

Abstract
Ischemia-reperfusion (I/R) injury is a process whereby an initial hypoxic insult and subsequent return of blood flow leads to the propagation of innate immune responses and organ injury. The necessity of the pattern recognition receptor, toll-like receptor (TLR)-4, for this innate immune response has been previously shown. However, TLR4 is present on various cell types of the liver, both immune and non-immune cells. Therefore, we sought to determine the role of TLR4 in individual cell populations, specifically parenchymal hepatocytes, myeloid cells including Kupffer cells, and dendritic cells following hepatic I/R. When hepatocyte specific (Alb-TLR4-/-) and myeloid cell specific (Lyz-TLR4-/-) TLR4 knockout mice were subjected to warm hepatic ischemia there was significant protection in these mice compared to wild-type (WT). However, the protection afforded in these two strains was significantly less than global TLR4 specific TLR4 knockout (TLR4-/-) mice. Dendritic cell specific TLR4-/- (CD11c-TLR4-/-) mice had significantly increased hepatocellular damage compared to WT mice. Circulating levels of high mobility group box-1 (HMGB1) were significantly reduced in the Alb-TLR4-/- mice compared to WT, Lyz-TLR4-/-, CD11c-TLR4-/- mice and equivalent to global TLR4-/- mice, suggesting that TLR4 mediated HMGB1 release from hepatocytes may be a source of HMGB1 after I/R. Hepatocytes exposed to hypoxia responded by rapidly phosphorylating the mitogen-activated protein kinases JNK and p38 in a TLR4-dependent manner; inhibition of JNK decreased the release of HMGB1 after both hypoxia in vitro and I/R in vivo.
Conclusion
These results provide insight into the individual cellular response of TLR4. It was found that the parenchymal hepatocyte is an active participant in the sterile inflammatory response after I/R through TLR4-mediated activation of pro-inflammatory signaling and release of danger signals such as HMGB1.
Keywords: Sterile inflammation, innate immunity, conditional knockout mice, danger-associated molecular pattern molecules, mitogen-activated protein kinases
INTRODUCTION
Thorough understanding of the pathophysiology of hepatic ischemia-reperfusion (I/R) is vital as it is commonly encountered clinically during elective liver surgical procedures, solid organ transplantation, trauma, and hypovolemic shock. The liver exhibits both direct cellular damage as the result of the ischemic insult and further dysfunction and damage resulting from activation of inflammatory pathways (1). Although the distal events involved in the inflammatory response resulting in liver damage after I/R injury have been well studied (2, 3), proximal events dictating the propagation of the inflammatory response and further tissue damage remain poorly understood.
The role of TLR4 in the recognition of endotoxin is well established; however, only recently has it become apparent that TLR4 also participates in the response to acute tissue injury (4-6). TLR4 has been found to be an essential constituent of I/R injury, with mice lacking intact TLR4 signaling being significantly protected from injury (5-8). The liver has a complex cellular composition consisting of parenchymal hepatocytes and non-parenchymal cells (NPC), such as Kupffer cells (KC), hepatic dendritic cells (DC), stellate cells, and natural killer cells, with functional TLR4 found ubiquitously (9, 10). Previous work has investigated whether the parenchymal hepatocytes or NPCs are the TLR4 responsive population in the sterile inflammatory response (5, 8, 11). Our studies with TLR4 chimeric mice demonstrate that in the setting of non-infectious I/R induced injury, bone marrow-derived cells are primarily responsible for TLR4-dependent hepatocellular injury (7). In contrast, other studies have also suggested a role for parenchymal/non-bone marrow derived cells contributing to TLR4-dependent injury (8, 11). Therefore, the role of TLR4 on specific cell types is still unclear.
The purpose of our study was to investigate the role of TLR4 on various cell types of the liver, both parenchymal and immune, during hepatic I/R using cellular specific TLR4 knockout mice. This is unique from other studies where the more global impact of TLR4 on the liver has been investigated. In this work, we have generated transgenic cell-specific TLR4 knockout mice to illustrate the dichotomous role of TLR4 after I/R. We find TLR4 on DCs contributes primarily a protective role while TLR4 on both hepatocytes and myeloid cells promotes injury. In addition to immune cells, hepatocytes are identified as one of the key cellular constituents in the innate immune response associated with I/R. These findings represent an advance over previous knowledge given the important cell specific findings.
MATERIAL AND METHODS
Animals
Male wild-type (TLR4loxP/loxP) mice, cell specific, and global TLR4-/- mice were bred at our facility and used at the age of 8-12 weeks. All mice developed were on a C57BL/6 genetic background. Animal protocols were approved by the Animal Care and Use Committee of the University of Pittsburgh and the experiments were performed in strict adherence to the NIH Guidelines for the Use of Laboratory Animals.
Generation of TLR4loxP/loxP and cellular specific TLR4-/- mice
In brief, the TLR4loxP allele was created by inserting loxP sites within intron 1 and intron 2, flanking exon 2 of TLR4. Overview of this construct is shown in Supplemental Figure 1. Mice homozygous for TLR4loxP were generated by Ozgene (Bentley, WA). TLR4loxP/loxP mice were interbred with stud males (TLR4loxP/-; Alb-cre, TLR4loxP/-; Lyz-cre, or TLR4loxP/-; CD11c-cre) to generate desired genotype. Mice homozygous for Cre recombinase linked to the albumin (alb), CD11c (cd11c), and lysozyme (lyz) promoter are commercially available from Jackson. Transgenic male mice used for experiments were confirmed to be desired genotype via standard genotyping techniques. WT mice used in this study were TLR4loxP/loxP mice without the introduction of Cre recombinase. Global TLR4-/- mice were globally lacking the loxP flanked exon 2, i.e. they were global homozygotes for the same mutation contained within the conditional knockout mice. Sodhi et al. have recently provided a detailed description of the novel TLR4-/- mice used in this study (12).
Genomic and functional characterization of transgenic TLR4-/- mice
For confirmation of TLR4 mRNA expression in WT, Alb-TLR4-/-, and TLR4-/- we first isolated HCs, NPCs, or tissue, using Qiagen RNeasy Mini Kit (Valencia, CA) to isolate RNA and Clontech Sprint™ RT Complete-Double PrePrimed (Mountain View, CA) to make cDNA. For confirmation of TLR4 expression in Lyz-TLR4-/- we first isolated peritoneal macrophages and performed positive selection using F4/80 beads (BD Bioscience). Specific primers were as follows: Forward 5’-TGCCACCAGTTACAGATCGTC-3’ and Reverse 5’-GAGTTTCTGATCCATGCATTGG-3’ for TLR4, and β-actin primers as described previously (5). The response of NPCs from WT, Alb-TLR4-/- or TLR4-/- mice and isolated macrophages from WT, Lyz-TLR4-/-, or TLR4-/- mice was determined by exposing cells to 10ng/mL of LPS (Sigma) for 6h and using TNF-α or IL-6 ELISA for quantification (R&D Systems). Confirmation that Kupffer cells in CD11c-TLR4-/- mice retained functional TLR4 was accomplished by isolating Kupffer cells from the NPCs by performing positive selections using F4/80 beads with subsequent exposure to LPS.
Liver ischemia/reperfusion
A nonlethal model of segmental (70%) hepatic warm ischemia and reperfusion was used as previously described (5). TLR4loxP/loxP mice were used as WT control for all in vivo experiments. The JNK inhibitor (SP600125; 10mg/kg; Calbiochem) and p38 inhibitor (SB203580; 10mg/kg; Calbiochem) were administered I.P. 1h prior to ischemia.
