexpressing cell lines (CCdl54, CCdl672). Total number of PI-stained nuclei (>100), and proportion of cleaved caspase 3 positive tumor cells was averaged for 3 optical fields scanned with a 20× objective. Differences in apoptotic cell yield between NS siRNA vs. BST2 siRNA-transfected cultures after treatment with each drug were significant (p<0.01). C . Growth reduction induced by BST2 knockdown measured by BrdU incorporation in primary tumor cells plated in Matrigel. Top panel - BrdU immunolocalization (yellow) in tumor nuclei (CCdl54) counterstained with PI (red) 7-day post transfection with BST2 siRNA. Bar - 25 µm. Middle panel - Data plots represent the fraction of BrdU positive proliferating cells in control (>100) and BST2 knockdown cultures averaged from 5 optical fields scanned with a 20× objective. Asterisk denotes a significant (p<0.01) difference. Bottom panel – Data plots demonstrate an increase in the number of small (4-cell) and medium (8- to 12-cell) colonies accompanied by a decrease in the number of large-sized colonies (>25-cell) in BST2 siRNA-treated Matrigel cultures. D. Stratification of hormone-treated ER+ breast cancer (n = 66) based on BST2 transcript levels. The Kaplan Meier plot suggests a trend whereby a relatively poor clinical outcome is conferred upon cases with moderate (red) or high (green) BST2 expression compared to those with low gene expression (blue).
Reduced BST2 transcript levels also led to decreased cell proliferation measured by BrdU incorporation and immunofluorescence localization of anti BrdU (Figure 5C, top panel). A decline in anti BrdU-stained cell populations (Figure 5C, middle panel) was accompanied by smaller colony size in BST2 overexpressing cell lines transfected with BST2 siRNA vs. nonspecific siRNA controls (p<0.01) (Figure 5C, bottom panel).
Association of BST2 Expression with Clinical Outcome of Breast Cancer Patients
Based on the demonstrated role for BST2 in apoptosis evasion in vitro, we used the GSE4922 gene expression dataset (http://www.ncbi.nlm.nih.gov/geo/) to explore BST2 stratification potential in breast cancer patients treated with pro apoptotic hormonal therapy. This dataset includes a total of 66 ER+ patients who received single agent hormone treatment. BST2 transcript values in these cases ranged from 6.4 to 10.3 relative units demonstrating that expression of this gene is variable in ER+ tumors. Using Martingale residuals as a measure of the influence of BST2 expression on the hazard ratio in a Cox Proportional hazards model, an increasing hazard ratio was noted for 55 cases where BST2 expression in the tumor ranged between 7.0 to 9.5 units. Twenty-two of these cases recurred within 9.5 yrs. The risk of recurrence doubled for each unit increase in BST2 expression (HR 2.00, CI: 0.95–4.26, p = 0.07). Other prognostic parameters, such as: histologic grade (HR 1.85, CI: 0.86–3.95, p = 0.11), age at diagnosis (HR 0.99, CI: 0.95–1.04, p = 0.96), tumor size (HR 1.01, CI: 0.97–1.05, p = 0.54), and lymph node status (HR 1.36, CI: 0.53–3.49, p = 0.51) were less or not predictive of outcome. Based on these findings, data were divided into increasing BST2 expression tertiles, and a Kaplan-Meier plot was generated (Figure 5D). A trend was evident, whereby patients in the top 2 tertiles of BST2 expression showed a shorter interval of disease-free survival compared to those with lower BST2 expression in the primary tumor (p = 0.12), supporting our in vitro molecular and functional studies on breast cancer cell lines. The predicted trend for hormonal therapy response observed with BST2 as a single gene classifier was consistent with the grade gene index (GGI)-predicted profile in this subgroup of ER+, hormone-resistant patients (HR 2.9, CI: 1.26–6.95, p = 0.01). However, unlike BST2 alone, the GGI predictor uses a multigene panel.
