

Abstract
Heart failure, a syndrome culminating the pathogenesis of many forms of heart disease, is highly prevalent and projected to be increasingly so for years to come. Major efforts are directed at identifying means of preventing, slowing, or possibly reversing the unremitting progression of pathological stress leading to myocardial injury and ultimately heart failure. Indeed, despite widespread use of evidence-based therapies, heart failure morbidity and mortality remain high. Recent work has uncovered a fundamental role of reversible protein acetylation in the regulation of many biological processes, including pathological remodeling of the heart. This reversible acetylation is governed by enzymes that attach (histone acetyltransferases, HAT) or remove (histone deacetylases, HDACs) acetyl groups. In the case of the latter, small molecule inhibitors of HDACs are currently being tested for a variety of oncological indications. Now, evidence has revealed that HDAC inhibitors blunt pathological cardiac remodeling in the settings of pressure overload and ischemia/reperfusion, diminishing the emergence of heart failure. Mechanistically, HDAC inhibitors reduce stress-induced cardiomyocyte death, hypertrophy, and ventricular fibrosis. Looking to the future, HDAC inhibitor therapy may emerge as a novel means of arresting the untoward consequences of pathological cardiac stress, conferring clinical benefit to the millions of patients with heart failure.
Keywords: heart failure, hypertrophy, remodeling, histone deacetylases
Introduction
Presently, five million Americans suffer from chronic heart failure, the final common pathway of many forms of heart disease and the most common discharge diagnosis in Medicare for several years running (Heidenreich et al., 2011). This syndrome carries a mortality approaching 50% at 5 years, and its incidence and prevalence are expanding rapidly around the globe. It is predicted that as our population ages, the direct medical costs of treatment of all cardiovascular diseases (including hypertension, coronary heart disease, stroke, and heart failure) will triple from $272 billion in 2010 to $818 billion in 2030 (Heidenreich et al., 2011). Although substantial efficacy of evidence-based therapies, including angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARB), aldosterone antagonists, and β-adrenergic receptors blockers (β-blockers) has been documented, heart failure morbidity and mortality remain unacceptably high and the prevalence of the syndrome continues to expand globally (Goldberg, 2010). To stem this enormous burden to individuals and society, comprehensive understanding of the biological processes participating in pathological ventricular remodeling is required.
Pathological ventricular remodeling leading to heart failure is the culmination of a complex series of transcriptional, signaling, structural, electrophysiological, and functional events occurring within the cardiac myocyte (Kehat and Molkentin, 2010). In addition, other cellular elements within the ventricle, including fibroblasts, the coronary vasculature and infiltrating inflammatory cells are involved in this preventable and reversible process.
Recent work has uncovered the fundamental role of reversible protein acetylation in the control of many biological processes, including pathological remodeling of the heart. This reversible acetylation, analogous to reversible phosphorylation, is governed by enzymes that covalently attach (histone acetyltransferases, HAT) or remove (histone deacetylases, HDACs) acetyl groups. Small molecule HDAC inhibitors have been developed and are currently being tested for a variety of oncological indications. Recent evidence has revealed that HDAC inhibitors are remarkably capable of blunting pathological cardiac remodeling in the settings of pressure overload and ischemia/reperfusion, diminishing the emergence of heart failure. Given this, HDAC inhibitor therapy may emerge as a novel means of arresting the untoward consequences of pathological cardiac stress, leading to clinical benefit to the millions of patients with heart failure.
HDACs and their functions in the heart
Many proteins are governed by reversible acetylation of ε-amino groups on lysine residues. Appreciated first in the case of histone proteins, where acetylation leads to chromatin relaxation and access of transcriptional effectors to bind DNA (Yang and Seto, 2008), this widespread post-translational modification takes place on numerous proteins beyond just histones. Thus, HDACs are increasingly, and more precisely, being termed lysine (K) DeACetylases (KDACs).
In mammalian cells, 18 HDACs have been described, grouped into four classes: Class I [HDACs 1, 2, 3 and 8], Class IIa [HDACs 4, 5, 7 and 9], Class IIb [HDACs 6, 10], Class III [also known as sirtuins, SirT1–7] and Class IV [HDAC11] (Gregoretti et al., 2004). The roles of several of these HDACs have been studied in the heart during the last decade. Gene deletion and over-expression studies have revealed important functions of these enzymes in the pathological processes of left ventricular remodeling, including hypertrophy, apoptosis, necrosis, metabolism, contractility, and fibrosis.