Isolation, culture, and treatment of hepatocytes and non-parenchymal cells
Hepatocytes and NPCs were isolated and plated as previously described (7). For experiments involving hypoxia, the medium was replaced with media equilibrated with 1% O2, 5% CO2, and 94% N2 and placed into a modular incubator chamber (Billups-Rothenberg), which was flushed with the same gas mixture. For experiments using JNK inhibitor (SP600125) or p38 inhibitor (SB203580), 25μM was added to the media 30min prior to treatment with hypoxia.
Serum ALT, HMGB1, and cytokine quantification
Serum alanine aminotransferase (sALT) levels were measured using the DRI-CHEM 4000 Chemistry Analyzer System (HESKA). HMGB1 was quantified using ELISA kit (IBL International). Serum cytokine quantification was performed using Cytometric Bead Array Mouse Inflammation Kit (BD Biosciences).
Immunoblotting
Western blot assay was performed using whole cell lysates from either liver tissue or HCs as previously described (13). Membranes were incubated overnight using the following antibodies: TLR4 (Imgenex), HMGB1 and HO-1 (Abcam); mouse monoclonal HMGB1 Ab and β-actin (Sigma); phospho-p38, p38, phospho-c-Jun, c-Jun, phospho-JNK, JNK, ERK, phospho-ERK, p65, and phospho-p65 (Cell Signaling).
Immunofluorescent and IHC staining
Immunofluorescent staining was performed using HMGB1 Ab (1:1000, Abcam) as previously described (14). IHC for neutrophil infiltration was accomplished using Anti-Neutrophil antibody [7/4] (Abcam).
Preparation and delivery of Adenoviral vectors
An E1- and E3-deleted adenoviral vector carrying AdTLR4 and AdLacZ cDNA were constructed and utilized in vivo as previously described (15).
SYBR green real-time reverse transcription polymerase chain reaction
SYBR green PCR was performed as previously described using β-actin as endogenous control (14). Specific primers were as follows: IL-10, Forward 5’-TACCTGGTAGAAGTGATGCC-3’ and Reverse 5’-CATCATGTATGCTTCTATGC-3’, and HO-1 is commercially available from Qiagen (Valencia, CA).
Statistical analysis
Results are expressed as either mean±standard error of the mean (SEM) or mean±standard deviation (SD). Group comparisons were performed using ANOVA and Student’s t test. A P≤0.05 was considered statistically significant.
RESULTS
Confirmation of TLR4 knockout mice
To investigate the role of TLR4 on individual cellular population, we generated hepatocyte, myeloid cell, and DC specific TLR4 knockout (Alb-TLR4-/-, Lyz-TLR4-/-, and CD11c-TLR4-/-, respectively) mice using Cre-loxP technology. Mice with loxP sites flanking exon 2 of TLR4 were interbred with mice that had Cre recombinase linked to the desired promoter. The WT mice used had loxP inserted without the expression of Cre recombinase, and TLR4-/- mice were globally lacking the loxP flanked exon 2. Both WT and TLR4-/- mice were born healthy and fertile, without any grossly apparent phenotypic differences.
Verification of the specificity of the TLR4 knockout in the Alb-TLR4-/- mice was accomplished by isolating both hepatocytes and NPCs, and analyzing these cells for the presence of TLR4 mRNA transcription using RT-PCR with primers specific for exon 2 of TLR4 (Fig.1A). TLR4 was present in both hepatocytes and NPCs of the WT mice, whereas the Alb-TLR4-/- mice had TLR4 expressed only in the NPCs. Global TLR4-/- had no detectable TLR4 in either cell population. Western blot analysis was performed to confirm that hepatocytes isolated from the Alb-TLR4-/- mice had TLR4 protein levels that were undetectable (Fig.1B).
Figure 1. Confirmation of specificity of TLR4 knockout (TLR4-/-).
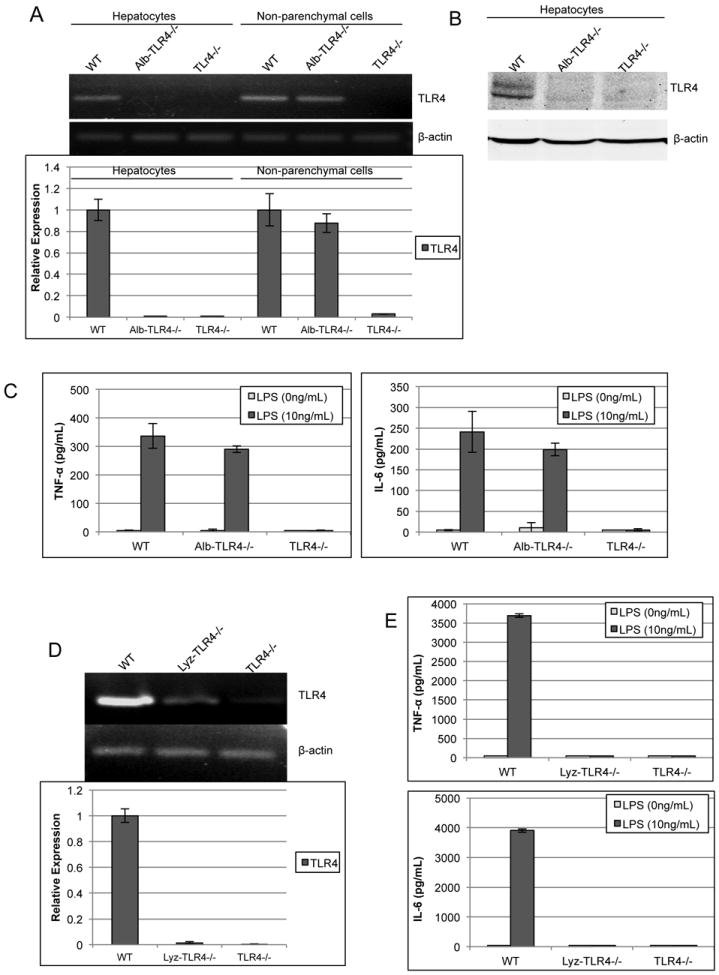
(A) RT-PCR was used to determine mRNA levels of TLR4 within either isolated hepatocytes or non-parenchymal cells (NPCs) from WT, Alb-TLR4-/-, and TLR4-/- mice. β-actin was used as endogenous control to determine relative expression compared against WT. (B) TLR4 protein levels were assessed by Western blot analysis. (C) NPCs were isolated and stimulated with LPS for 6h and the supernatant was analyzed by ELISA for either TNF-α or IL-6. (D) RT-PCR analysis of peritoneal macrophages to determine TLR4 mRNA expression in Lyz-TLR4-/-. (E) LPS response was determined in peritoneal macrophages from WT, Lyz-TLR4-/-, and TLR4-/- mice. Data represent mean ± SD. N=2 samples per group run in duplicate. Figure is representative of two experiments with similar results.
It has previously been shown that the immune cells of the liver play a major role in I/R injury (7, 8, 11). Although the presence of TLR4 was seen in the NPC population of cells in the Alb-TLR4-/- mice, we also wanted to confirm that the functional characteristics of TLR4 in these cells was unchanged. Therefore, we studied the response of isolated NPCs to LPS by determining the level of TNF-α and IL-6 in the supernatant using ELISA (Fig.1C). There was no significant difference between the NPCs of the WT and Alb-TLR4-/- mice, while the TLR4-/- NPCs failed to respond to LPS, as expected. Additional confirmation of the functional deletion of TLR4 in Alb-TLR4-/- mice was demonstrated by lack of Alexa-488 labeled LPS uptake in Alb-TLR4-/- hepatocytes similar to global TLR4-/- (data not shown). It was also seen that hepatic intracellular downstream signaling to TLR4 activation was concordant with the above results, with p65 (NF-κβ) activation attenuated in response to LPS in both the Alb-TLR4-/- and global TLR4-/- mice (Supplemental Fig.2A).
