

Abstract
DNA methylation is an important regulatory mechanism for gene expression that involved in the biological processes of development and differentiation in plants. To investigate the association of DNA methylation with heterosis in Brassica, a set of intraspecific hybrids in Brassica rapa and B. napus and interspecific hybrids between B. rapa and B. napus, together with parental lines, were used to monitor alterations in cytosine methylation at 5′-CCGG sites in seedlings and buds by methylation-sensitive amplification polymorphism analysis. The methylation status of approximately a quarter of the methylation sites changed between seedlings and buds. These alterations were related closely to the genomic structure and heterozygous status among accessions. The methylation status in the majority of DNA methylation sites detected in hybrids was the same as that in at least one of the parental lines in both seedlings and buds. However, the association between patterns of cytosine methylation and heterosis varied among different traits and between tissues in hybrids of Brassica, although a few methylation loci were associated with heterosis. Our data suggest that changes in DNA methylation at 5′-CCGG sites are not associated simply with heterosis in the interspecific and intraspecific hybridizations derived from B. rapa and B. napus.
Introduction
Although heterosis has been utilized widely to improve seed yield potential in crops, the underlying biological mechanisms remain poorly understood. Some efforts have been made to investigate the relationship between heterosis and parental diversity [1], and to dissect heterosis by mapping quantitative trait loci in segregating populations [2]–[5].
The differences that exist between hybrids and their parents at the level of gene expression have received considerable attention in recent years. It has been proposed that nonadditive patterns of gene expression are predominant in hybrids [6]–[8]. However, reports showed that the majority of genes that are expressed differentially in inbred parents have an additive pattern of expression in hybrids, and that novel patterns of gene expression in hybrids are rare [9], and that the proportion of additively expressed genes is correlated positively with heterosis and yield in maize [10]. Moreover, Swanson-Wagner et al. [11] observed all possible modes of gene action in a global comparison of gene expression in a maize F1 hybrid and its inbred parents [11]. The varied results of those studies led Birchler et al. [12] to propose that heterosis is a complex phenotype, and that the pattern of gene expression in hybrids is not simply an additive effect of the parental alleles.
DNA methylation, which involves the conversion of cytosine to 5-methylcytosine, is an epigenetic change that does not affect DNA sequence but does influence gene expression, especially when it occurs in the promoter region of a gene [13]. DNA methylation is involved in the biological processes of development and differentiation in plants, such as plant embryogenesis, seed viability, floral development [14]–[16], as well as stress response [17].
Given the widespread and important effects of DNA methylation on gene expression, some researchers have tried to relate heterosis to the alterations of DNA methylation in hybrids relative to their parental lines [18]–[19]. Alterations in the pattern of cytosine methylation relative to the parents are evident in interspecific hybrids, including newly formed allopolyploid and introgression lines [20]–[24], and in intraspecific hybrids [25]–[26]. However, there is a fundamental problem in these studies in that it is difficult to validate the association of the alterations of DNA methylation with the observed heterosis in statistics. Comparative studies across a variety of hybrids exhibiting various degrees of heterosis were required rather than comparison of inbred plants with their corresponding hybrids [27].
Brassica napus (AACC, rapeseed) and B. rapa (AA) are two important oilseed crops in which heterosis has been utilized widely. It is easy to develop interspecific hybrids between B. napus and B. rapa without the requirement of tissue culture [28], and strong heterosis for biomass production [28], and seed yield [29]–[30] has been documented among combinations derived from B. napus and B. rapa.
To associate the changes in DNA methylation with heterosis, a set of interspecific and intraspecific hybrids and four parental accessions each of B. napus and B. rapa was evaluated for agronomic traits in two years, and the alterations of DNA methylation at 5′-CCGG sites in seedlings and buds among hybrids and parents were monitored using methylation-sensitive amplification polymorphism analysis in this study. Our data suggest that the status in most methylation sites in hybrids was the same as that in at least one of the parental lines in both seedlings and buds, and that the alterations of DNA methylation during development were associated with the genomic structure and heterozygous status among parents and hybrids. However, no direct correlation between heterosis and the alteration of DNA methylation at 5′-CCGG sites could be found in the both interspecific and intraspecific hybrids derived from B. rapa and B. napus.