Class I HDACs in the heart
The class I HDACs, HDAC1 and HDAC2, are expressed ubiquitously, and systemic deletion of either results in embryonic lethality (Montgomery et al., 2007). HDAC1 and HDAC2 are functionally redundant in heart, as cardiomyocyte-specific inactivation of either one individually does not impair the hypertrophic phenotype elicited by isoproterenol or transverse aortic constriction (TAC) (Montgomery et al., 2007). However, homozygous inactivation of both genes together results in heart failure, arrhythmia, and neonatal lethality (Montgomery et al., 2007). Cardiomyocyte-specific over-expression of HDAC2 provokes severe cardiac hypertrophy (Trivedi et al., 2008). HDAC3 over-expression in the heart results in cardiomyocyte hyperplasia (Trivedi et al., 2008). Systemic HDAC8 deletion results in neonatal lethality due to skull instability; cardiomyocyte-specific knockout has not been studied (Haberland et al., 2009). Based on these data, class I HDACs are generally considered detrimental to cardiac function.
Class II HDACs in the heart
Among the two subclasses of class II HDACs, class IIa HDACs have been studied extensively. Class IIa HDACs bind MEF2 (myocyte enhancer factor 2) transcription factors and suppress hypertrophic growth (Han et al., 2003; McKinsey et al., 2002). MEF2 is a transcription factor that responds to pathologic cardiac stress to mediate hypertrophic responses (Han et al., 2003). MEF2's interaction with all four class IIa HDACs results in suppression of downstream targets and blunted hypertrophic remodeling (Han et al., 2005; Han et al., 2003). Over-expression of HDAC4 (Backs et al., 2006), HDAC5 (Bush et al., 2004; Vega et al., 2004; Zhang et al., 2002) or HDAC9 (Zhang et al., 2002) in cardiomyocytes suppresses MEF2 expression and pathological stimuli-induced hypertrophy. Silencing of HDAC9 (Zhang et al., 2002) or HDAC5 (Chang et al., 2004) results in an exaggerated hypertrophic response to pressure overload and spontaneous hypertrophy at old age. Thus, class IIa HDACs are generally considered cardioprotective. However, the functions of class IIb HDACs in the heart are largely unknown.
Class III HDACs in the heart
In contrast to the zinc-dependent class I and class II HDACs, class III HDACs (sirtuins) require nicotinamide adenine dinucleotide (NAD+) in order to function. Class III HDACs are insensitive to the most commonly used HDAC inhibitors, which function largely as zinc chelators (McKinsey, 2012). In mice over-expressing SIRT1 in the heart, oxidative stress-induced apoptosis is suppressed (Alcendor et al., 2007). Inhibition of SIRT1 activity by a small molecule inhibitor results in enhanced apoptosis (Alcendor et al., 2004), and cardiomyocyte-specific SIRT7 loss of function results in severe cardiomyocyte apoptosis and cardiac hypertrophy with fibrosis (Vakhrusheva et al., 2008). SIRT7 suppresses apoptosis by deacetylating p53 to inhibit its activity (Vakhrusheva et al., 2008). SIRT3 protects cardiomyocytes by deacetylating Ku70, which subsequently interacts with Bax to suppress apoptosis (Sundaresan et al., 2008). Based on these data, class III HDACs are generally considered cardioprotective.
HDAC inhibition in cardiovascular pathology
Small molecule inhibitors of HDACs are divided into four structurally distinct groups: hydroxamic acids (e.g. Trichostatin A [TSA], suberoylanilide hydroxamic acid [SAHA]); short chain fatty acids (e.g. phenylbutyrate, valproic acid); benzamides (e.g. MS-275); and cyclic peptides (e.g. depsipeptides) (Bradner et al., 2010). Two are FDA approved for treatment of cutaneous T cell lymphoma. In tumor cells, HDAC inhibitors induce cell lysis by triggering apoptosis; they also induce autophagy, a response which may be a secondary, prosurvival reaction (Rikiishi, 2011).