By linking Cre recombinase expression with the lyz promoter, mice were generated with TLR4-/- specific to the myeloid cell lineage, including Kupffer cells (16). We confirmed that the peritoneal macrophages lack both TLR4 expression (Fig.1D) in addition to functional response to LPS (Fig.1E). Cre recombinase linked to the cd11c promoter was use to generate DC specific TLR4-/- mice. This has previously been shown by both others (17) and also our lab (unpublished data) to be an effective method for developing DC specific transgenic mice. Additionally, we confirmed that Kupffer cells isolated from CD11c-TLR4-/- mice retained functional TLR4 (Supplemental Fig.2B and C).
Hepatocyte and myeloid cell TLR4 is required for maximal hepatic I/R injury
Although our previous studies with TLR4 chimeric mice highlight the importance of bone marrow-derived cells in mediating TLR4-dependent inflammation in response I/R (7), the role of TLR4 on individual cell types can now be investigated with the use of transgenic mice. To better clarify whether TLR4 on hepatocytes, myeloid cells, and DCs impacts the inflammatory response during I/R, transgenic mice were subjected to hepatic I/R. In WT control mice, the sALT level was significantly greater than both the Alb-TLR4-/- mice and Lyz-TLR4-/- mice; however, the global TLR4-/- mice had significant protection compared to both Alb-TLR4-/- and Lyz-TLR4-/- mice (Fig.2A). Interestingly, the CD11c-TLR4-/- mice had significantly increased hepatocellular injury compared to WT (Fig.2A). The degree of liver damage on histologic analysis was concordant with the sALT results (Fig.2B). Sham livers demonstrated no histologic evidence of hepatocellular injury (data not shown). These results demonstrate that lack of TLR4 on both hepatocytes and myeloid cells results in protection from I/R, whereas TLR4 on DCs may have a protective role following liver I/R.
Figure 2. Cellular specific role of TLR4 on hepatocellular injury after I/R.
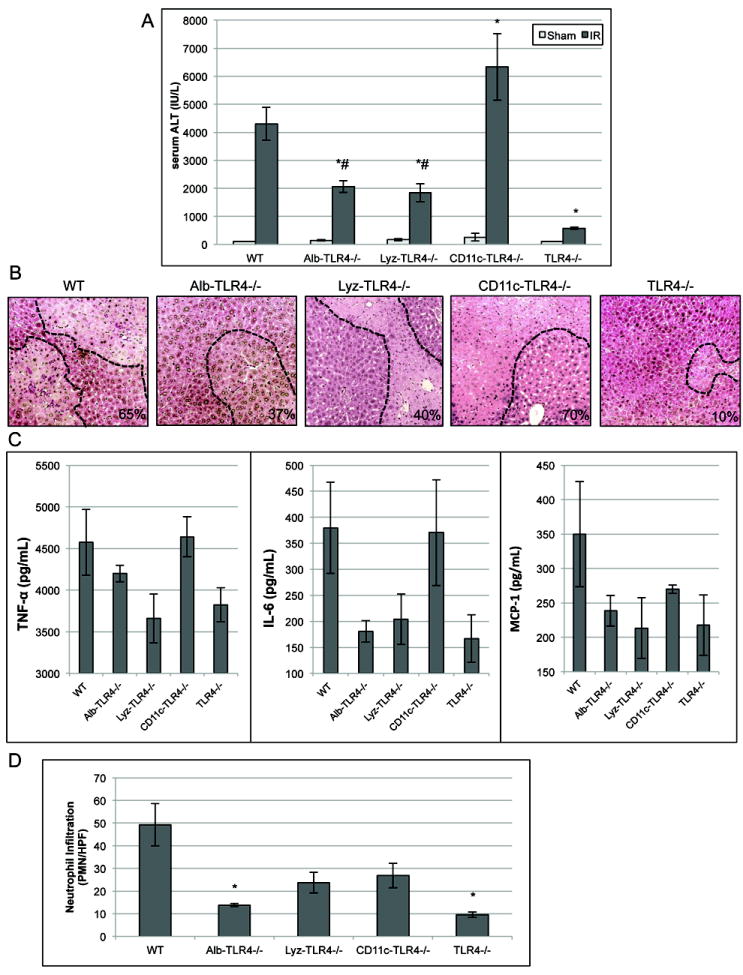
(A) Serum ALT levels were analyzed in WT, cell specific and global TLR4-/- mice after either sham laparotomy or 1h of ischemia and 6h of reperfusion. (B) H&E stained liver sections from obtained from mice 6h after reperfusion (original magnification ×100). Images are representative liver sections from six mice per group. Necrotic area is outlined in black dashed line and percent of area that is necrotic is indicated. (C) Serum cytokine analysis after I/R was performed. (D) The number of neutrophils infiltrating into liver tissue after I/R as assessed by IHC of neutrophils per HPF (×200). Data represents mean ± SEM; n≥6 mice per group; *P<0.05 when compared against WT; #P<0.05 when compare against global TLR4-/-.
Additionally, serum levels of TNF-α, IL-6, and MCP-1 were quantified (Fig.2C). Cytokines generally reflected the serum ALT levels with intermediate cytokine decrease in the cell-specific KO mice compared to the global TLR4-/- mice. Quantification of neutrophil infiltration was also determined (Fig.2D). Interesting, the number of neutrophils was significantly decreased in not only the global TLR4-/-, but also the Alb-TLR4-/- mice. These results again demonstrate the importance of hepatocyte TLR4 in the I/R inflammatory response.
Serum HMGB1 release after hepatic I/R is dependent on TLR4
HMGB1 is an evolutionarily conserved protein present in the nucleus of almost all eukaryotic cells where it functions to stabilize nucleosomes and acts as a transcription factor (18). HMGB1 is also rapidly mobilized and released in the setting of hepatic I/R to act as key DAMP molecule (5, 19). TLR4 and HMGB1 are intimately related, with TLR4 both functioning as a receptor for HMGB1 in addition to mediating its nucleocytoplasmic shuttling and subsequent release (7, 19). Thus, we sought to determine the role of cell specific TLR4-/- in the release of HMGB1 following hepatic I/R. When serum HMGB1 levels after I/R were analyzed, Alb-TLR4-/- transgenic mice had significantly lower serum HMGB1 levels compared to WT (Fig.3A). Lyz-TLR4-/- also had lower serum HMGB1 levels but did not reach statistical significance (Fig.3A). Alb-TLR4-/- and global TLR4-/- mice had HMGB1 levels that were similar and significantly lower than Lyz-TLR4-/- mice (Fig.3A). CD11c-TLR4-/- mice, on the other hand, did not have any significant difference in HMGB1 levels compared to WT.
Figure 3. Cellular specific role of TLR4 on HMGB1 release after I/R.