Materials and Methods
Parents and Combinations
We used eight elite inbred lines of Brassica that are widely sown in China: four accessions each of B. napus (Xiangyou 15, Shuyou 1, Youyan 2, and Zhongshuan 9) and B. rapa (Hangzou Changcai, Changge Youcai, Qixingjian, and Daye Youcaibai). A complete diallel cross was performed to develop 56 combinations: 24 intraspecific hybridizations in B. napus and B. rapa, and 32 interspecific hybridizations between B. napus and B. rapa. The parental lines were considered to be pure lines because they had been selfed successively for at least seven generations before crossing.
The parents and hybrids were sown in the experimental station of Southwest University, Chongqing, China, with a randomized complete block design, 3 m2 per plot, in two growth seasons, 2007 and 2008. Plant density was in accordance with farming practice in the region of the Yangtze River, i.e. a row spacing of 0.3 m and 10 plants grown in short 2.5-m rows. The field was managed in the same manner as normal agricultural production fields.
Methylation-sensitive Amplified Polymorphism Analysis
Seedlings are considered to be associated with vegetative growth, and buds with the conversion from vegetative to reproductive growth [31]. Therefore, 1-month-old seedlings and buds just before flowering were collected to monitor alterations in cytosine methylation in the global genome in Brassica, using methylation-sensitive amplified polymorphisms (MSAPs). These tissues were harvested and pooled from at least 10 individuals for each accession. Genomic DNA was isolated from fresh tissue with cetyltrimethyl ammonium bromide. The MSAP method was developed from technology based on amplification fragment length polymorphisms and uses a pair of isoschizomers, MspI/HpaII, that display differential sensitivity to methylation at the 5′-CCGG sequence sites, full methylation of the internal cytosine (cleavage by MspI but not HpaII), or hemimethylation of the external cytosine (cleavage by HpaII but not MspI) [26]. Ten combinations of MSAP primers of EcoR I and MspI/HpaII were randomly selected to detect changes in cytosine methylation at 5′-CCGG. The products of selective amplification were visualized on 6.5% polyacrylamide sequencing gels on a Licor-4300 DNA analyzer.
To explore the genetic mechanism for the alteration of cytosine methylation at 5′-CCGG during development in Brassica, a matrix that described the differential methylation of cytosine among accessions (with ‘1’ for alteration and ‘0’ for no change in cytosine methylation pattern from seedling to bud) was used for principal component analysis (PCA) and analysis of molecular variance (AMOVA). PCA was performed using the software NTSYS-PC [32], to evaluate genetic diversity with respect to the alterations in cytosine methylation among accessions of Brassica, with the genetic distance calculated on the basis of the Dice genetic similarity coefficient [33]. AMOVA was performed using the ARLEQUIN software [34], to test the genetic structure for the alterations in cytosine methylation within or between groups. The groups were sorted according to genomic structure, for example B. rapa, B. napus and interspecific hybrids, and according to heterozygous status, for example inbred line and hybrid.
Association of Heterosis with Alterations in DNA Methylation
The DNA methylation status of the hybrids was compared with that of their parental lines with regard to the sites that were altered. Five patterns of DNA methylation status were identified. These included P1 = P2 = F1, which signified that the same alteration in DNA methylation occurred in a hybrid and both its parents, P1 = F1≠P2, P2 = F1≠P1, and P1 = P2≠F1 (i.e. the hybrid showed hypomethylation or hypermethylation). To investigate the relationship between methylation patterns in the hybrids and mid-parental heterosis (MPH), the methylation sites that were associated with heterosis were selected by using single-marker analysis in seedlings and buds. Stepwise regression analysis of the methylation patterns of these loci selected against MPH was performed at the default 0.150 level with phenotypic trait as a dependent variable and five methylation patterns as independent variables. MPH was calculated on the basis of the performance of the parents and their hybrids. Analysis of variance (ANOVA) and Pearson correlation analysis were performed using the SAS software for the traits of interest [35].
Results
Cytosine Methylation Status in Brassica
We used 22 interspecific and 23 intraspecific hybrids for the MSAP analysis, together with eight parental lines. The number of available hybrids was lower than the potential total due to failure to cross or limitation of the number of seeds for some crosses. We used 10 combinations of MSAP primers to detect changes in cytosine methylation at 5′-CCGG in seedlings and buds (Fig. 1). In total, 252 polymorphism fragments were clearly detected among the 53 accessions. Of the 252 polymorphism fragments, 46 (18.2%) were amplified differentially on average in each tissue for each accession after the DNA was digested with MspI and HpaII.