Inhibitors of class I and class II HDACs harbor a stereotypical three-part structure, comprising a zinc-binding “warhead” group that docks in the catalytically active site, a linker, and a surface recognition domain that interacts with residues near the active site (Bradner et al., 2010). Class I HDAC activity is more sensitive to zinc concentration than that of class II HDACs. Nevertheless, the strong zinc-chelating properties of the hydroxamic acid warhead confers potent (nM - M) class I and class II HDAC inhibitory properties (Bradner et al., 2010). By contrast, the short chain fatty acid HDAC inhibitors are weak (mM) zinc chelators, with modest selectivity towards class I HDACs when used at low concentrations (Bradner et al., 2010). Benzamide HDAC inhibitors are generally highly selective for HDACs 1, 2, and 3, as are the cyclic peptide HDAC inhibitors (Bradner et al., 2010). Of note, most preclinical data obtained in animal models of heart disease employed a potent class I–II HDAC inhibitor, TSA.
HDAC inhibition in pressure-overload cardiac hypertrophy
As class I and IIa HDACs have been implicated in the pathogenesis of cardiac hypertrophy, studies to test small molecule HDAC inhibitors have emerged (Figure 1). In cardiomyocytes cultured in vitro, the HDAC inhibitors TSA and sodium butyrate block phenylephrine-induced myocyte hypertrophy (Antos et al., 2003). As the deacetylase catalytic activity of class IIa HDACs is modest (Bradner et al., 2010), it is thought that the inhibition of class I HDAC activity by hydroxamic acid-based compounds is dominant (Zhang et al., 2002). Both TSA and valproic acid abolished the development of cardiac hypertrophy in transgenic mice that over-express an HDAC2-dependent SRF inhibitor, Hop (Kook et al., 2003). Similarly, TSA blunts isoproterenol-induced (Kook et al., 2003), angiotensin II-induced (Kee et al., 2006), and TAC-induced (Kee et al., 2006) (Kong et al., 2006) ventricular hypertrophy. TSA treatment was also capable of regressing established TAC-induced cardiac hypertrophy (Kee et al., 2006). TSA and scriptaid, another hydroxamic acid-based HDAC inhibitor, blunted cardiac hypertrophy in a pressure-overload TAC model; cardiomyocyte cross-sectional area was reduced, accompanied by significant improvements in systolic function (Kong et al., 2006). Long-term (9 week) treatment with TSA was well tolerated, and improvements in systolic function were enduring (Kong et al., 2006). Together, these results have opened the possibility of HDAC inhibition as a viable therapeutic intervention in hypertensive patients at risk of developing left ventricular hypertrophy or even with established hypertrophy.
Figure 1. HDACs in cardiac hypertrophy.
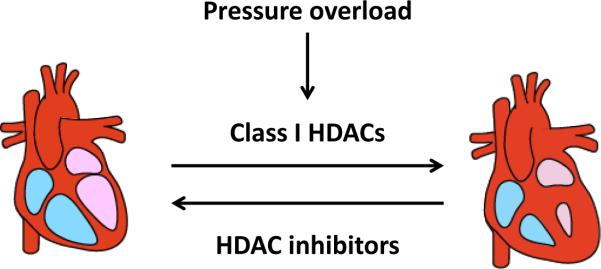
Class I HDACs participate in pressure overload-induced pathological cardiac hypertrophy. HDAC inhibition can prevent load-induced hypertrophy and reverse pre-existing hypertrophy.
HDAC inhibitors increase the expression of the anti-hypertrophic transcription factor Krüppel-like factor 4 (KLF4) (Kee and Kook, 2009; Liao et al., 2010), at least in part by promoting acetylation of histones near its gene regulatory elements (Kee and Kook, 2009). Genetic silencing of KLF4 in mice resulted in exaggerated cardiac hypertrophy and fibrosis in response to TAC (Liao et al., 2010).
We have tested whether the pathway of protein and organelle degradation, autophagy, is involved in the anti-hypertrophic actions of HDAC inhibition. Autophagy is an intracellular catabolic pathway evolutionarily conserved from yeast to human. By degradation of cellular contents, autophagy replenishes depleted stores of building blocks required for cell growth and energy for cell survival (Mizushima, 2007). Autophagic activity increases during hypertrophic growth in preclinical models and in failing human hearts (Zhu et al., 2007). Left ventricular assist device-based mechanical unloading reduces up-regulated autophagy in heart failure (Xie et al., 2011). However, when autophagy is eliminated completely, cell death occurs (Nakai et al., 2007). Thus, current thinking holds that maintenance of autophagic flux, not too much and not too little, is required for cardiomyocyte homeostasis (Xie et al., 2011).
