

Abstract
Although complement is a known contributor to biomaterial-induced complications, pathological implications and therapeutic options remain to be explored. Here we investigated the involvement of complement in the inflammatory response to polypropylene meshes commonly used for hernia repair. In vitro assays revealed deposition of complement activation fragments on the mesh after incubation in plasma. Moreover, significant mesh-induced complement and granulocyte activation was observed in plasma and leukocyte preparations, respectively. Pretreatment of plasma with the complement inhibitor compstatin reduced opsonization >2-fold, and compstatin and a C5a receptor antagonist (C5aRa) impaired granulocyte activation by 50 and 67%, respectively. We established a clinically relevant mouse model of implantation and could confirm deposition of C3 activation fragments on mesh implants in vivo using immunofluorescence. In meshes extracted after subcutaneous or peritoneal implantation, the amount of immune cell infiltrate in mice deficient in key complement components (C3, C5aR), or treated with C5aRa, was approximately half of that observed in wild-type littermates or mice treated with inactive C5aRa, respectively. Our data suggest that implantation of a widely used surgical mesh triggers the formation of an inflammatory cell microenvironment at the implant site through complement activation, and indicates a path for the therapeutic modulation of implant-related complications.—Kourtzelis, I., Rafail, S., DeAngelis, R. A., Foukas, P. G., Ricklin, D., Lambris, J. D. Inhibition of biomaterial-induced complement activation attenuates the inflammatory host response to implantation.
Keywords: C5a anaphylatoxin, immune cells, compstatin, C3, polypropylene
The clinical use of biomaterials has profoundly shaped modern medicine and greatly facilitated applications ranging from drug delivery to tissue repair and implant surgery (1–3). Yet biomaterials, either embedded in tissue or circulating in blood, usually stay in contact with body fluids and are thus prone to defensive responses. Our immune system is formidably trained to recognize and combat nonself intruders, and the complement system plays a crucial role in the initial recognition of foreign material, as well as in orchestrating subsequent immune and inflammatory responses (2, 4). In view of this interplay between foreign surfaces, complement, and inflammation, it is evident that complement can act as a key contributor to adverse effects that have been observed for many biomaterials (4–6). The complement response may be triggered by pattern recognition molecules or by the direct binding of complement components to nonself surfaces. Recruitment and activation of immune cells, proinflammatory signaling by anaphylatoxins and crosstalk with other systems are considered essential for immune surveillance by complement (6). In the case of biomaterials, a direct and continuous activation of the alternative pathway is believed to be the driving force behind the observed complement-related effects (5, 7), yet contributions of the classical pathway (8, 9) and the lectin pathway (10) have also been reported. Given their structural and physicochemical diversity (11), each biomaterial may initiate distinct activators and effectors that define its overall biocompatibility. Although complement is considered a key contributor to biomaterial-induced adverse effects, other systems, such as immune cells, the cytokine network, and the contact/coagulation cascades, also contribute to the outcome, both by direct activation after contact with biomaterial surfaces (5, 7) and via extensive crosstalk between the pathways (6, 12, 13). For example, we have previously demonstrated that modern hemodialysis filters still induce strong complement activation, which not only increases inflammatory markers but may also foster thrombotic complications through crosstalk with the coagulation system (14). The same study also indicated that complement-targeted therapeutics are capable of impairing such adverse effects and bear the potential for clinical use (14, 15). In view of the profound knowledge about complement-mediated reactions to biomaterials in model systems, blood, and extracorporeal settings, it is surprising that very little is known about the effect of surgical implants on the complement system. Here we extend previous biomaterial-related studies to investigate the involvement of complement in the inflammatory response to surgical implants and explore options for therapeutic modulation of complement-mediated inflammation.
Nonabsorbable mesh implants for hernia repair are among the most frequently implanted biomaterials worldwide, and chronic inflammation has been observed with surgical meshes (16, 17). In addition to covering materials with hydrophilic coatings (to prevent protein binding), the administration of angiogenic and/or anti-inflammatory drugs is currently considered a viable therapeutic option for implant-related complications. However, long-term administration of anti-inflammatory drugs such as steroids causes severe adverse effects, and a more effective treatment to prevent the initial triggers of inflammation is highly desired (18). Whereas complement may present a suitable target for therapeutic intervention, specific information about the effect of complement on the initiation of and mediation between implant-induced inflammation is still scarce. In addition, there are only a few relevant in vivo models available to assess the underlying processes and evaluate potential therapeutic candidates. In this report, we address those issues by applying relevant mouse models of subcutaneous and deep-tissue biomaterial implantation, investigating the effect of complement activation by a clinically used surgical mesh in vitro and in vivo, and evaluating the modulatory potential of two established complement inhibitors. Our findings reveal, for the first time, the complement-mediated inflammatory reactions on implantation of biomaterials used for hernia repair, and suggest that inhibiting the complement system represents a potentially novel treatment strategy for attenuating such adverse immune reactions in clinical situations.
MATERIALS AND METHODS
Materials
The polypropylene (PP) monofilament mesh material, Prolene (Ethicon, Inc., Somerville, NJ, USA), which is widely used in hernia repair, was utilized for these studies. Lepirudin-anticoagulated (50 μg/ml) blood samples were obtained from healthy donors (n=6), and informed consent was obtained from all persons involved. The research was performed in compliance with institutional guidelines and in accordance with the Declaration of Helsinki.