Figure 1. Changes in methylation-sensitive polymorphisms in bud using EcoR I + AAC/HpaII vs MspI+TAG.

Arrows indicate polymorphic methylation-sensitive fragments.
Tissue Specificity of Cytosine Methylation
Among the accessions in Brassica, seedlings and buds showed differential methylation patterns at 5′-CCGG sites, as detected by MSAP [Fig. S1]. In general, the number of sites that were amplified differentially was higher in seedlings than in buds. For seedlings, significant differences in the proportion of differentially methylated sites were detected among the 53 accessions of Brassica. The B. rapa parental lines exhibited the highest proportion of differentially methylated sites, with an average of 31.9%, followed by the B. napus parental lines with an average of 22.7%. The intraspecific combinations for B. rapa had an average value of 22.4%, whereas interspecific combinations between B. napus and B. rapa had an average value of 20.5%. The lowest proportion of differentially methylated sites was found in the intraspecific combinations for B. napus, with an average value of 19.9%. However, no difference was observed in the proportion of differentially methylated sites in seedlings between the interspecific and intraspecific combinations. For buds, the average proportion of differentially methylated sites was 14.5%, with no significant differences among the 53 accessions (F test, P = 0.49). And some sites that displayed the same methylation status were detected among the accessions. For example, 15 sites in seedlings and 11 in buds were detected in all eight parental lines.
Seedlings and buds are two important tissues in plant development, and are associated closely with vegetative growth and the conversion from vegetative to reproductive growth, respectively [31]. We found that the methylation status differed at approximately a quarter of the methylation sites between seedlings and buds among the 53 accessions of Brassica, except for the parental lines of B. rapa in which the status at almost half of the sites changed, and that the predominant pattern of alterations in cytosine methylation from seedlings to buds was hypomethylation over hypermethylation, except for intraspecific hybrids of B. napus (Table S1).
To explore the genetic alterations of cytosine methylation at 5′-CCGG sites during development, a matrix that described the differential status of cytosine methylation between seedlings and buds was used for PCA (Fig. 2). The total variation explained by the first and second principal components was 42.2% and 29.8%, respectively. The Brassica accessions could be clustered into three groups that were in accordance with the diversity of their genomic structure: B. napus, B. rapa, and interspecific hybrids between B. napus and B. rapa. Moreover, within B. napus and B. rapa, an obvious difference between hybrids and parental lines was found (Fig. 2). This indicated that heterozygous status (homozygotic vs. heterozygotic) influenced the alteration of cytosine methylation during development. No obvious differences in cytoplasmic effects were found in the interspecific hybrids between B. napus and B. rapa, because it was difficult to separate clearly the reciprocal crosses between B. napus and B. rapa in Fig. 2.
Figure 2. Association among eight parental lines and 45 hybrids of Brassica with respect to alterations in differentially methylated sites between seedlings and buds as revealed by principal component analysis.
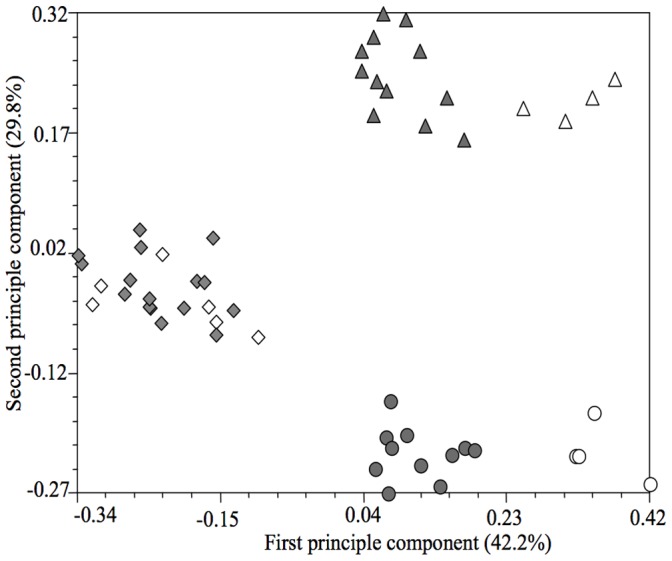
Intraspecific hybrids, B. rapa × B. rapa and B. napus × B. napus, are represented by closed triangles and closed circles in light grey, respectively. Interspecific hybrids, B. napus ×B. rapa and B. rapa × B. napus are represented by rhombuses in light grey and open rhombuses, respectively. Parental lines of B. napus and B. rapa are represented by open triangles and circles, respectively.