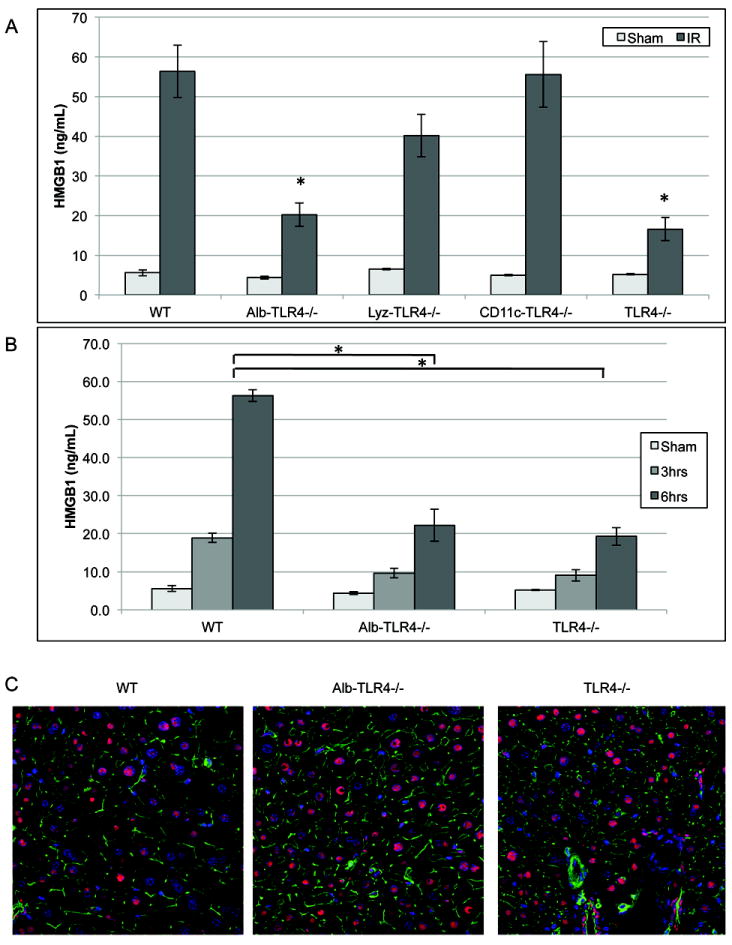
(A) Serum HMGB1 ELISA after 6h of reperfusion. *P<0.05 when compared against WT. (B) Serum HMGB1 ELISA in WT, Alb-TLR4-/-, and TLR4-/- after either 3h or 6h of reperfusion. *P<0.05 when compared against WT. (C) Immunofluorescence of liver tissue after 3h of reperfusion. Red, HMGB1; green, F-actin; blue, nuclei (original magnification ×600). Data represents mean ± SEM; n≥6 mice per group mice per group.
Since TLR4 on hepatocytes appeared to be the main contributor to TLR4 mediated HMGB1 release after I/R, we next further investigated HMGB1 release in Alb-TLR4-/- and global TLR4-/- mice. These mice had decreased levels of circulating HMGB1 following both 3 and 6h of reperfusion when compared to WT mice (Fig.3B). Immunofluorescent staining of liver sections of these mice confirmed the role that TLR4 plays in the release of HMGB1 following I/R. Both Alb-TLR4-/- and global TLR4-/- mice livers had retained nuclear and decreased cytoplasmic HMGB1 when compared to the WT mice (Fig.3C). Our findings show that TLR4 on parenchymal cells are the main contributors to circulating HMGB1 release during liver I/R.
IL-10 and HO-1 expression is altered in Lyz-TLR4-/- and CD11c-TLR4-/- mice
It has been found previously that decreased expression of hepatoprotective factors HO-1 and IL-10 from Kupffer cells and decreased IL-10 from DCs resulted in increased I/R injury (20-22). Therefore, we investigated IL-10 and HO-1 expression in Lyz-TLR4-/- and CD11c-TLR4-/- mice. When compared to WT mice, Lyz-TLR4-/- mice had both IL-10 and HO-1 up-regulated after I/R, possibly leading to the protection seen in these mice (Fig.4A, C). This expression pattern was confirmed at the protein level as well (Fig.4B, D). Additionally, expression of IL-10 was decreased in CD11c-TLR4-/- mice after I/R, suggesting a mechanism for the increased hepatocellular injury seen in these mice (Fig.4C, D). Alb-TLR4-/- did not show any notable differences in either IL-10 or HO-1 expression when compared to WT (data not shown). Therefore, TLR4-mediated modulation of the expression of IL-10 and HO-1 in myeloid cells and IL-10 in DCs may contribute to the differences seen in hepatic damage.
Figure 4. HO-1 and IL-10 mRNA and protein expression is altered in Lyz-TLR4-/- and CD11c-TLR4-/- mice after I/R.
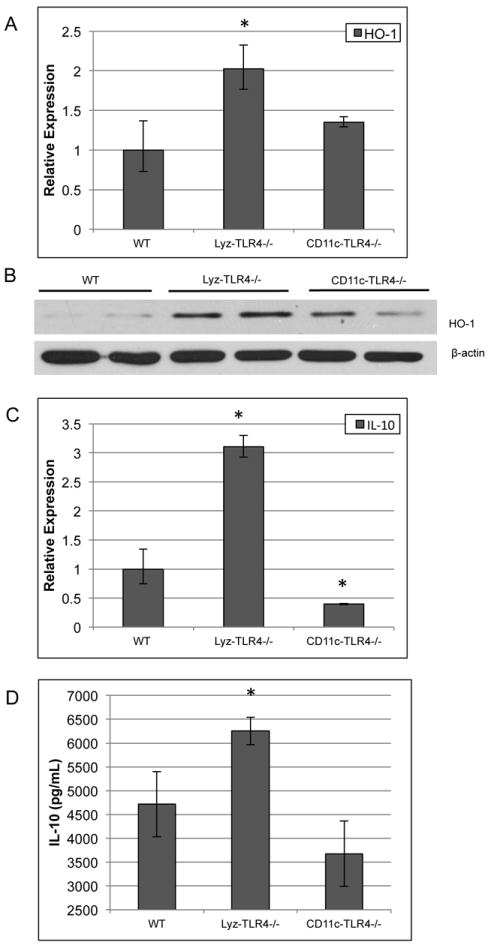
Real-time PCR was used to determine (A) HO-1 and (C) IL-10 expression in liver tissue after I/R in Lyz-TLR4-/- and CD11c-TLR4-/- mice. Protein quantification was determined using Western blot analysis for HO-1 (B) and Cytometric Bead Array assay for IL-10 (D). β-actin was used as endogenous control to determine relative expression compared against WT. *P<0.05 when compared against WT. Figures A and C are representative from triplicate experiments with pooled RNA from 6 animals per group. Figure B shows two representative animals for each group.
Hepatocyte TLR4 mediates inflammatory signaling following I/R
Since the Alb-TLR4-/- mice were significantly protected, we further investigated the mechanisms by which this was taking place. Among the most proximal inflammatory signaling events following I/R is the activation of mitogen-activated protein (MAP) kinases (23, 24). To determine if hepatocyte TLR4 was involved in the activation of MAP kinase signaling, we performed Western blot analysis on liver lysates from WT, Alb-TLR4-/-, and global TLR4-/- mice after I/R. The phosphorylation of the MAP kinases, c-Jun-N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) were substantially reduced at 3h of reperfusion in both Alb-TLR4-/- and global TLR4-/- mice when compared to WT mice (Fig.5). We found no role for hepatocyte TLR4 in p38 phosphorylation at this time point; however, p38 phosphorylation occurs very early after reperfusion (23). This may account for the lack of difference seen at 3h of reperfusion. Notably, we did not see major differences in MAP kinase activation at either 1h or 6h time points. To confirm that these findings were related to the local effects of I/R and not systemic inflammatory mediators, we demonstrated no increased phosphorylation of these proteins in the non-ischemic lobes (Fig.5). Therefore, hepatocyte TLR4 seems to be an important mediator of MAP kinase activation following I/R.
Figure 5. Mitogen-activated protein (MAP) kinase activation is decreased in Alb-TLR4-/- mice after I/R.