Surface deposition of complement activation fragments
Human plasma (100 μl) was incubated with a sterile piece of PP mesh (5×5 mm) in a well of a 96-well polystyrene plate that had been pretreated with 1% BSA blocking solution for 60 min. Afterward, 10 mM ethylenediaminetetraacetic acid (EDTA) was added to stop any further complement activation. The treated pieces of the mesh biomaterial were placed in clean BSA-treated wells and washed with PBS. Blocking solution was then added for 60 min and followed by incubation with anti-C3b/iC3b/C3c mAb C3-9 (2 μg/ml; ref. 19) for 60 min. Wells with the mesh were washed twice with PBS, and anti-mouse immunoglobulin G–horseradish peroxidase (IgG-HRP; 1:1000) was added. After 3 washes with PBS, sodium citrate/H2O2/2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonicacid) (ABTS) was added to the wells. The pieces of the mesh material were removed, and the optical density of the remaining solution was measured at 405 nm.
For complement inhibition studies, plasma samples were incubated with 20 μM of Cp40 (yI[CVW(Me)QDWSarAHRC]mI-NH2; ref. 20), an analog of the highly specific and potent complement inhibitor compstatin, or with a corresponding inactive, scrambled control peptide (yI[CSarVDWAHW(Me)QRC]mI-NH2)for 5 min prior to addition of the mesh.
Complement and granulocyte activation in whole blood
Lepirudin-anticoagulated blood was incubated with shredded mesh material (20 mg/ml) for 60 min at 37°C. For the complement inhibition studies, blood samples were preincubated with 20 μM of the compstatin analog Cp20 (AcI[CVW(Me)QDWSarAHRC]mI-NH2), linear inactive peptide (IAVVQDWGHHRAT-NH2; ref. 21, 22), 10 μM of the C5a receptor antagonist PMX53 (AcF-[OPdChaWR]; synthesized by GL Biochem, Shanghai, China; ref. 23), or its inactive control [AcdChaPWFRO-NH2; synthesized in house], for 5 min prior to biomaterial addition. Activation of complement and granulocytes was determined in plasma by enzyme-linked immunosorbent assay (ELISA) and in granulocytes by measuring the expression levels of surface cluster of differentiation (CD)11b by flow cytometry, as described previously (14). All inhibitors that were used for the purposes of this study were endotoxin free.
Mouse model of biomaterial implantation
This study employed 12- to 16-wk-old mice deficient in C3 (C3−/−) or C5a receptor (C5aR−/−), along with their wild-type (WT) littermates (C3+/+, C5aR+/+), all of which were on a C57BL/6 background and obtained from our colony, maintained by the Jackson Laboratory (Bar Harbor, ME, USA). C57BL/6J mice ordered from Jackson Laboratory production facilities were also used (for studies testing complement inhibitors). Animals were housed in an animal facility of the University of Pennsylvania (Philadelphia, PA, USA), within a barrier, on a 12-h light-dark cycle. Mouse colonies were tested for the most common rodent infections. Water and standard rodent diet were provided ad libitum. All mice were used with the approval of the University of Pennsylvania Institutional Animal Care and Use Committee and according to criteria outlined in the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals.
To examine the effect of complement on the host immune response after biomaterial implantation, we inserted biomaterial either subcutaneously or intraperitoneally. Sterile PP mesh normally used for hernia repair was used for implantation into mice. This biomaterial mesh provides a large implanted material surface area for tissue contact. The original mesh material was cut to the desired shape and size under sterile conditions. During the implantation procedure, sterile conditions and equipment were utilized.
For the subcutaneous implantation procedure, a modified protocol from Higgins et al. (24) was utilized. Briefly, the dorsal area was shaved and disinfected, and two incisions of 1 cm each at a distance of 2 cm apart were made in the skin, thereby creating 2 subcutaneous pockets. Sterile mesh pieces (5×5 mm) were inserted into both pockets, and incisions were then closed using surgical staples.
For the deep-tissue implantation studies, midventral laparotomy was performed, with an incision of ∼1–2 cm made in both the skin and muscle. A 5- × 7.5-mm piece of mesh was placed inside the abdomen beneath the incision site. A 5-0 silk suture was used to attach each corner of the mesh to the underside of the muscle. The suture was placed through the muscle, then the top right corner of the mesh, crossed over to the other side, inserted through the top left corner of the mesh and then the muscle, and tied to close the top part of the incision. This procedure was then repeated for the bottom of the incision. A 5-0 silk suture was also used to close the skin incision.
To exclude possible nonspecific results due to the implantation procedure, sham-treated control mice were used. Incisions were made and closed as described above, but no implant materials were inserted. Mice were observed until they awoke from anesthesia, then on a half-hourly basis for the first 3 h after implantation, and every 12 h thereafter. Inflammation at the implantation site, behavioral changes, and other adverse reactions to the implant were monitored for the duration of the experiment.
The implantation procedures were performed on both untreated complement-deficient mice and on C57BL/6J mice treated with either the C5aR antagonist PMX-53 or a scrambled control peptide. For these studies, injections were performed intraperitoneally at a dose of 2 mg/kg mouse body weight every 2 d, beginning 1 d before the initiation of the implantation procedure, as described previously (25, 26).
Implant retrieval and tissue analyses
Two weeks after subcutaneous implantation or 5 d after deep-tissue implantation, mice were euthanized, and the implant along with associated surrounding tissue was excised for cell population analysis. The morphology of the tissue surrounding the biomaterial was assessed in a blinded fashion by light microscopy (Nikon Eclipse E400; Nikon, Tokyo, Japan) of hematoxylin and eosin (H&E)-stained, 5-mm paraffin sections. Cell densities were determined by counting the number of cells surrounding each section of the implant. More specifically, cell nuclei were measured per high-power field (HPF; ×10/22 eyepiece, ×60 objective) in the stroma surrounding the empty spaces indicative of the implant area as well as at the interface (walls of empty spaces) using a straight eyepiece micrometer. Four to 5 stromal areas and interfaces were counted per sample.