Resveratrol Mediated Abrogation of Endogenous BST2 Expression
Resveratrol is known to modulate AP2 binding within promoters of putative oncogenes, and included in over 60 human trials for a variety of diseases (clinicaltrials.gov). As a pragmatic approach for reversing gene expression in the clinical setting, we tested the in vitro effects of resveratrol on BST2. Overexpressing tumor cultures treated with increasing doses of resveratrol for 24 hr demonstrated a significant reduction in BST2 transcript levels (p<0.01) (Figure 6A). In 3-dimensional Matrigel cultures treated with resveratrol, this change in expression effectively reproduced the downstream pro apoptotic effects of gene silencing observed with BST2 siRNA transfection (Figure 6B). The synergistic effect of concurrent exposure to resveratrol and tamoxifen on apoptosis induction measured by anti cleaved caspase 3 immunolocalization was over 10-fold that of either treatment alone (p<0.01). Closely analogous to the consequences of BST2 knockdown on the cell cycle, resveratrol treatment of tumor cultures induced a marked decline in S-phase cells accompanied by an increase in the G1 fraction (Figure 6C).
Figure 6. Resveratrol attenuates endogenous BST2 expression and disrupts downstream functional effects in high grade breast cancer cells.

A. Tumor cells (CCdl54) treated with resveratrol for 24 hrs displayed a significant decline in BST2 expression denoted by asterisks (p<0.01). QPCR analysis performed in triplicate, and normalized to ACTB expression. B. Enhancement of apoptotic cell death induced by 24 hr tamoxifen treatment of 6-day resveratrol (20 µM) supplemented tumor cultures plated in Matrigel. Top panel – immunolocalization of cleaved caspase 3 (green) in apoptotic cells; nuclei counterstained with PI (red). Bar - 25 µm. Bottom panel – Immunostaining data plotted as apoptotic index represents proportion of PI-stained nuclei (>100) vs. immunopositive cells in multiple microscopic fields scanned with a 20× objective, and averaged. Asterisk denotes significant (p<0.01) differences in the apoptotic index in the presence of tamoxifen alone vs. resveratrol pretreatment followed by tamoxifen. C. Resveratrol-mediated suppression of the cell cycle in high grade tumor cells (CCdl54, CCdl672) is similar to the effects of BST2 silencing. A statistically significant reduction in S-phase resulting from both approaches for BST2 attenuation is accompanied by an increase in the G1-fraction in independent cell lines. The data derived from BrdU labeling of 2 independent cell lines represents an average of 2 separate FACS runs on each cell line, with each treatment performed in triplicate sets. D. Induction of AP2 expression in high grade tumor cells (CCdl675, CCdl54) treated with increasing concentrations of resveratrol. QPCR analysis performed in triplicate, and normalized to ACTB expression. Asterisks indicate significant differences (p<0.01) between control and treated samples.
To evaluate whether BST2 attenuation was mediated by the mode of promoter regulation characteristic of TGFβ responsive tumor cells described above, AP2 expression was measured in resveratrol-treated cells. Accompanying the suppressive effects on BST2 expression, increasing concentrations of resveratrol led to markedly increased AP2 levels in 2 independent grade 3 cell lines (Figure 6D). This suggests the possibility of resveratrol induced BST2 promoter repression by AP2 in TGFβ-resistant cells analogous to that occurring normally in TGFβ-responsive breast cancer cells.
Discussion
BST2 overexpression in cancerous breast tissue specimens demonstrated here poses critical questions regarding usurpation of its normal host protective function through aberrant gene regulation. We have defined an oncogenic framework for BST2 in breast cancer by correlative and functional analysis of clinical tissue and novel in vitro model systems, respectively. We show that the overexpression of this gene is characteristic of high histopathologic grade tumors known to display a higher mitotic index, and greater clinical aggressiveness. Generally, most high grade (poorly differentiated or grade 3) breast cancer is associated with poor clinical outcome. Aberrant signaling pathways that undermine the histological differentiation of such cancer cells could shed light on the functional underpinnings of breast cancer aggressiveness, and its timely control. We describe approaches and consequences of in vitro manipulation of BST2 towards the overall goal of suppressing functional hallmarks of tumor aggressiveness.