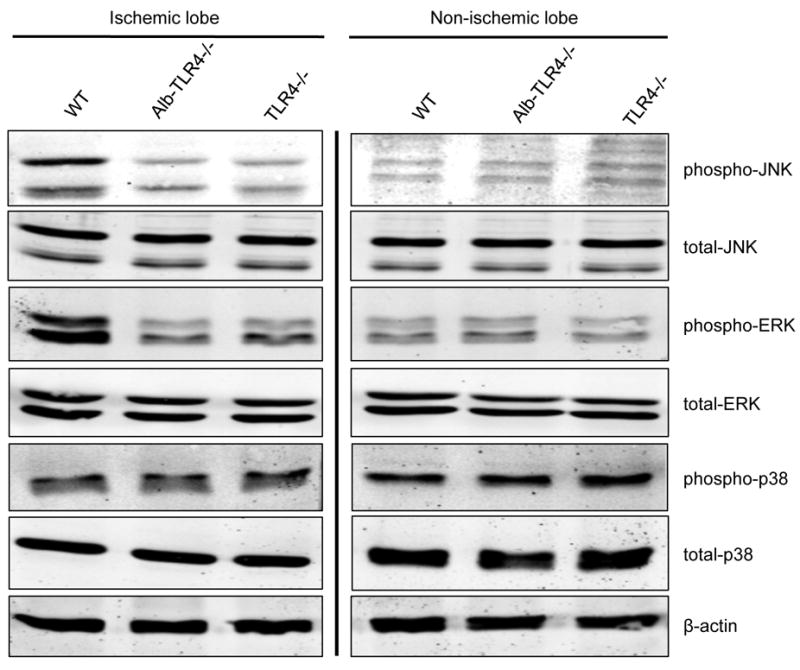
Western blot of phosphorylated or total MAP kinases from protein isolated from either ischemic lobe or non-ischemic lobe after 1h of ischemia and 3h of reperfusion. Figure is representative from 3 experiments of 4 animals each with similar results.
Hypoxia induced JNK and p38 activation in hepatocytes is dependent on TLR4
Our above experiments found that hepatocyte TLR4 was involved in the activation of JNK signaling in the liver following I/R. JNK is activated by exposure of cells to cytokines and environmental stress and has previously been demonstrated to be activated in hepatocytes by both hypoxia and liver I/R (25, 26). Therefore, we exposed WT hepatocytes to hypoxia and rapidly saw increased phosphorylation of JNK and p38 compared to normoxia (Fig.6A). When TLR4-/- hepatocytes were exposed to hypoxia the phosphorylation of JNK, c-Jun (the downstream target of JNK) and p38 was substantially reduced compared to WT hepatocytes (Fig.6B). We saw no increase in NF-κβ (p65) or ERK phosphorylation with hypoxia exposure (Fig.6B). To confirm this response was in fact due to the lack of functional TLR4 and not some other mechanism, hepatocytes from TLR4-/- mice were then transfected with either a control adenoviral vector (AdLacZ) or recombinant adenovirus encoding TLR4 (AdTLR4). TLR4 expression using AdTLR4 was confirmed by Western blot (Fig.6C). Transfection of TLR4-/- hepatocytes with AdTLR4 restored JNK and p38 phosphorylation in response to hypoxia (Fig.6D), indicating that this is in fact a TLR4-dependent response. Thus, these results demonstrate that hepatocytes respond to hypoxic stress with a rapid activation of JNK and p38 in a TLR4-dependent manner.
Figure 6. Hypoxia induced JNK and p38 phosphorylation is TLR4-depedent.
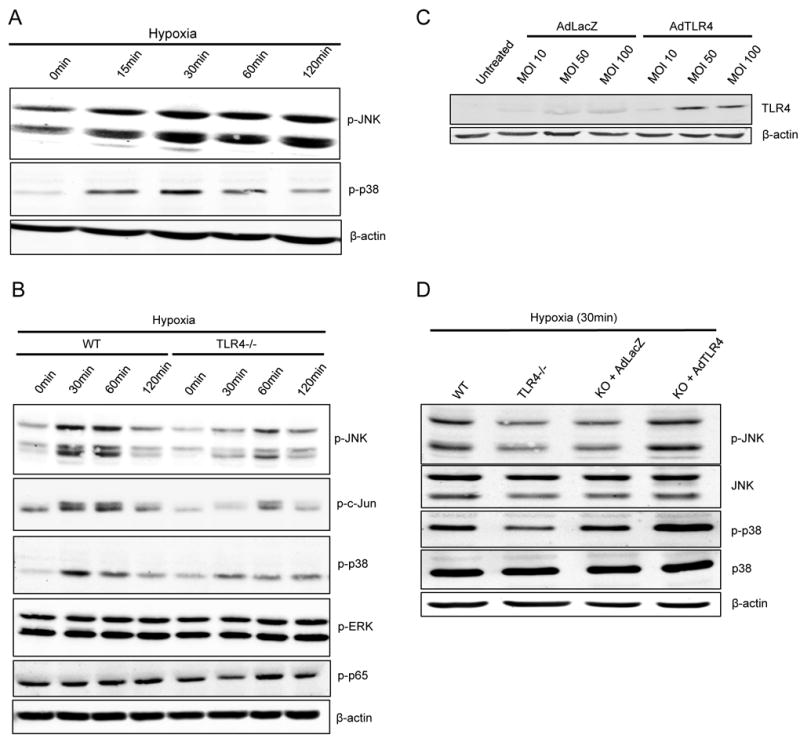
(A) Western blot of p-JNK and p-p38 after WT hepatocytes were exposed to hypoxia for the indicated time period. (B) Western blot of phosphorylated and total MAP kinase levels during hypoxia with comparison between WT and TLR4-/-. (C) Western blot analysis of TLR4 expression after TLR4-/-hepatocytes were transfected with AdTLR4 or the control AdLacZ. (D) Western blot analysis of p-JNK, and p-p38 after WT, TLR4-/-, KO+AdLacZ, or KO+AdTLR4 hepatocytes (MOI 100) were exposed to hypoxia for 30min. Figures are representative of three trials with identical results.
HMGB1 release from hepatocytes under hypoxic stress is JNK-dependent
We next sought to determine whether the release of HMGB1 was mediated by JNK phosphorylation. Therefore, we added the JNK inhibitor (SP600125) to the media of hepatocytes exposed to hypoxia. The phosphorylation of the target of JNK, c-Jun, was inhibited with addition of the JNK inhibitor (Fig.7A). With addition of the JNK inhibitor and 24h exposure to hypoxia there was substantial reduction of the release of HMGB1 into the hepatocyte supernatant (Fig.7B). Additionally, we determined that this difference was not due to decreased hepatocyte death and passive HMGB1 release by determining supernatant levels of LDH and β-actin (Fig. 7B).
Figure 7. HMGB1 release from hepatocytes under hypoxic stress is JNK-dependent.