Deposition of C3 activation fragments was examined in paraffin sections from tissue fixed in formalin-free immunohistochemistry (IHC) zinc fixative (BD Pharmingen, San Diego, CA, USA) using a polyclonal rabbit anti-C3c fluorescein isothiocyanate (FITC)-conjugated antibody (F0201; Dako, Carpinteria, CA, USA) for immunofluorescence (IF) staining. Fluorescence was evaluated by standard fluorescence microscopy (Nikon Eclipse E600).
Isolation of cells and immunophenotyping
Tissue adhering to the explanted biomaterial was incubated in RPMI 1640/collagenase type IV at 37°C for 1 h with occasional agitation and then removed from the mesh using razor blades. The tissue was passed through a 70-mm cell strainer (BD Falcon, Bedford, MA, USA) and the cells were washed with PBS. They were incubated for 15 min at 4°C with a mAb recognizing mouse CD16/CD32 (553142; BD Biosciences) to block Fcγ receptors (FcγRs), and then for 30 min at 4°C with a fluorochrome-conjugated mAb recognizing mouse CD45 (557659; BD Biosciences) as well as Viaprobe (555815; BD Biosciences). They were then similarly incubated with fluorochrome-conjugated mAbs recognizing Gr-1 and CD11b to identify granulocytes (Gr-1+/CD11b+) and macrophages (Gr-1−/CD11b+). Abs and Viaprobe were used according to the manufacturer's instructions. Stained cells were analyzed by flow cytometry on a FACS Canto II instrument (BD Biosciences) with FlowJo software (TreeStar, Ashland, OR, USA).
Statistical analysis
Data are presented as means ± se. Unpaired or paired t tests were used to assess statistical significance between 2 groups. For experiments with ≥3 groups, 1-way ANOVA was used with the Bonferroni posttest to confirm differences between individual groups. All statistical analyses were performed with GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA), and values of P ≤ 0.05 were considered significant.
RESULTS
Surgical hernia repair mesh activates complement and is marked by opsonins
The recognition and opsonization of biomaterials by complement marks a critical first step that shapes the subsequent immune response. Although PP is considered a comparatively weak initiator (27), several studies have found considerable complement activation of PP-based materials (e.g., hollow fibers in oxygenators) that even varied between different makes (28). We therefore carefully evaluated the possible opsonization of a clinically used PP mesh product (Prolene) with complement activation fragments by incubating the mesh in plasma. Indeed, activation fragments of the central complement component C3 (i.e., C3b, iC3b) were detected on the mesh surface (Fig. 1), thereby indicating that the mesh exerts relevant complement-inducing activity. The detected opsonin signal was clearly elevated when compared to background activation in control plasma caused by the polystyrene surface of the assay plate; inhibition with EDTA confirmed that the increased opsonization was complement dependent. Notably, the activation was completely suppressed when the plasma was preincubated with a compstatin analog, while an inactive control peptide had no effect (Fig. 1).
Figure 1.

Cleavage products of C3 deposit on PP mesh after plasma-biomaterial interaction. Lepirudin-anticoagulated plasma (100 μl) isolated from healthy donors was incubated with mesh (5×5 mm) for 60 min, and deposition of C3 cleavage products was measured by ELISA. Treatment of plasma with the compstatin analog Cp40 (20 μM) before mesh addition abolished complement deposition when compared to plasma samples treated with an inactive control peptide (20 μM; n=5). Incubation of plasma alone or incubation of plasma with mesh in the presence of EDTA (10 mM) served as negative control. *P < 0.05; 1-way ANOVA.
We validated and further examined the ability of PP to activate complement by utilizing an established and more clinically relevant human whole-blood model (14). The mesh was incubated with whole blood, and the amount of complement activation in plasma was examined. The presence of the mesh resulted in a profound increase in C3 cleavage products (as a measure of complement activation) in the plasma fraction when compared to the background activation by the sample containers (Fig. 2A). As observed in the plasma experiment above, this effect was abolished when the blood was pretreated with a compstatin analog, but the use of an inactive form of compstatin had no effect on the process (Fig. 2A).
Figure 2.
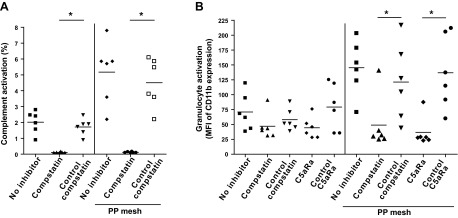
PP mesh induces complement and granulocyte activation upon contact with blood, which can be blocked by complement inhibitors. A) Amounts of C3 cleavage products in plasma isolated from blood incubated at 37°C for 60 min without treatment (no inhibitor) or with shredded 20 mg/ml PP mesh (no inhibitor–PP mesh), and blood pretreated with 20 μM of a compstatin analog (compstatin) or an inactive control peptide (control compstatin) in the presence or absence of shredded PP mesh, as determined by ELISA (n=6). Results are expressed as percentage activation seen in plasma treated with cobra venom factor, which results in 100% complement activation. B) Induction of CD11b expression on the surface of granulocytes obtained from lepirudin-anticoagulated blood incubated at 37°C for 60 min without treatment (no inhibitor) or treated with 20 mg/ml shredded PP mesh implant, and blood incubated with or without the mesh implant and pretreated with either 20 μM compstatin, 20 μM control compstatin, 10 μM C5aRa (C5aRa), or 10 μM C5aRa control peptide (control C5aRa). The results are presented as ratios of the mean fluorescence intensity (MFI) of granulocytes stained with CD11b mAb to cells stained with the isotype control, as determined by flow cytometry (n=6). Granulocytes were gated according to their forward scatter channel/side scatter channel characteristics. *P < 0.05, 1-way ANOVA.