In cultured neonatal rat ventricular cardiomyocytes (NRVMs), TSA attenuates phenylephrine (PE)-induced hypertrophic growth and abolishes the associated activation of autophagy (Cao et al., 2011). RNAi-mediated knockdown of either Atg5 or Beclin 1, two essential autophagy effectors, was similarly capable of suppressing ligand-induced autophagy and myocyte growth. RNAi experiments uncovered the class I isoforms, HDAC1 and HDAC2, as required for the autophagic response. In cardiomyocyte-specific Beclin 1 over-expressing mice, the autophagic response elicited by TAC is increased four-fold; yet TSA abolished TAC-induced increases in autophagy and blunted TAC-induced hypertrophy (Cao et al., 2011). These studies also confirmed that TSA is capable of reversing preexisting hypertrophy, even in cardiomyocyte-specific Beclin 1 over-expressing mice with augmented autophagy (Cao et al., 2011).
In light of our findings of HDAC-dependent regulation of autophagy and its participation in cardiac plasticity, we believe that autophagy is a plausible therapeutic target in heart disease. Looking to the future, additional studies are required to determine which HDAC isoforms are responsible for the anti-hypertrophic effects of HDAC inhibition (McKinsey, 2012). A combination of isoform-specific knockout mouse lines and small molecules specific for individual HDAC isoforms will be required (Bradner et al., 2010). Meanwhile, a number of other therapeutic agents specifically targeting autophagy are being developed (Rubinsztein et al., 2012).
HDAC inhibition in ischemic heart disease
Given the robust beneficial effects of HDAC inhibition in models of pressure overload, HDAC inhibitors have been tested in another prevalent form of pathological stress, cardiac ischemia (Figure 2). The first report was in a rat infarct model, where a coronary artery is surgically occluded permanently. The HDAC inhibitors valproic acid (VPA) and tributyrin significantly reduced cardiomyocyte hypertrophy and collagen deposition in both the remote and border zones of the infarcted left ventricle (Lee et al., 2007). Systolic function was also preserved, and these protective effects were abolished by theophylline, an HDAC activator (Lee et al., 2007).
Figure 2. HDACs in ischemic heart disease.

Both ischemia and ischemia/reperfusion trigger cell death via mechanisms that involve class I HDACs. Inhibition of HDAC activity reduces infarct size. Post-infarct stresses induce cardiomyocyte hypertrophy, cell death (e.g. apoptosis) and fibrosis. Again, HDAC inhibitors are capable of blocking elements of these detrimental biological processes and preserve cardiac function.
In most clinically relevant instances of tissue ischemia, reperfusion occurs – either spontaneously or therapeutically – leading to restoration of oxygen and nutrients, and triggering a second wave of cell death termed reperfusion injury. In light of this, two studies tested HDAC inhibitors in a murine ischemia/reperfusion (I/R) model. Using an ex vivo Langendorff isolated mouse heart system, both perfusion of the explanted heart with TSA or intraperitoneal injection of TSA before surgery reduced infarct size and preserved cardiac systolic function (Zhao et al., 2007). These effects were tied to TSA-induced p38 activation and acetylation of p38 at lysine residues (Zhao et al., 2007). Another study employed a mouse in vivo I/R model and reported that HDAC activity increased significantly after I/R (Granger et al., 2008). Administration of TSA or scriptaid by intraperitoneal injection reduced infarct size and preserved systolic function (Granger et al., 2008). Of note, administration of the HDAC inhibitor even one hour after the ischemic insult reduced infarct size to the same extent as drug pretreatment prior to the surgical injury (Granger et al., 2008). These findings suggest that HDAC inhibition blunts reperfusion injury specifically, raising the intriguing prospect that HDAC inhibitor therapy might reduce infarct size if delivered at the time of reperfusion, for example, when a patient presenting with a myocardial infarction undergoes percutaneous coronary intervention. This study went on to report that HDAC inhibition reduces hypoxia inducible factor-1 protein levels, diminishes cell death and vascular permeability and suggested that HDAC4 is a mediator of reperfusion injury (Granger et al., 2008).