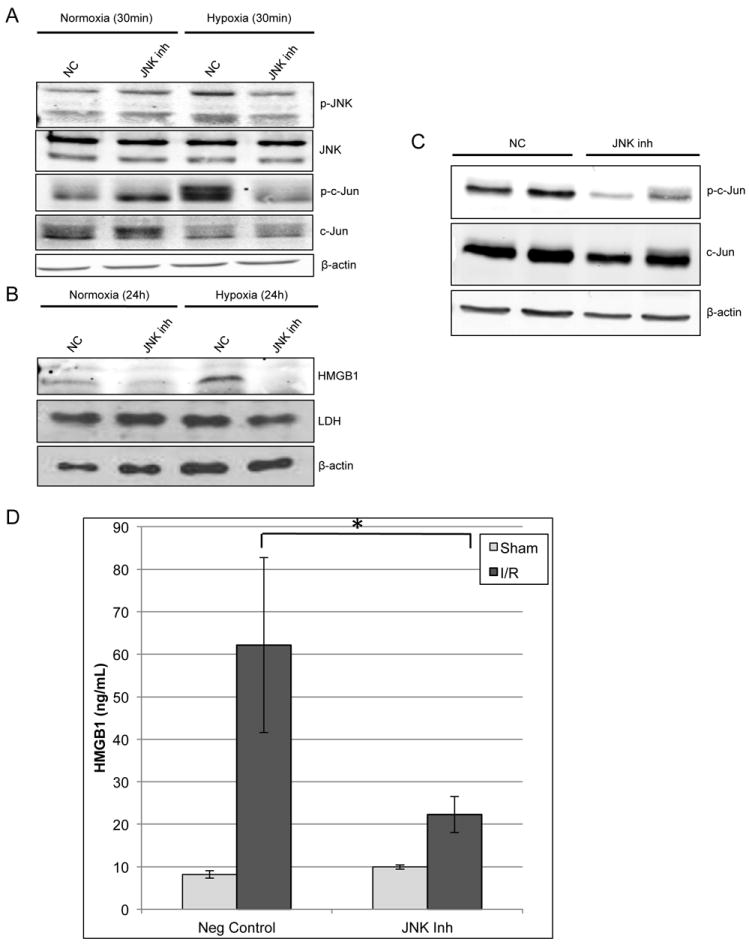
JNK inhibitor (SP600125; 25μM) or negative control (NC) was added to the media of hepatocytes and they were either exposed to hypoxia or normoxia for 30min. (A) Western blot analysis of JNK and c-Jun activation in absence or presence of JNK inhibitor. (B) Western blot of hepatocyte supernatant for HMGB1 after 24h of either normoxia or hypoxia. Western blot of the supernatant was also performed for β-actin and LDH to estimate cellular death. (C) Western blot for in vivo confirmation of JNK inhibitor efficacy after 1h of ischemia and 3h of reperfusion. Representative samples from 2 mice of each group are shown. (D) ELISA of serum HMGB1 levels after either negative control or JNK inhibitor were given prior to I/R. Data represents mean ± SEM; n≥6 mice per group; *P<0.05.
The effect of the JNK inhibitor on HMGB1 release in vivo after I/R was also investigated. The efficacy of the JNK inhibitor was first confirmed by decreased phosphorylation of c-Jun compared to vehicle control on Western blot analysis (Fig.7C). With administration of the inhibitor given prior to I/R there was a significant decrease in serum levels of HMGB1 after I/R (Fig.7D). We again confirmed that this decrease in HMGB1 was not due solely to decreased hepatocellular injury with JNK inhibition by determining that sALT levels were unchanged at 3h of reperfusion (Supplemental Fig.3) in addition to histologic analysis (data not shown).
The p38 inhibitor, SB203580, was also studied both in vitro and in vivo similar to the JNK inhibitor. With administration of the p38 inhibitor prior to hypoxia exposure in vitro and prior to I/R in vivo there was no inhibitory effect seen on HMGB1 release (data not shown), suggesting that p38 does not play a major role in TLR4 mediated HMGB1 release. Therefore, it seems that activation of JNK but not p38 is required for the extracellular release of HGMB1 both after hypoxic stress in vitro and I/R in vivo.
DISCUSSION
Hepatic I/R is dependent on the PRRs to sense and initiate the sterile inflammatory response. While the central role of the PRR, TLR4, in this process had been previously demonstrated (5, 6) the role of TLR4 on individual cell types, specifically parenchymal versus NPC, during the sterile inflammatory response was conflicted. Therefore, in this study we describe the novel use of Cre-loxP technology to knockout TLR4 in hepatocytes, myeloid cells, and DCs and elucidate their individual role in I/R injury. The key and novel findings include: 1) Both hepatocyte and myeloid cell TLR4 is required for maximal I/R associated injury; 2) DC TLR4-/- worsens injury after I/R and is associated with decreased IL-10 expression;3) Hepatocytes are a major source of circulating HGMB1 after I/R; 4) Hepatocytes respond to hypoxia with increased phosphorylation of MAP kinases, JNK and p38, in a TLR4-dependent fashion; 5) Hypoxia induced HMGB1 release from hepatocytes is dependent on the function of JNK.
Previous work to define the function of TLR4 on individual cellular populations was limited to the use of chimeras. Although we have shown that there was not a significant difference in hepatic I/R induced injury with lack of TLR4 on non-bone marrow derived cells, there was a trend towards an effect and others have subsequently shown that both bone marrow and non-bone marrow derived populations have a role in mediating I/R injury (7, 11). A substantial weakness of the studies using chimeric mice is that there are many individual cell types affected in each experimental group. For example, with chimeric mice it is impossible to differentiate the role of TLR4 on hepatocytes versus endothelial cells or myeloid cells versus DCs. The use of Cre-loxP technology to generate transgenic mice has major advantages in helping to elucidate the precise role of receptors on individual cellular populations. Notably, Cre recombinase linked to lyz is highly expressed in all myeloid derived cells including KCs, neutrophils, and monocytes, but not within DCs (16). This model is not perfect, however, and deletion of TLR4 may occur within a small portion of CD11c+ DCs in these mice, though our functional studies suggest this spillover is negligible. Additionally, while the albumin promoter is active in immature cells that can differentiate into either hepatocytes or cholangiocytes, only the hepatocytes continue to express albumin (27-29). Therefore, it may be possible that some cholangiocytes have some deletion of TLR4, but this is likely negligible since it has been shown to take 6 weeks for maximal Alb-Cre mediated recombination to take place (30). While other methods exist for targeting hepatocytes specifically, such as the AAV8-Ttr-Cre model (28), this is not useful against the other cell types considered here. Therefore, although this technology is not perfect, it is useful here in that it allows for meaningful comparison between parenchymal and non-parenchymal cell specific knockouts. Our characterization along with the previous reports have demonstrated that Cre expression linked to alb, lyz, and cd11c promoter is an efficient and specific way of developing cellular specific knockouts (16, 17, 31).
Hepatic DCs are thought to primarily be anti-inflammatory. Consistent with this, Loi et al. have previously shown that although hepatic I/R leads to DC maturation, they preferentially produce inhibitory cytokines IL-10 and TGF-β (32). Interestingly, our results indicate that DC TLR4 plays a protective role with lack of functional TLR4 in DCs associated with a decrease in IL-10 expression and worsening of hepatocellular injury. Our results mirror the TLR9 results of Bamboat et al. (22), where TLR9 activation by hepatocyte DNA led to the production of IL-10 and hepatoprotection from I/R, leading us to hypothesize that DC TLR9 and TLR4 function similarly after I/R possibly in a redundant fashion. Kupffer cells, on the other hand, have traditionally been thought to be a major mediator of I/R associated injury (1). Our results confirm this finding and further demonstrate this effect to be dependent on TLR4 expression in these cell types. However, other studies in addition to our unpublished data using liposomal clodronate for KC depletion show a decrease in IL-10 and HO-1 expression and increase in hepatocellular injury after I/R suggesting KCs may also provide a protective role, in addition to the pro-inflammatory role driven by TLR4 (20, 21). Additionally, the role of TLR4 ligands in the protective response of KCs has been suggested by Ellet et al. using a model of total hepatic I/R with bowel congestion (20, 33), a model with direct TLR4 activation through LPS release. In TLR4-/- mice, KC depletion leads to increased hepatocellular injury and decreased IL-10 response (33).
We have shown previously that TLR4 signaling is necessary for the hepatic I/R response, and that this response is in part mediated by HMGB1 (5, 19). Here we demonstrate the role hepatocytes play in this response, with release of HMGB1 significantly reduced with lack of functional hepatocyte TLR4 signaling, approximately equal to mice with global TLR4 deficiency. Furthermore, there was an intermediate decrease in serum HMGB1 with lack of TLR4 in myeloid cells even though the hepatocellular injury was not significantly different in these mice. These findings suggest that hepatocytes are the primary cell type responsible for TLR4 dependent HMGB1 release after I/R which is a novel finding and contrary to the current thought that HMGB1 release is primarily dependent on immune cells (34). However, it certainly seems plausible that hepatocytes may be the primary producer of HMGB1 early in I/R, since in our previous work we have shown that hepatocytes can actively release HMGB1 in a response to oxidative stress in a regulated process (15, 19, 35).