Mesh-induced blood reaction activates granulocytes through C5aR signaling
In an attempt to assess the effects of the mesh-induced complement activation in blood cells, we examined the levels of expression of integrin αM (CD11b), an established marker of granulocyte activation. As complement-mediated attraction and priming of immune cells is primarily exerted by C5a, which is released from C5 during the complement activation process, we evaluated the effect of a C5aR antagonist (C5aRa) in addition to C3 inhibition by compstatin. Whole blood was incubated with PP mesh in the presence of inhibitors or corresponding control compounds, followed by evaluation of granulocyte activation by flow cytometry. Granulocytes from blood incubated with PP exhibited a strong up-regulation of CD11b when compared to untreated cells; this effect was potently abolished by pretreating the blood with either of the inhibitors (Fig. 2B). The attenuating effects of the C5aRa strongly suggest that the anaphylatoxin C5a is involved in the proinflammatory activation of granulocytes after exposure to the PP mesh. Treating blood cells with inactive compounds in the presence of the mesh resulted in stimulation levels comparable to those of untreated blood, confirming the specificity of these findings (Fig. 2B).
Biomaterial-induced inflammatory cell recruitment is complement dependent
Whereas implantation of biomaterial into tissues is expected to trigger responses similar to those observed in the ex vivo studies described above, the microenvironment and the timeframe are entirely different. To examine the role of C3 in the postimplantation immune reaction of the host, we utilized an animal model of biomaterial implantation, in which we subcutaneously implanted the PP mesh into the dorsal area of both C3+/+ and C3−/− mice. The animals were euthanized 2 wk after implantation, and the mesh and attached tissue were retrieved and analyzed morphologically and histologically. H&E staining of tissue sections revealed a significantly lower number of inflammatory cells in the stroma and at the mesh interface in C3−/− mice than in their C3+/+ littermates (Fig. 3), thereby indicating that the genetic absence of C3 attenuates immune cell recruitment to the mesh in this model.
Figure 3.

Reduced recruitment of inflammatory cells to subcutaneously implanted PP mesh in C3−/− mice. PP mesh (5×5 mm) was implanted subcutaneously in the dorsal area of C3−/− or C3+/+ mice, and the biomaterial was retrieved 2 wk after implantation. A) Representative H&E-stained samples of PP meshes retrieved from C3−/− or C3+/+ mice are depicted, with surrounding stroma (diamonds) showing leukocytic infiltration and the presence of multinucleated giant cells surrounding the empty spaces indicative of the implant area (interface). B, C) Assessment of inflammatory cell density in the stroma (B) or at the mesh interface (C) in C3−/− and C3+/+ mice (n=7/strain). Cell density is expressed as the number of inflammatory nuclei per ×60 HPF (B) or per 100 length units in a ×40 field (C). *P < 0.05; unpaired t test.
Complement modulates the innate inflammatory microenvironment at the implantation site
Even though the subcutaneous implantation of a mesh material represents a valid and relevant model with low surgical invasiveness, it may only partially reflect the clinical condition of deep-tissue implantation that is faced during hernia repair in humans. We therefore developed an additional model in which the mesh was implanted into the peritoneal space. Having shown the involvement of C5a in granulocyte activation in blood (see above), we further examined the role of this complement effector by using C5aR+/+ and C5aR−/− mice in our deep-tissue model. Given that the effects of C5a in granulocytes occur relatively quickly and that we wished to assess the immediate host response to the biomaterial implantation, we retrieved the mesh and surrounding tissue 5 d postimplantation (Fig. 4A). Isolation and flow cytometric analysis of the cells surrounding the retrieved mesh showed a decreased number of immune cells in C5aR−/− mice (Fig. 4B, C), confirming our previous findings. In an attempt to therapeutically reproduce these results, we treated C57BL/6J mice with C5aRa or its inactive control. Indeed, we found that C5aR inhibition resulted in decreased immune cell accumulation (Fig. 4D–G), especially at the mesh interface, that was similar to the results seen in the C5aR−/− mice. Of note, markers of complement activation (i.e., C3 fragments) were detected in the tissue around the mesh using IF (Fig. 4H), thereby confirming that the observed effect was initiated as a reaction to the presence of the biomaterial. As a consequence, these results verify the role of C5a in modulating the inflammatory microenvironment at the implantation site and indicate that C5aRa treatment could potentially be used to alleviate adverse inflammatory effects in implant surgery.
Figure 4.
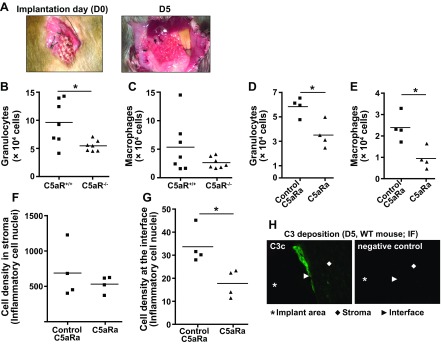
C5aR signaling is involved in shaping the host immune response after deep-tissue implantation of PP mesh. A) Image of a PP mesh on the day of implantation into the peritoneal space (D0) and on the day of its retrieval (D5). B, C) Levels of granulocytes (B) and macrophages (C) isolated from the tissue surrounding the PP mesh, 5 d after implantation in the peritoneal space of C5aR−/− and C5aR+/+ mice, as assessed by flow cytometry (n=7/strain). D, E) Effect of C5aR antagonism on granulocyte (D) and macrophage (E) accumulation around the implanted mesh in WT mice treated with either C5aRa or an inactive analog (control C5aRa) (n=4/treatment). F, G) Histological analysis of the meshes retrieved from WT mice treated with either C5aRa or inactive control compound. Cell density in the stroma (F) and mesh interface (G) was evaluated in H&E-stained sections (n=4/treatment); cell density is expressed as described for Fig. 3B, C. H) Immunofluorescent staining for C3 cleavage products in tissue sections retrieved from WT mice 5 d after implantation. Bright green staining indicates the presence of deposited C3c fragments at the interface. Incubation of slides with PBS, instead of anti-C3c Ab, was used as a negative control. *P < 0.05; unpaired t test.