In a murine model of myocardial infarction (permanent coronary artery occlusion), TSA stimulated c-kit+ cardiac stem cell (CSC) proliferation (Zhang et al., 2012). In c-kit-null mice, TSA's beneficial effects on left ventricular remodeling were abolished, and re-introduction of TSA-treated wild-type c-kit+ CSC into c-kit-null mice restored the beneficial effects of TSA on myocardial function and cardiac repair (Zhang et al., 2012). After TSA treatment, the abundance of c-kit+ CSC-derived myocytes was significantly increased (Zhang et al., 2012). This study raises the possibility that HDAC inhibition could be used initially as an infarct-size reducing strategy and subsequently as long-term treatment to blunt post-infarct remodeling by mobilizing cardiac stem cells.
HDAC inhibition in cardiac fibrosis
Ventricular fibrosis due to accumulation of collagen and other extracellular matrix elements is a cardinal feature of left ventricular hypertrophy and post-infarct remodeling. Other than aldosterone antagonists, there are no clinically available therapeutic agents that effectively reduce cardiac fibrosis (Dooley et al., 2011). Small molecule inhibitors of class I–II HDACs reduce fibrosis in models of both hypertrophic and ischemic heart disease (Kong et al., 2006). In cultured rat cardiac fibroblasts, TSA blocks transforming growth factor- (TGF-)-induced collagen synthesis (Kong et al., 2006). The relevant downstream targets seem to be ERK, AKT and PI3K, but not the transcription factor SMAD (Barter et al., 2010; Yoshikawa et al., 2010). SAHA reduced fibrosis in both heart and vessels in deoxycorticosterone acetate (DOCA)-salt hypertensive rats (Iyer et al., 2010).
Atrial fibrosis is a significant contributor to supraventricular arrhythmia, especially atrial fibrillation. In a model of Hop over-expression, TSA treatment reduced atrial fibrosis, normalized connexin 40 levels, and reduced vulnerability to atrial arrhythmia (Liu et al., 2008).
Potential clinical applications
Decades of research into the epigenetic regulation of cellular processes have culminated in the development and eventual clinical investigation of HDAC inhibitors for use in the field of cancer biology. Today, two HDAC inhibitors, Zolinza® (SAHA or vorinostat, structurally similar to TSA) and Istodax® (romidepsin, cyclic peptide) have received FDA approval, while clinical trials with several other compounds are underway in a number of tumor types (McKinsey, 2012). More recently, HDAC inhibition has demonstrated promising efficacy to block maladaptive left ventricular remodeling in the clinically prevalent scenarios of pressure overload and ischemia/reperfusion injury. Given the pressing clinical void of drugs to treat pressure overload and I/R-induced left ventricular remodeling, the need to test currently available FDA-approved HDAC inhibitors in humans is compelling.
In an effort to translate these basic discoveries into the clinical domain, we tested vorinostat in a large animal model of cardiac I/R injury. We observed that this FDA-approved HDAC inhibitor conferred robust cardioprotection from I/R injury; indeed, the extent of protection was similar to that observed in mouse models of I/R (unpublished observations). Specifically, vorinostat reduced infarct size by about 40% when the drug was administrated prior to I/R surgery. Plasma concentrations of vorinostat were comparable to those achieved in patients during phase I clinical trials. Importantly and more clinically relevant, the declines in infarct size were similar even when the drug was administered at the time of reperfusion (unpublished observations). In other words, the cardioprotective effects of HDAC inhibition – in both mouse and rabbit models of I/R injury – were identical, such that pharmacological treatment at the time of reperfusion conferred robust protection. Of course, in the clinical context, pharmacotherapy well into the ischemic phase of injury is the only possible option, and a therapeutic intervention efficacious at the reperfusion phase of the process is required. The cardioprotective effects of vorinostat derive, at least in part, from re-activation of I/R-dependent suppression of cardiomyocyte autophagic flux (unpublished observations).
Given the encouraging results in a large animal model of I/R injury, which phenocopied precisely those reported by us and others in mice, we are moving ahead to test the safety profile of vorinostat in patients with stable coronary artery disease. In this safety study, we will monitor for symptoms, ECG changes including QT prolongation, changes in blood pressure, serum electrolytes, renal and liver function, and blood cell counts. If this compound is found to be safe in patients with structural heart disease, the prospect of moving these exciting preclinical data to a first-in-human clinical trial is exciting. This would involve a double-blinded placebo-controlled pilot efficacy study. We would consider enrolling patients presenting with ST-segment elevation myocardial infarction who are hemodynamically stable. Patients would be randomized into treatment versus placebo arms, with therapy delivered at the time of percutaneous coronary intervention on the infarct-related artery. The primary endpoint will be infarct size measured by cardiac MRI both in the immediate post-infarct phase and at 3 months, a protocol for which there is precedent (Piot et al., 2008). If the results are positive, they would require additional investigation in a large scale, multicenter clinical trial.