Critical roles for BST2 have been previously established in the innate immune response. While its expression is induced by interferon (IFN) in the event of initial viral infection, BST2 itself negatively regulates IFN to minimize undesirable effects of prolonged IFN exposure, such as autoimmune diseases [20]. In terms of tissue distribution, it appears that different Transcription Factor Binding Sequences in the BST2 promoter region might be activated in a cell type specific manner [6]. In dendritic cells, interaction of BST2 with its cognate receptor, immunoglobulin-like transcript 7 (ILT7) provides the negative signal to control IFN release [21]. As we have shown here, BST2 regulatory mechanisms in breast epithelium appear to be distinctive from those of immune cell types. Likewise, paracrine signaling phenotypes are known to be remarkably different from tissue to tissue. Genomic profiling of stromal-epithelial crosstalk identifying the signature of fibroblast triggered expression in tumor (FTExT) cells of the breast [22] is in stark contrast to that between bone marrow-derived fibroblasts and lymphoma cells [23].
Here, we have uncovered a causal link between the disruption of TGFβ signaling in high grade breast cancer and the constitutive activation of BST2. Our data suggest that aberrant BST2 overexpression promotes the consequences of obliterated TGFβ-mediated tumor suppressive effects in breast cancer, and subsequent loss of differentiation programs. Members of the TGFβ family are pluripotent cytokines exerting an array of biologic effects in a wide variety of cell types. Several studies have implicated the TGFβ pathway in the pathophysiology of cancer [24]. We show that the transcription factor, AP2 is a critical component of TGFβ-mediated regulation of BST2 in breast cancer. Our data suggests that impaired TGFβ signaling leads to the inability of AP2 to bind the BST2 promoter resulting in failed repression of this putative oncogene in high grade tumor cells. The role of AP2 in TGFβ-mediated signaling has been suggested by other studies [25]. Closely parallel to our observations of the negative regulation of BST2 in breast cancer, AP2 is involved in the repression of promoter sequences of the oncogene, ERBB2 [26], evident as an inverse correlation between AP2 and ERBB2 immunolocalization in archival tumor specimens [27]. In the latter study, AP2 positive tumors displayed a reduced mitotic index and a lower tumor grade, consistent with our findings of intact AP2 regulation of BST2 in low grade breast cancer.
Predictive markers of therapeutic drug resistance are of significant utility towards the consideration of alternate treatment approaches in the clinic but rarely available. Antibody based targeting of BST2 upregulation in B cell lymphoma lesions delayed tumor growth [28] supporting our observations of a BST2-mediated role in breast cancer outcome. Although the TGFβ signaling axis has been implicated in endocrine response [29], molecular mediators have not been clearly identified. BST2 overexpression could serve to identify patients likely to relapse after pro apoptotic cancer therapies. Our data demonstrates a direct functional role of BST2 in the establishment of therapeutic resistance to an ER antagonist. Among changes in the expression of several other genes, profiling studies of tamoxifen-treated normal human mammary epithelial cell cultures have noted induction of BST2 [30]. Further work is needed to decipher the molecular mechanism of BST2-mediated drug resistance, for example how BST2 interacts with, and influences the ER signaling cascade, could be illuminating. Our findings suggest the possibility of engaging resveratrol treatment as a long-term preventive modality, and as an adjunct to conventional therapeutic strategies, particularly since the anti carcinogenic effects of resveratrol are well known. At the molecular level, resveratrol is involved in the inhibition of oncogenic processes, including increased oxidoreductase activity, and oncogenic phenotypes such as BCL2, AKT, FAK, NF-kB, MMP9, and positive cell cycle regulators [31]. Specifically in the context of breast cancer, resveratrol inhibits TGFα resulting in the elevated expression of TGFβ2 [32]. Resveratrol mediated BST2 repression in the model systems studied here can be attributed to the induction of TGFβ regulated AP2. AP2 binding regions within promoters of putative oncogenes, such as Na(+1)/H(+1) exchanger-1 (NHE-1), were previously identified targets of resveratrol [33].