DISCUSSION
This study reveals important new insight into the hitherto scarcely described role of complement in the inflammatory reaction to biomaterial implants. We demonstrated the intrinsic ability of a clinically important PP mesh to induce complement activation and found that this complement activation leads to the activation of immune cells through C5aR signaling. Notably, our study also identifies inhibitors acting at the level of both C3 and C5aR as potential candidates for therapeutic modulation of implant-induced inflammation.
Despite progress in material design, adverse effects related to activation of defense pathways by biomaterial contact with blood or interstitial fluid remain common and often lead to complications, such as inflammation and thrombosis. Complement has moved into the spotlight in biocompatibility research, as its activation, induced by the binding of components such as C3 or C1q to the material surface or an attached plasma protein layer (29, 30), results in the production of inflammatory mediators and influences other key systems ranging from coagulation and platelets to cytokine networks (4–7). In the case of extracorporeal systems, the benefit of therapeutically modulating complement has been repetitively demonstrated by ourselves and other groups (4, 7); examples include the attenuation of adverse reactions during cardiopulmonary bypass surgery (5), the reduction of inflammatory and thrombotic markers during hemodialysis by compstatin and C5aRa (14), and the protection of material and cell surfaces by coatings that recruit host complement regulators (31, 32). In contrast, our knowledge about the impact of complement and complement-targeted therapeutics on the fate of surgical implants remains comparatively scarce. This is particularly important since complement is likely involved in the initial foreign body reaction, the modulation of inflammatory responses, and, as newer reports suggest, in wound healing processes (33).
In the present study, we therefore utilized in vitro assays and newly established in vivo models to monitor complement activation and inflammatory markers induced by a clinically used implant material. PP-based meshes for hernia repair have previously been found to induce chronic inflammation, causing a persistent T-cell response and accumulation of macrophages (16). In addition, the up-regulation of inflammatory gene expression as a result of mesh implantation has also been reported (34). However, the underlying mechanisms of these adverse reactions and their potential association to the complement system have not yet been investigated. The contact of PP-based biomaterials, such as hollow fibers, membranes, or sutures with blood components has previously been indicated to initiate complement, but the degree of activation appears to vary considerably between the application and even the make of the PP material (28, 35, 36). In fact, PP tubes are often used in complement-related studies due to their comparatively low background activation when compared to other polymers (37). Our findings that the clinically used Prolene mesh for hernia repair induces considerable opsonization with C3 activation products and triggers ∼5% complement activation in whole blood is therefore an important indication of a potential involvement of complement in mesh-induced complications. Of note, given the high plasma levels of the central complement component C3 (∼1 mg/ml), even an activation level of 5% has great potential effect. The notable potency of the C3 inhibitor compstatin to prevent mesh-induced complement activation confirms the involvement of C3 convertase-mediated activation pathways.
The activation of C3 by biomaterials typically results in the downstream cleavage of C5 and generation of the anaphylatoxin C5a, which attracts immune cells to sites of ongoing complement attack and modulates their activity through C5aR-mediated signaling (6). Indeed, we observed increased expression levels of CD11b, a well-recognized activation marker of immune cells and building block of phagocytic complement receptors (e.g., CD11b/CD18), in granulocytes from blood that was in contact with the mesh. Together with the suppression of this CD11b expression by both compstatin and C5aRa, our findings confirm mesh-induced activation of immune cells in vitro that is complement dependent and mediated through the C5aR.
The intensity of immune activation in response to a biomaterial varies depending on the type of material, the time of exposure, and the site of contact (38). To elucidate the potential effect of mesh-induced complement activation in a clinical setting, relevant in vivo models are needed to validate and extend the findings of the whole-blood model. Currently, there are few relevant animal models for analyzing inflammatory consequences of implanted biomaterials reported (24), and, to our knowledge, none of them have been used in the context of complement-mediated reactions. With the description of a deep-tissue implant model in mice, the present study therefore helps to fill an important gap in the field. Notably, this model provides high translational value by utilizing biomaterials used in the clinic that are surgically embedded in the tissue of the model animals in a way that mimics implantation in human patients.
Using transgenic mice deficient in C3 or C5aR in our implant models, we have confirmed that both of these complement components are involved in the biomaterial-induced host immune reaction. An increase in the number of infiltrating immune cells was seen in the implantation site in WT mice, and we have demonstrated that their presence is a result of the chemoattractant properties of C5a. The persistent presence of a nonphagocytosable foreign body drives a physiological continuum in which professional phagocytes are accumulated at the site of implantation. More specifically, granulocytes are considered a hallmark of the acute-phase inflammation that occurs after biomaterial implantation, and they are capable of releasing inflammatory mediators and producing reactive oxygen species, thus affecting the biocompatibility outcome (39, 40). Chronic inflammation develops as inflammatory stimuli persist at the implant site, with macrophages representing the driving force in perpetuating immune responses (41, 42). This cell population fosters the invasion of additional inflammatory cells by secreting a variety of chemokines, thus exerting key controlling influences on the ensuing wound-healing responses (41). Interestingly, based on our in vivo implantation models, we have found that complement inhibition is able to shape the character and degree of immune cell infiltration and may therefore promote faster and safer integration of biomaterials (43).