There are a number of cellular pathways involved with hypoxia-induced HMGB1 release by hepatocytes, all of which are actively regulated (15, 19, 35, 36). The hyperacetylation of HMGB1, which is largely regulated by histone deacetylases (HDACs), leads to the shuttling of nuclear HMGB1 into the cytoplasm (35, 36). Additionally, HMGB1 translocation and subsequent extracellular release is also dependent on calcium/calmodulin-dependent kinases (CaMKs) and also on functional interferon regulatory factor-1 (IRF-1) (15, 19). JNK has recently been shown to be able to induce expression of IRF-1 (37), substantiating our hypothesis that JNK is upstream of other known pathways leading to HMGB1 release. While JNK inhibition has been shown to be protective in I/R, these effects are seen at time points >6h despite JNK activation occurring much earlier. Therefore, we hypothesized that JNK activation may be responsible for release of a pro-inflammatory mediator rather than directly contributing to injury. Here, we provide evidence that the role of JNK includes facilitation of HMGB1 release from hepatocytes both in vitro and in vivo, thus providing one possible solution.
In summary, we use cellular specific TLR4-/- transgenic mice to establish that TLR4 may either worsen or alleviate hepatocellular injury after I/R depending on cell type. The role of TLR4 on both myeloid and hepatocytes is primarily pro-inflammatory, compared to DCs in which TLR4 plays a primarily anti-inflammatory role (Fig.8). We are intrigued that parenchymal cells, such as hepatocytes, are not mere passive recipients of injury during I/R, but active participants in the sterile inflammatory process. In addition to the cell types studied here, there are certainly other cellular populations that have influential roles in sterile inflammation, such as endothelial cells, and we are currently investigating these. The novel findings of this study help to dissect out the role of the complex cellular interactions occurring in I/R, with the potential to develop therapeutic interventions to abrogate the sterile inflammatory response.
Figure 8.
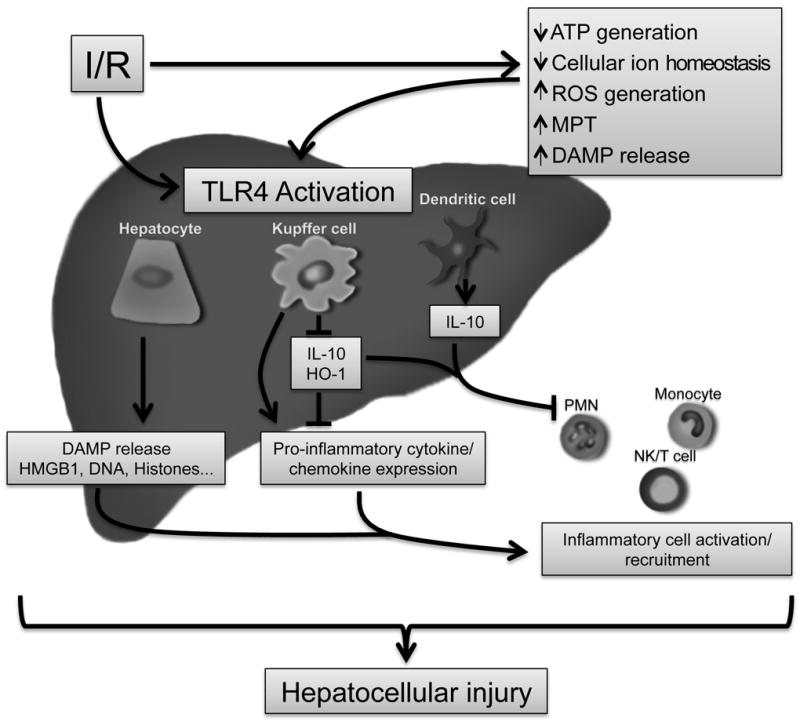
Schematic summary highlighting role TLR4 plays on individual cell populations after hepatic ischemia-reperfusion. (MPT, mitochondrial permeability transition; ROS, reactive oxygen species)
Supplementary Material
Illustration describing generation of TLR4loxP construct used to create transgenic mice. Figure is not to scale.
(A) Hepatocyte TLR4 activation by LPS was determined by quantifying p65 (NF-κβ) nuclear translocation using Array scan after 60 minutes of LPS stimulation. As can be seem, hepatocytes from both Alb-TLR4-/- mice and global TLR4-/- mice lack the normal response to LPS stimulation. *P<0.05 when compared against WT. Additionally, NPCs were isolated from WT, CD11c-TLR4-/-, and TLR4-/- mice and macrophages were purified using positive selection with F4/80 beads. These cells were then exposed to LPS for 6hrs and ELISA used to quantify (B) TNF-α and (C) IL-6 production within the supernatant. Each data point is mean of ࣙ3 independent experiments.
Serum ALT levels were analyzed after mice had intraperitoneal administration of either vehicle (“Neg Control”) or JNK inhibitor prior to 1h of ischemia and 3h of reperfusion. Data represents mean ± SEM; n≥6 mice per group.
Acknowledgments
We thank Nicole Hays and Junda Chen for technical assistance in preparing this manuscript.
Support: This work was supported by Howard Hughes Medical Institute Physician-Scientist Award (A.T.), R01-GM95566 (A.T.), R01-GM50441(T.B.), Association of Academic Surgery Foundation Research Fellowship Award (G.N.), ACS Resident Research Scholarship (J.K.), and American Heart Association Predoctoral Fellowship (B.R.)
Abbreviations
- I/R
ischemia-reperfusion
- TLR
toll-like receptor
- HMGB1
high mobility box-1
- PAMP
pathogen-associated molecular pattern
- DAMP
damage-associated molecular pattern
- PRR
pattern recognition receptor
- NPC
non-parenchymal cell
- DC
dendritic cell
- KC
Kupffer cell
- sALT
serum alanine aminotransferase
- MAP
mitogen-activated protein
- JNK
c-Jun-N-terminal kinase
- ERK
extracellular signal-regulated kinase
Footnotes
Disclosure: The authors declare that they have nothing to disclose.
Contributor Information
Gary W Nace, Email: nacegw@upmc.edu.
Hai Huang, Email: huangh2@upmc.edu.
John R Klune, Email: klunejr@upmc.edu.
Raymond E Eid, Email: eidre@upmc.edu.
Brian R Rosborough, Email: rosborough.brian@medstudent.pitt.edu.
Sebastian Korff, Email: korffs@upmc.edu.
Shen Li, Email: li.shen@medstudent.pitt.edu.
Richard A Shapiro, Email: rashapir@pitt.edu.
Donna B Stolz, Email: dstolz@pitt.edu.
Chhinder P Sodhi, Email: Sodhi.Chhinder@chp.edu.
David J Hackam, Email: hackamd@upmc.edu.
David A Geller, Email: gellerda@upmc.edu.
Timothy R Billiar, Email: billiartr@upmc.edu.
Allan Tsung, Email: tsunga@upmc.edu.