The importance of genomic structure and heterozygous status in the changes in cytosine methylation during development was supported by AMOVA (Table 1). Highly significant variances were found among and within genomic structures and heterozygous status (P≤0.001). Genomic structure accounted for 42.62% of the variation and heterozygous status for 37.79%, which indicated that both of these factors played important roles in the alteration of cytosine methylation from seedling to bud.
Table 1. Analysis of molecular variation of 53 accessions with different genomic structure and heterozygous status with respect to alterations in differentially methylated sites between seedlings and buds.
| Group/source | df | Variance component | Variance accounted for (%) |
| Genomic structure | |||
| Among genomic structures | 2 | 11.81* | 42.62 |
| Within genomic structures | 50 | 15.90* | 57.38 |
| Total | 52 | 27.71 | |
| Heterozygous status | |||
| Among heterozygous status | 1 | 10.75* | 33.79 |
| Within heterozygous status | 51 | 21.06* | 66.21 |
| Total | 52 | 31.80 | |
: Significant at P = 0.001.
Differential Cytosine Methylation among Parents and Hybrids
Cytosine methylation status at 5′-CCGG sites in seedlings and buds was compared between parents and hybrids. The parents exhibited more differentially methylated sites in seedlings than the hybrids did, whereas there were no significant differences between parental lines and hybrids in buds (SI Fig. 1). In general, the proportion of differentially methylated sites in a hybrid correlated positively to some extent with the average value of its two parents in both seedlings (r = 0.356, P = 0.008) and buds (r = 0.24, P = 0.056).
The patterns and extent of the alterations in cytosine methylation in hybrids relative to their parents are shown in Table 2. Approximately 85% and 91% of sites detected by MSAP in hybrids shared their cytosine methylation status with at least one of their parents in seedlings and buds, respectively. This indicated that hybrids were associated closely with their parents with regard to the alteration of cytosine methylation. The pattern of alterations in cytosine methylation at 5′-CCGG sites in interspecific hybrids between B. napus and B. rapa, especially in seedlings, showed more similarities to the pattern for parental B. napus than that for parental B. rapa, regardless of whether the former acted as the female or male parent (Table 2). This indicated that there were no obvious cytoplasm effects with respect to the extent of alteration of cytosine methylation at 5′-CCGG sites in the interspecific hybrids.
Table 2. Pattern and extent of alterations at differentially methylated 5′-CCGG sites among hybrids relative to parental lines in Brassica.
| Combinations | P1 = P2 = F1 (%) | P1 = F1≠P2 (%) | P2 = F1≠P1 (%) | Hypomethylation (%) | Hypermethylation (%) |
| Seedling | |||||
| B. rapa × B. napus | 56.1 | 9.7 | 22 | 9.6 | 2.5 |
| B. napus × B. rapa | 55.6 | 23 | 9.1 | 10.6 | 1.6 |
| B. rapa × B. rapa | 60 | 12.2 | 11.8 | 13.3 | 2.7 |
| B. napus × B. napus | 77 | 6.7 | 6.2 | 8.4 | 1.6 |
| Bud | |||||
| B. rapa × B. napus | 80.1 | 7.2 | 7.3 | 4.1 | 1.4 |
| B. napus × B. rapa | 78 | 9.9 | 6.7 | 4 | 1.4 |
| B. rapa × B. rapa | 78.6 | 7 | 7.1 | 3.9 | 3.4 |
| B. napus × B. napus | 81.8 | 5.1 | 5.3 | 3.1 | 4.6 |
Considering the patterns for the differentially methylated sites in hybrids relative to their parental lines, hypomethylation predominated over hypermethylation in both interspecific hybrids (10.3 vs. 1.9%) and intraspecific hybrids (10.8 vs. 2.1%) in seedlings, and in interspecific hybrids in buds (4.1 vs. 1.4%) (Table 2).
Association of Cytosine Methylation with Heterosis
Seven agronomic traits were observed among the 53 accessions of Brassica over 2 years, and a positive correlation was found between most of these traits (Table S2). Strong MPH was seen among the hybrids for the traits observed, namely plant height (7.4%), main inflorescence length (7.1%), number of branches (6.8%), number of pods per plant (22.7%), number of seeds per pod (5.4%), seed yield (21.1%), and biomass (28.3%) (Table S3).