The inhibition of complement activation on PP meshes may be achieved by preventive or therapeutic approaches. Modification of the mesh using complement-targeting coatings similar to those described in our recent work (31, 32) could result in a decreased inflammatory microenvironment, thus avoiding or minimizing long-term inflammation. In addition, a broad variety of complement-targeted drugs for local and/or systemic administration are either on the market or in clinical trials (44). Although complement drugs that were clinically evaluated thus far have shown beneficial safety profiles, the long-term use of systemic complement inhibitors is still regarded with some reservation, as it might potentially affect the antimicrobial actions of complement (44). In implant surgery, however, a time-restricted use of soluble inhibitors following implantation may be sufficient for suppressing complement activation and beneficially shaping the immune and inflammatory environment. Besides the direct effect on the implant microenvironment, recent studies indicate that complement inhibition may also influence other parameters associated with the implantation procedure such as wound healing (33). Compstatin appears as a promising candidate, as it inhibits both C3-mediated opsonization and generation of effector molecules independent of the initiation pathway (which may vary between materials) and, as a peptidic drug, is expected to be comparatively cost-effective (45, 46). Moreover, compstatin has been successfully used in many disease models, including biomaterial applications, and a compstatin analog is in active clinical development for age-related macular degeneration (44). Of note, compstatin specifically inhibits complement only in humans and nonhuman primates and was therefore not used in our mouse models. Alternatively, and in particular when long-term inhibition of complement would be advantageous, complement inhibitors that are specifically targeted to surfaces under complement attack (44) or that exclusively block proinflammatory signaling without affecting complement activation and opsonization may be considered. For the latter case, several potent C5aR antagonists, including PMX53 that was used in this study, have shown efficacy in counteracting complement-mediated inflammation in experimental and preclinical models (44). The selection of appropriate therapeutic options will certainly benefit from further elucidation of initiation pathways and critical effector functions in future studies on the involvement of complement in implant surgery.
In summary, our findings demonstrate that PP-based meshes that are widely used in the clinic for hernia repair induce a considerable complement response, both in vitro and in vivo, with direct effects on immune cells and the inflammatory microenvironment at the implant site. This insight is particularly important and novel in view of the limited knowledge about the influence of complement on surgical implants, and provides a valuable extension to the vibrant field of complement-related biomaterial and biocompatibility research. The results of our work may thus be relevant to other clinical applications that involve PP-based biomaterials [e.g., glaucoma valves (47), sutures (48), nanoparticles (49), and dentures (50)]. Although biomaterial-induced complement activation may vary in severity and involved initiation pathways between different polymers (11, 27, 36), the resulting effector functions (i.e., opsonization, release of C5a) are expected to be the same; as a consequence, the insight gained for the PP mesh may at least partially translate to other biomaterial implants. The newly introduced model of deep-tissue implantation is expected to serve as a valuable tool in future studies of immune and inflammatory reactions to implants. Given the key role of complement and the promising effects of compstatin in biomaterial models, the use of therapeutic complement inhibition may pave the way to improved biomaterial applications with better, faster, and safer healing processes.
Acknowledgments
This work was supported by grants from the U.S. National Institutes of Health: AI068730, AI030040, EY020633, GM097747, and DE021685 (J.D.L.) and AI097805 (D.R.).
J.D.L. and D.R. are the inventors of patents describing compstatin analogs and their clinical use. J.D.L. is the founder of Amyndas Pharmaceuticals, which develops complement-targeted therapeutics. The authors thank Dr. Stergios Rafail (General Regional Hospital George Papanikolaou, Thessaloniki, Greece) for providing the mesh polypropylene biomaterials, Dr. Hongchang Qu (University of Pennsylvania, Philadelphia, PA, USA) for synthesis of complement inhibitors, and Dr. Deborah McClellan for her help in editing the manuscript. D.R. and J.D.L. shared the supervision of this work.