References
- 1.Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury--a fresh look. Experimental and Molecular Pathology. 2003;74:86–93. doi: 10.1016/s0014-4800(03)00008-x. [DOI] [PubMed] [Google Scholar]
- 2.Clavien PA, Rudiger HA, Selzner M. Mechanism of hepatocyte death after ischemia: apoptosis versus necrosis. Hepatology. 2001;33:1555–1557. doi: 10.1053/jhep.2001.0103306le02. [DOI] [PubMed] [Google Scholar]
- 3.Fan C, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver. Journal of molecular medicine. 1999;77:577–592. doi: 10.1007/s001099900029. [DOI] [PubMed] [Google Scholar]
- 4.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 5.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. The Journal of experimental medicine. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. Journal of immunology. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 7.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 8.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. The Journal of clinical investigation. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu S, Gallo DJ, Green AM, Williams DL, Gong X, Shapiro RA, Gambotto AA, et al. Role of Toll-Like Receptors in Changes in Gene Expression and NF- B Activation in Mouse Hepatocytes Stimulated with Lipopolysaccharide. Infection and Immunity. 2002;70:3433–3442. doi: 10.1128/IAI.70.7.3433-3442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su GL, Goyert SM, Fan MH, Aminlari A, Gong KQ, Klein RD, Myc A, et al. Activation of human and mouse Kupffer cells by lipopolysaccharide is mediated by CD14. American journal of physiology. Gastrointestinal and liver physiology. 2002;283:G640–645. doi: 10.1152/ajpgi.00253.2001. [DOI] [PubMed] [Google Scholar]
- 11.Hui W, Jinxiang Z, Heshui W, Zhuoya L, Qichang Z. Bone marrow and non-bone marrow TLR4 regulates hepatic ischemia/reperfusion injury. Biochemical and biophysical research communications. 2009;389:328–332. doi: 10.1016/j.bbrc.2009.08.149. [DOI] [PubMed] [Google Scholar]
- 12.Sodhi CP, Neal MD, Siggers R, Sho S, Ma C, Branca MF, Prindle T, Jr, et al. Intestinal epithelial toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology. 2012;143:708–718. e705. doi: 10.1053/j.gastro.2012.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhai Y, Qiao B, Shen XD, Gao F, Busuttil RW, Cheng G, Platt JL, et al. Evidence for the pivotal role of endogenous toll-like receptor 4 ligands in liver ischemia and reperfusion injury. Transplantation. 2008;85:1016–1022. doi: 10.1097/TP.0b013e3181684248. [DOI] [PubMed] [Google Scholar]
- 14.Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, Liao X, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through toll-like receptor 9. Hepatology. 2011 doi: 10.1002/hep.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhupar R, Klune JR, Evankovich J, Cardinal J, Zhang M, Ross M, Murase N, et al. Interferon regulatory factor 1 mediates acetylation and release of high mobility group box 1 from hepatocytes during murine liver ischemia-reperfusion injury. Shock. 2011;35:293–301. doi: 10.1097/SHK.0b013e3181f6aab0. [DOI] [PubMed] [Google Scholar]
- 16.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 17.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Javaherian K, Liu JF, Wang JC. Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science. 1978;199:1345–1346. doi: 10.1126/science.628842. [DOI] [PubMed] [Google Scholar]
- 19.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellett JD, Atkinson C, Evans ZP, Amani Z, Balish E, Schmidt MG, van Rooijen N, et al. Murine Kupffer cells are protective in total hepatic ischemia/reperfusion injury with bowel congestion through IL-10. J Immunol. 2010;184:5849–5858. doi: 10.4049/jimmunol.0902024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devey L, Ferenbach D, Mohr E, Sangster K, Bellamy CO, Hughes J, Wigmore SJ. Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Molecular therapy : the journal of the American Society of Gene Therapy. 2009;17:65–72. doi: 10.1038/mt.2008.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bamboat ZM, Ocuin LM, Balachandran VP, Obaid H, Plitas G, DeMatteo RP. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest. 2010;120:559–569. doi: 10.1172/JCI40008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi M, Takeyoshi I, Yoshinari D, Matsumoto K, Morishita Y. P38 mitogen-activated protein kinase inhibition attenuates ischemia-reperfusion injury of the rat liver. Surgery. 2002;131:344–349. doi: 10.1067/msy.2002.121097. [DOI] [PubMed] [Google Scholar]
- 24.Onishi I, Shimizu K, Tani T, Hashimoto T, Miwa K. JNK activation and apoptosis during ischemia-reperfusion. Transplant Proc. 1999;31:1077–1079. doi: 10.1016/s0041-1345(98)01912-5. [DOI] [PubMed] [Google Scholar]
- 25.Bradham CA, Stachlewitz RF, Gao W, Qian T, Jayadev S, Jenkins G, Hannun Y, et al. Reperfusion after liver transplantation in rats differentially activates the mitogen-activated protein kinases. Hepatology. 1997;25:1128–1135. doi: 10.1002/hep.510250514. [DOI] [PubMed] [Google Scholar]
- 26.McCloskey CA, Kameneva MV, Uryash A, Gallo DJ, Billiar TR. Tissue Hypoxia Activates Jnk in the Liver during Hemorrhagic Shock. Shock. 2004;22:380–386. doi: 10.1097/01.shk.0000140660.78744.bf. [DOI] [PubMed] [Google Scholar]
- 27.Shiojiri N. Enzymo- and immunocytochemical analyses of the differentiation of liver cells in the prenatal mouse. J Embryol Exp Morphol. 1981;62:139–152. [PubMed] [Google Scholar]
- 28.Malato Y, Naqvi S, Schurmann N, Ng R, Wang B, Zape J, Kay MA, et al. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J Clin Invest. 2011;121:4850–4860. doi: 10.1172/JCI59261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benhamouche S, Curto M, Saotome I, Gladden AB, Liu CH, Giovannini M, McClatchey AI. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes & development. 2010;24:1718–1730. doi: 10.1101/gad.1938710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 31.Pinkert CA, Ornitz DM, Brinster RL, Palmiter RD. An albumin enhancer located 10 kb upstream functions along with its promoter to direct efficient, liver-specific expression in transgenic mice. Genes & development. 1987;1:268–276. doi: 10.1101/gad.1.3.268. [DOI] [PubMed] [Google Scholar]
- 32.Loi P, Paulart F, Pajak B, Nagy N, Salmon I, Moser M, Goldman M, et al. The fate of dendritic cells in a mouse model of liver ischemia/reperfusion injury. Transplant Proc. 2004;36:1275–1279. doi: 10.1016/j.transproceed.2004.05.052. [DOI] [PubMed] [Google Scholar]
- 33.Ellett JD, Atkinson C, Evans ZP, Amani Z, Balish E, Schmidt MG, Schnellmann RG, et al. Toll-like receptor 4 knockout mice are protected from endothelial overactivation in the absence of Kupffer cells after total hepatic ischemia/reperfusion. Liver transplantation : official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2011;17:1089–1098. doi: 10.1002/lt.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 35.Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. The Journal of biological chemistry. 2010;285:39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. The EMBO journal. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Zhang W, Zhang Y, Chen Y, Zou B, Jiang B, Pang R, et al. c-Jun N-terminal kinase (JNK1) upregulates XIAP-associated factor 1 (XAF1) through interferon regulatory factor 1 (IRF-1) in gastrointestinal cancer. Carcinogenesis. 2009;30:222–229. doi: 10.1093/carcin/bgn271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Illustration describing generation of TLR4loxP construct used to create transgenic mice. Figure is not to scale.
(A) Hepatocyte TLR4 activation by LPS was determined by quantifying p65 (NF-κβ) nuclear translocation using Array scan after 60 minutes of LPS stimulation. As can be seem, hepatocytes from both Alb-TLR4-/- mice and global TLR4-/- mice lack the normal response to LPS stimulation. *P<0.05 when compared against WT. Additionally, NPCs were isolated from WT, CD11c-TLR4-/-, and TLR4-/- mice and macrophages were purified using positive selection with F4/80 beads. These cells were then exposed to LPS for 6hrs and ELISA used to quantify (B) TNF-α and (C) IL-6 production within the supernatant. Each data point is mean of ࣙ3 independent experiments.
Serum ALT levels were analyzed after mice had intraperitoneal administration of either vehicle (“Neg Control”) or JNK inhibitor prior to 1h of ischemia and 3h of reperfusion. Data represents mean ± SEM; n≥6 mice per group.