The cellular models used here define an important role for the TGFβ axis as a major contributor to the well-known biological and clinical heterogeneity of breast cancer. Generally, in breast tumors classified as grade 1, and in a proportion of grade 2 cases, TGFβ acts as a potent tumor suppressor by repressing putative oncogenes [17]. In grade 3 and some grade 2 breast tumors, impairment of TGFβ-mediated signaling intermediates, often due to inactivating mutations [34], results in the derepression of these oncogenes, enabling an escape from TGFβ-dependent growth modulation, and culminating in an overall aggressive tumor phenotype. For direct cancer targeting, cell surface markers, such as BST2, are the proverbial “silver bullet” particularly if overexpressed in tumors prone to therapeutic resistance. Breast cancer therapies that target cell surface proteins, such as EGFR and ERBB2 are rare examples of successful biologically informed cancer treatments. Overall, BST2 is a strong candidate for consideration as a theranostic in the target discovery pipeline for breast cancer.
Funding Statement
This work was supported by the National Institutes of Health (NIH) under grant CA109325. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bieniasz PD (2004) Intrinsic immunity: A front-line defense against viral attack. Nat Immunol 5: 1109–1115. [DOI] [PubMed] [Google Scholar]
- 2. Bartee E, McCormack A, Früh K (2006) Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog 2: e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Neil SJ, Zang T, Bieniasz PD (2008) Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451: 425–430. [DOI] [PubMed] [Google Scholar]
- 4. Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, et al. (2009) Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139: 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao W, Bover L (2010) Signaling and ligand interaction of ILT7: receptor-mediated regulatory mechanisms for plasmacytoid dendritic cells. Immunol Rev 234: 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bego MG, Mercier J, Cohen EA (2012) Virus-activated interferon regulatory factor 7 upregulates expression of the interferon-regulated BST2 gene independently of interferon signaling. J Virol 86: 3513–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, et al. (2008) The interferon induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3: 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kupzig S, Korolchuk V, Rollason R, Sugden A, Wilde A, et al. (2003) Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4: 694–709. [DOI] [PubMed] [Google Scholar]
- 9. Andrew AJ, Kao S, Strebel K (2011) C-terminal hydrophobic region in human bone marrow stromal cell antigen 2 (BST-2)/tetherin protein functions as second transmembrane motif. J Biol Chem 286: 39967–39981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, et al. (2006) Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol 177: 3260–3265. [DOI] [PubMed] [Google Scholar]
- 11. Ohtomo T, Sugamata Y, Ozaki Y, Ono K, Yoshimura Y, et al. (1999) Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem Biophys Res Commun 258: 583–591. [DOI] [PubMed] [Google Scholar]
- 12. Cai D, Cao J, Li Z, Zheng X, Yao Y, et al. (2009) Up-regulation of bone marrow stromal protein 2 (BST2) in breast cancer with bone metastasis. BMC Cancer 9: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, et al. (2008) Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res 10: R65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dairkee SH, Ji Y, Ben Y, Moore DH, Meng Z, et al. (2004) A molecular ‘signature’ of primary breast cancer cultures; patterns resembling tumor tissue. BMC Genomics 5: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dairkee SH, Nicolau M, Sayeed A, Champion S, Ji Y, et al. (2007) Oxidative stress pathways highlighted in tumor cell immortalization: association with breast cancer outcome. Oncogene 26: 6269–6279. [DOI] [PubMed] [Google Scholar]