Footnotes
- ABTS
- 2,2′-azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid)
- C5aR
- C5a receptor
- C5aRa
- C5a receptor antagonist
- Cp40
- yI[CVW(Me)QDWSarAHRC]mI-NH2)
- CD
- cluster of differentiation
- EDTA
- ethylenediaminetetraacetic acid
- ELISA
- enzyme-linked immunosorbent assay
- FcγR
- Fcγ receptor
- FITC
- fluorescein isothiocyanate
- HPF
- high-power field
- HRP
- horseradish peroxidase
- H&E
- hematoxylin and eosin
- IgG
- immunoglobulin G
- IF
- immunofluorescence
- IHC
- immunohistochemistry
- PP
- polypropylene
- WT
- wild-type
REFERENCES
- 1. Huebsch N., Mooney D. J. (2009) Inspiration and application in the evolution of biomaterials. Nature 462, 426–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peppas N. A., Langer R. (1994) New challenges in biomaterials. Science 263, 1715–1720 [DOI] [PubMed] [Google Scholar]
- 3. Langer R., Tirrell D. A. (2004) Designing materials for biology and medicine. Nature 428, 487–492 [DOI] [PubMed] [Google Scholar]
- 4. Ekdahl K. N., Lambris J. D., Elwing H., Ricklin D., Nilsson P. H., Teramura Y., Nicholls I. A., Nilsson B. (2011) Innate immunity activation on biomaterial surfaces: a mechanistic model and coping strategies. Adv. Drug Deliv. Rev. 63, 1042–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nilsson B., Ekdahl K. N., Mollnes T. E., Lambris J. D. (2007) The role of complement in biomaterial-induced inflammation. Mol. Immunol. 44, 82–94 [DOI] [PubMed] [Google Scholar]
- 6. Ricklin D., Hajishengallis G., Yang K., Lambris J. D. (2010) Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nilsson B., Korsgren O., Lambris J. D., Ekdahl K. N. (2010) Can cells and biomaterials in therapeutic medicine be shielded from innate immune recognition? Trends Immunol. 31, 32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nilsson U. R. (2001) Deposition of C3b/iC3b leads to the concealment of antigens, immunoglobulins and bound C1q in complement-activating immune complexes. Mol. Immunol. 38, 151–160 [DOI] [PubMed] [Google Scholar]
- 9. Lhotta K., Würzner R., Kronenberg F., Oppermann M., König P. (1998) Rapid activation of the complement system by cuprophane depends on complement component C4. Kidney Int. 53, 1044–1051 [DOI] [PubMed] [Google Scholar]
- 10. Mares J., Thongboonkerd V., Tuma Z., Moravec J., Matejovic M. (2009) Specific adsorption of some complement activation proteins to polysulfone dialysis membranes during hemodialysis. Kidney Int. 76, 404–413 [DOI] [PubMed] [Google Scholar]
- 11. Engberg A. E., Rosengren-Holmberg J. P., Chen H., Nilsson B., Lambris J. D., Nicholls I. A., Ekdahl K. N. (2011) Blood protein-polymer adsorption: Implications for understanding complement-mediated hemoincompatibility. J. Biomed. Mater. Res. A 97, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Markiewski M. M., Nilsson B., Ekdahl K. N., Mollnes T. E., Lambris J. D. (2007) Complement and coagulation: strangers or partners in crime? Trends Immunol. 28, 184–192 [DOI] [PubMed] [Google Scholar]
- 13. Oikonomopoulou K., Ricklin D., Ward P. A., Lambris J. D. (2012) Interactions between coagulation and complement–their role in inflammation. Semin. Immunopathol. 34, 151–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kourtzelis I., Markiewski M. M., Doumas M., Rafail S., Kambas K., Mitroulis I., Panagoutsos S., Passadakis P., Vargemezis V., Magotti P., Qu H., Mollnes T. E., Ritis K., Lambris J. D. (2010) Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood 116, 631–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DeAngelis R. A., Reis E. S., Ricklin D., Lambris J. D. (2012) Targeted complement inhibition as a promising strategy for preventing inflammatory complications in hemodialysis. Immunobiology 217, 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosch R., Junge K., Schachtrupp A., Klinge U., Klosterhalfen B., Schumpelick V. (2003) Mesh implants in hernia repair. Inflammatory cell response in a rat model. Eur. Surg. Res. 35, 161–166 [DOI] [PubMed] [Google Scholar]
- 17. Schachtrupp A., Klinge U., Junge K., Rosch R., Bhardwaj R. S., Schumpelick V. (2003) Individual inflammatory response of human blood monocytes to mesh biomaterials. Br. J. Surg. 90, 114–120 [DOI] [PubMed] [Google Scholar]
- 18. Morais J. M., Papadimitrakopoulos F., Burgess D. J. (2010) Biomaterials/tissue interactions: possible solutions to overcome foreign body response. AAPS J. 12, 188–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hammel M., Sfyroera G., Ricklin D., Magotti P., Lambris J. D., Geisbrecht B. V. (2007) A structural basis for complement inhibition by Staphylococcus aureus. Nat. Immunol. 8, 430–437 [DOI] [PubMed] [Google Scholar]
- 20. Qu H., Ricklin D., Bai H., Chen H., Reis E. S., Maciejewski M., Tzekou A., Deangelis R. A., Resuello R. R. G., Lupu F., Barlow P. N., Lambris J. D. (2013) New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology 218, 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sahu A., Kay B. K., Lambris J. D. (1996) Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J. Immunol. 157, 884–891 [PubMed] [Google Scholar]
- 22. Qu H., Magotti P., Ricklin D., Wu E. L., Kourtzelis I., Wu Y.-Q., Kaznessis Y. N., Lambris J. D. (2011) Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol. Immunol. 48, 481–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Finch A. M., Wong A. K., Paczkowski N. J., Wadi S. K., Craik D. J., Fairlie D. P., Taylor S. M. (1999) Low-molecular-weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. J. Med. Chem. 42, 1965–1974 [DOI] [PubMed] [Google Scholar]