Several hurdles need to be overcome before these novel agents can be used in humans. Efficacy in large animal models must be tested, as we have done. This is particularly important, given the large number of failed clinical trials targeting reperfusion injury based on mouse models alone (Schwartz Longacre et al., 2011). Second, safety of FDA-approved HDAC inhibitors in patients with structural heart disease must be evaluated. Early clinical trials using the cyclic peptide HDAC inhibitor, depsipetitde, reported QT prolongation (Piekarz et al., 2006). However, subsequent clinical trials of hydroxamic acid-based HDAC inhibitors did not reveal significant effects on QT duration (Munster et al., 2009). In the case of treating ventricular hypertrophy, where longer term therapy with HDAC inhibitors would be required, the side-effects of leukopenia, anemia, bone marrow suppression become relevant, even though doses lower than those used in cancer would be tested. Importantly, side effects on the hematopoietic system vary among class I-specific HDAC inhibitors. Finally, more detailed mechanistic studies are needed. Presently, our understanding of the HDAC isoform(s) involved is emerging, but little is known of the molecular substrates of those enzymes and the relevant protein(s) which become hyperacetylated in the setting of HDAC inhibition (Figure 3) (Cao et al., 2011). Together, elucidation of these critical knowledge gaps will do much to propel this field toward clinical application.
Figure 3. Unanswered questions regarding the actions of HDACs in the pathogenesis of heart failure.

Strong evidence implicates class I HDACs in the pathogenesis of heart failure. However, data defining the roles of specific HDAC isoforms is just beginning to emerge. Beyond that, little is known regarding the protein target(s) which is/are reversibly acetylated in these processes. Whether those proteins govern transcriptional, signaling, or other events, is similarly unknown.
Acknowledgements
We thank members of the Hill lab for helpful discussions and critique.
Source of Funding
This work was supported by grants from the NIH (HL-075173; HL-080144; HL-090842), AHA (0640084N), and the AHA-Jon Holden DeHaan Foundation (0970518N).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest Disclosures
None
References
- Alcendor RR, Gao S, Zhai P, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circulation research. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circulation research. 2004;95:971–980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- Antos CL, McKinsey TA, Dreitz M, et al. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. The Journal of biological chemistry. 2003;278:28930–28937. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. The Journal of clinical investigation. 2006;116:1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter MJ, Pybus L, Litherland GJ, et al. HDAC-mediated control of ERK- and PI3K-dependent TGF-beta-induced extracellular matrix-regulating genes. Matrix Biol. 2010;29:602–612. doi: 10.1016/j.matbio.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Bradner JE, West N, Grachan ML, et al. Chemical phylogenetics of histone deacetylases. Nature chemical biology. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush E, Fielitz J, Melvin L, et al. A small molecular activator of cardiac hypertrophy uncovered in a chemical screen for modifiers of the calcineurin signaling pathway. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2870–2875. doi: 10.1073/pnas.0308723101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao DJ, Wang ZV, Battiprolu PK, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Molecular and cellular biology. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley R, Harvey BJ, Thomas W. The regulation of cell growth and survival by aldosterone. Frontiers in bioscience : a journal and virtual library. 2011;16:440–457. doi: 10.2741/3697. [DOI] [PubMed] [Google Scholar]
- Goldberg LR. Heart failure. Ann Intern Med. 2010;152:ITC61–15. doi: 10.7326/0003-4819-152-11-201006010-01006. quiz ITC616. [DOI] [PubMed] [Google Scholar]
- Granger A, Abdullah I, Huebner F, et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008;22:3549–3560. doi: 10.1096/fj.08-108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. Journal of molecular biology. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Haberland M, Mokalled MH, Montgomery RL, Olson EN. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes & development. 2009;23:1625–1630. doi: 10.1101/gad.1809209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han A, He J, Wu Y, Liu JO, Chen L. Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor-2. Journal of molecular biology. 2005;345:91–102. doi: 10.1016/j.jmb.2004.10.033. [DOI] [PubMed] [Google Scholar]
- Han A, Pan F, Stroud JC, Youn HD, Liu JO, Chen L. Sequence-specific recruitment of transcriptional co-repressor Cabin1 by myocyte enhancer factor-2. Nature. 2003;422:730–734. doi: 10.1038/nature01555. [DOI] [PubMed] [Google Scholar]
- Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933–944. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- Iyer A, Fenning A, Lim J, et al. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats. Br J Pharmacol. 2010;159:1408–1417. doi: 10.1111/j.1476-5381.2010.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee HJ, Kook H. Kruppel-like factor 4 mediates histone deacetylase inhibitor-induced prevention of cardiac hypertrophy. J Mol Cell Cardiol. 2009;47:770–780. doi: 10.1016/j.yjmcc.2009.08.022. [DOI] [PubMed] [Google Scholar]
- Kee HJ, Sohn IS, Nam KI, et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation. 2006;113:51–59. doi: 10.1161/CIRCULATIONAHA.105.559724. [DOI] [PubMed] [Google Scholar]
- Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation. 2010;122:2727–2735. doi: 10.1161/CIRCULATIONAHA.110.942268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Tannous P, Lu G, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation. 2006;113:2579–2588. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kook H, Lepore JJ, Gitler AD, et al. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. The Journal of clinical investigation. 2003;112:863–871. doi: 10.1172/JCI19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TM, Lin MS, Chang NC. Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am J Physiol Heart Circ Physiol. 2007;293:H968–977. doi: 10.1152/ajpheart.00891.2006. [DOI] [PubMed] [Google Scholar]
- Liao X, Haldar SM, Lu Y, et al. Kruppel-like factor 4 regulates pressure-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:334–338. doi: 10.1016/j.yjmcc.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Levin MD, Petrenko NB, et al. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J Mol Cell Cardiol. 2008;45:715–723. doi: 10.1016/j.yjmcc.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annual review of pharmacology and toxicology. 2012;52:303–319. doi: 10.1146/annurev-pharmtox-010611-134712. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends in biochemical sciences. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes & development. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Montgomery RL, Davis CA, Potthoff MJ, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes & development. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster PN, Rubin EH, Van Belle S, et al. A single supratherapeutic dose of vorinostat does not prolong the QTc interval in patients with advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:7077–7084. doi: 10.1158/1078-0432.CCR-09-1214. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nature medicine. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Piekarz RL, Frye AR, Wright JJ, et al. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:3762–3773. doi: 10.1158/1078-0432.CCR-05-2095. [DOI] [PubMed] [Google Scholar]
- Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. The New England journal of medicine. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- Rikiishi H. Autophagic and apoptotic effects of HDAC inhibitors on cancer cells. Journal of biomedicine & biotechnology. 2011;2011:830260. doi: 10.1155/2011/830260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature reviews. Drug discovery. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz Longacre L, Kloner RA, Arai AE, et al. New horizons in cardioprotection: recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation. 2011;124:1172–1179. doi: 10.1161/CIRCULATIONAHA.111.032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Molecular and cellular biology. 2008;28:6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi CM, Lu MM, Wang Q, Epstein JA. Transgenic overexpression of Hdac3 in the heart produces increased postnatal cardiac myocyte proliferation but does not induce hypertrophy. The Journal of biological chemistry. 2008;283:26484–26489. doi: 10.1074/jbc.M803686200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakhrusheva O, Smolka C, Gajawada P, et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circulation research. 2008;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- Vega RB, Harrison BC, Meadows E, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Molecular and cellular biology. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Morales CR, Lavandero S, Hill JA. Tuning flux: autophagy as a target of heart disease therapy. Current opinion in cardiology. 2011;26:216–222. doi: 10.1097/HCO.0b013e328345980a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Molecular cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa M, Hishikawa K, Idei M, Fujita T. Trichostatin a prevents TGF-beta1-induced apoptosis by inhibiting ERK activation in human renal tubular epithelial cells. Eur J Pharmacol. 2010;642:28–36. doi: 10.1016/j.ejphar.2010.05.055. [DOI] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chen B, Zhao Y, et al. Inhibition of Histone Deacetylase-induced Myocardial Repair Is Mediated by c-kit in Infarcted Hearts. The Journal of biological chemistry. 2012;287:39338–39348. doi: 10.1074/jbc.M112.379115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res. 2007;76:473–481. doi: 10.1016/j.cardiores.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Zhu H, Tannous P, Johnstone JL, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. The Journal of clinical investigation. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]