- 16. Dairkee SH, Sayeed A, Luciani G, Champion S, Meng Z, et al. (2009) Immutable functional attributes of histologic grade revealed by context-independent gene expression in primary breast cancer cells. Cancer Res 69: 7826–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sayeed A, Meng Z, Luciani G, Chen LC, Bennington JL, et al. (2010) Negative regulation of UCP2 by TGFβ signaling characterizes low and intermediate-grade primary breast cancer. Cell Death Dis 1: e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dairkee SH, Paulo EC, Traquina P, Moore DH, Ljung BM, et al. (1997) Partial enzymatic degradation of stroma allows enrichment and expansion of primary breast tumor cells. Cancer Res 57: 1590–1596. [PubMed] [Google Scholar]
- 19. Ivshina AV, George J, Senko O, Mow B, Putti TC, et al. (2006) Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res 66: 10292–102301. [DOI] [PubMed] [Google Scholar]
- 20. Gota C, Calabrese L (2003) Induction of clinical autoimmune disease by therapeutic interferon-alpha. Autoimmunity 36: 511–518. [DOI] [PubMed] [Google Scholar]
- 21. Cao W, Bover L, Cho M, Wen X, Hanabuchi S, et al. (2009) Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J. Exp. Med 206: 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luciani MG, Seok J, Sayeed A, Champion S, Goodson WH, et al. (2011) Distinctive responsiveness to stromal signaling accompanies histologic grade programming of cancer cells. PLoS One 6: e20016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, et al. (2010) Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med 16: 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ikushima H, Miyazono K (2010) TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer 10: 415–424. [DOI] [PubMed] [Google Scholar]
- 25. Koinuma D, Tsutsumi S, Kamimura N, Taniguchi H, Miyazawa K, et al. (2009) Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Mol Cell Biol 29: 172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perissi V, Menini N, Cottone E, Capello D, Sacco M, et al. (2000) AP-2 transcription factors in the regulation of ERBB2 gene transcription by oestrogen. Oncogene 19: 280–288. [DOI] [PubMed] [Google Scholar]
- 27. Gee JM, Robertson JF, Ellis IO, Nicholson RI, Hurst HC (1999) Immunohistochemical analysis reveals a tumour suppressor-like role for the transcription factor AP-2 in invasive breast cancer. J. Pathol 189: 514–520. [DOI] [PubMed] [Google Scholar]
- 28. Schliemann C, Roesli C, Kamada H, Borgia B, Fugmann T, et al. (2010) In vivo biotinylation of the vasculature in B-cell lymphoma identifies BST-2 as a target for antibody-based therapy. Blood 115: 736–744. [DOI] [PubMed] [Google Scholar]
- 29. Buck MB, Pfizenmaier K, Knabbe C (2004) Antiestrogens induce growth inhibition by sequential activation of p38 mitogen-activated protein kinase and transforming growth factor-beta pathways in human breast cancer cells. Mol Endocrinol 18: 1643–1657. [DOI] [PubMed] [Google Scholar]
- 30. Schild-Hay LJ, Leil TA, Divi RL, Olivero OA, Weston A, et al. (2009) Tamoxifen induces expression of immune response-related genes in cultured normal human mammary epithelial cells. Cancer Res 69: 1150–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Athar M, Back JH, Tang X, Kim KH, Kopelovich L, et al. (2007) Resveratrol: a review of preclinical studies for human cancer prevention. Toxicol Appl Pharmacol 224: 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Serrero G, Lu R (2001) Effect of resveratrol on the expression of autocrine growth modulators in human breast cancer cells. Antioxid Redox Signal 3: 969–979. [DOI] [PubMed] [Google Scholar]
- 33. Jhumka Z, Pervaiz S, Clément MV (2009) Resveratrol regulates the expression of NHE-1 by repressing its promoter activity: critical involvement of intracellular H2O2 and caspases 3 and 6 in the absence of cell death. Int J Biochem Cell Biol 41: 945–956. [DOI] [PubMed] [Google Scholar]
- 34. Wang D, Kanuma T, Mizunuma H, Takama F, Ibuki Y, et al. (2000) Analysis of specific gene mutations in the transforming growth factor-beta signal transduction pathway in human ovarian cancer. Cancer Res 60: 4507–4512. [PubMed] [Google Scholar]