- 24. Higgins D. M., Basaraba R. J., Hohnbaum A. C., Lee E. J., Grainger D. W., Gonzalez-Juarrero M. (2009) Localized immunosuppressive environment in the foreign body response to implanted biomaterials. Am. J. Pathol. 175, 161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Strey C. W., Markiewski M., Mastellos D., Tudoran R., Spruce L. A., Greenbaum L. E., Lambris J. D. (2003) The proinflammatory mediators C3a and C5a are essential for liver regeneration. J. Exp. Med. 198, 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manthey H. D., Thomas A. C., Shiels I. A., Zernecke A., Woodruff T. M., Rolfe B., Taylor S. M. (2011) Complement C5a inhibition reduces atherosclerosis in ApoE−/− mice. FASEB J. 25, 2447–2455 [DOI] [PubMed] [Google Scholar]
- 27. McLeod B. C., Viernes A., Sassetti R. J. (1983) Complement activation by plasma separator membranes. Transfusion 23, 143–147 [DOI] [PubMed] [Google Scholar]
- 28. Hussain M. A., Murali C. V., Willi P., Sharma C. P., Bhuvneshwar G. S., Sreekumar R. (1998) Comparative complement activation study of polypropylene hollow fibres of two different makes in static condition. J. Biomater. Appl. 12, 300–320 [DOI] [PubMed] [Google Scholar]
- 29. Andersson J., Ekdahl K. N., Lambris J. D., Nilsson B. (2005) Binding of C3 fragments on top of adsorbed plasma proteins during complement activation on a model biomaterial surface. Biomaterials 26, 1477–1485 [DOI] [PubMed] [Google Scholar]
- 30. Andersson J., Ekdahl K. N., Larsson R., Nilsson U. R., Nilsson B. (2002) C3 adsorbed to a polymer surface can form an initiating alternative pathway convertase. J. Immunol. 168, 5786–5791 [DOI] [PubMed] [Google Scholar]
- 31. Wu Y.-Q., Qu H., Sfyroera G., Tzekou A., Kay B. K., Nilsson B., Nilsson Ekdahl K., Ricklin D., Lambris J. D. (2011) Protection of nonself surfaces from complement attack by factor H-binding peptides: implications for therapeutic medicine. J. Immunol. 186, 4269–4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nilsson P. H., Ekdahl K. N., Magnusson P. U., Qu H., Iwata H., Ricklin D., Hong J., Lambris J. D., Nilsson B., Teramura Y. (2013) Autoregulation of thromboinflammation on biomaterial surfaces by a multicomponent therapeutic coating. Biomaterials 34, 985–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cazander G., Jukema G. N., Nibbering P. H. (2012) Complement activation and inhibition in wound healing. Clin. Dev. Immunol. 2012, 534291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Asarias J. R., Nguyen P. T., Mings J. R., Gehrich A. P., Pierce L. M. (2011) Influence of mesh materials on the expression of mediators involved in wound healing. J. Invest. Surg. 24, 87–98 [DOI] [PubMed] [Google Scholar]
- 35. De Vroege R., Rutten P. M., Kalkman C., Out T. A., Jansen P. G., Eijsman L., De Mol B. J., Wildevuur C. R. (1997) Biocompatibility of three different membrane oxygenators: effects on complement, neutrophil and monocyte activation. Perfusion 12, 369–375 [DOI] [PubMed] [Google Scholar]
- 36. Böhler J., Donauer K., Köster W., Schollmeyer P. J., Wieland H., Hörl W. H. (1991) Biocompatibility of four plasmapheresis membranes in patients treated for hypercholesterolemia. Am. J. Nephrol. 11, 479–485 [DOI] [PubMed] [Google Scholar]
- 37. Nielsen E. W., Waage C., Fure H., Brekke O. L., Sfyroera G., Lambris J. D., Mollnes T. E. (2007) Effect of supraphysiologic levels of C1-inhibitor on the classical, lectin and alternative pathways of complement. Mol. Immunol. 44, 1819–1826 [DOI] [PubMed] [Google Scholar]
- 38. Anderson J. M., Rodriguez A., Chang D. T. (2008) Foreign body reaction to biomaterials. Semin. Immunol. 20, 86–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Labow R. S., Meek E., Santerre J. P. (2001) Neutrophil-mediated biodegradation of medical implant materials. J. Cell. Physiol. 186, 95–103 [DOI] [PubMed] [Google Scholar]
- 40. Patel J. D., Krupka T., Anderson J. M. (2007) iNOS-mediated generation of reactive oxygen and nitrogen species by biomaterial-adherent neutrophils. J. Biomed. Mater. Res. A 80, 381–390 [DOI] [PubMed] [Google Scholar]
- 41. Xia Z., Triffitt J. T. (2006) A review on macrophage responses to biomaterials. Biomed. Mater. 1, R1–9 [DOI] [PubMed] [Google Scholar]
- 42. Kao W. J. (1999) Evaluation of protein-modulated macrophage behavior on biomaterials: designing biomimetic materials for cellular engineering. Biomaterials 20, 2213–2221 [DOI] [PubMed] [Google Scholar]
- 43. McNally A. K., Anderson J. M. (2011) Macrophage fusion and multinucleated giant cells of inflammation. Adv. Exp. Med. Biol. 713, 97–111 [DOI] [PubMed] [Google Scholar]
- 44. Ricklin D., Lambris J. (2013) Complement in immune and inflammatory disorders: therapeutic interventions. [E-pub ahead of print] J. Immunol. doi: 10.4049/jimmunol.1203200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bray. B. L. (2003) Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat. Rev. Drug Discov. 2, 587–593 [DOI] [PubMed] [Google Scholar]
- 46. Ricklin D., Lambris J. D. (2008) Compstatin: a complement inhibitor on its way to clinical application. Adv. Exp. Med. Biol. 632, 273–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mackenzie P. J., Schertzer R. M., Isbister C. M. (2007) Comparison of silicone and polypropylene Ahmed glaucoma valves: two-year follow-up. Can. J. Ophthalmol. 42, 227–232 [PubMed] [Google Scholar]
- 48. Sönmez K., Bahar B., Karabulut R., Gülbahar O., Poyraz A., Türkyilmaz Z., Sancak B., Basaklar A. C. (2009) Effects of different suture materials on wound healing and infection in subcutaneous closure techniques. B-ENT 5, 149–152 [PubMed] [Google Scholar]
- 49. Thomas S. N., Van der Vlies A. J., O'Neil C. P., Reddy S. T., Yu S. S., Giorgio T. D., Swartz M. A., Hubbell J. A. (2011) Engineering complement activation on polypropylene sulfide vaccine nanoparticles. Biomaterials 32, 2194–2203 [DOI] [PubMed] [Google Scholar]
- 50. Schneider R. L. (1987) Using clear polypropylene splints in the fabrication of anterior porcelain-fused-to-metal fixed restorations. Quintessence Dent. Technol. 11, 93–96 [PubMed] [Google Scholar]