To reveal the relationship between methylation patterns and MPH, single-marker analysis at the 0.05 significance level was used to select 72 (28.6%) of the 252 methylation loci that were associated with heterosis (Table S4). The percentage of methylation loci associated with MPH for the seven traits was 14.4% in seedlings and 11.2% in buds on average. Of the 72 methylation loci associated with heterosis, 55 (76.4%) influenced more than one trait simultaneously in the same or reverse direction. For two traits that exhibited a high positive correlation, there were more loci that overlapped in the same direction and fewer that overlapped in a different direction (Table S4). Unfortunately, we could not obtain additional information about the association of biomass and seed yield with the other traits due to the limited amount of research samples, i.e. the intraspecific hybrids were used to evaluate seed yield rather than biomass, whereas the interspecific hybrids were used to evaluate biomass rather than seed yield.
The loci associated with heterosis were used for stepwise regression analysis of methylation patterns against heterosis (Table 3). All five methylation patterns were found to contribute to heterosis in seedlings and buds. Each methylation pattern significantly affected one or more traits, whereas each trait was associated with one or more methylation patterns. The methylation patterns that were associated with heterosis varied according to traits and tissues, and no unified methylation pattern could explain heterosis for all seven traits in both tissues (Table 3). This suggests that heterosis might not be associated simply with genome-wide alterations in DNA methylation.
Table 3. Impact of five methylation patterns on agronomic traits in seedlings and buds.
| Pattern | Plant Height | Main Inflorescence Length | No. of Branch | No. of Podper Plant | No. of Seedper Pod | Seed Yield | Biomass |
| Seedling | |||||||
| P1 = P2 = F1 | 0.36/0.26* | ||||||
| P1 = F1≠P2 | 0.09/0.11* | 0.13/0.17** | 0.10/0.07 | 1.64/0.4*** | 0.47/0.08 | ||
| P2 = F1≠P1 | −2.4 | −18.3333 | 0.53/0.66*** | ||||
| Hypomethylation | 1.40/0.17** | ||||||
| Hypermethylation | 4.1/0.36*** | 5.79/0.05 | |||||
| Bud | |||||||
| P1 = P2 = F1 | 1.03/0.15** | −0.89/0.57*** | |||||
| P1 = F1≠P2 | 0.76/0.09 | ||||||
| P2 = F1≠P1 | 0.49/0.07 | ||||||
| Hypomethylation | −2.03/0.33** | ||||||
| Hypermethylation | −4.14286 | −0.48/0.15* | −13.0909 | −3.95/0.19** | |||
The loci in association with heterosis detected by using single marker analysis were used for stepwise regression analysis of methylation patterns against heterosis at the default 0.150 level. The numbers to the left and right of each slash represent the regression coefficient and coefficient of determination, respectively.
,**,***: p = 0.05, 0.01 and 0.001, respectively.
Discussion
Alterations of DNA Methylation
DNA methylation is an important aspect of epigenetics that regulates gene expression, and is involved in the biological processes of development and differentiation. In the present study, on average, a quarter of cytosine methylation sites throughout the global genome were observed to be methylated differentially between seedlings and buds among 53 accessions of Brassica, and that hypomethylation predominated over hypermethylation from seedlings to buds, except for intraspecific hybrids of B. napus. Similar tissue-specific cytosine methylation has been reported in other plants [36]–[38]. This implies that DNA methylation is tissues specific, and possibly is influenced by hybridization.
In the study reported herein, most cytosine methylation sites in interspecific and intraspecific hybrids of Brassica shared the same status as that of at least one of the parents. This observation was in accordance with Zhang et al. [39], who have shown that the profile of cytosine methylation in hybrids of sorghum deviates from those of their parents at a rate of only 1.69–3.22% of sites. These results have indicated that the methylation status at most cytosine methylation sites displays stable inheritance from inbred parents to hybrids. Moreover, we found that alterations in cytosine methylation status between seedling and bud were associated closely with genomic structure and heterozygous status in Brassica, which indicates that changes in cytosine methylation do not occur randomly during development. Similar observations have also been reported in humans by Bjornsson et al [40], who found that methylation changes over time, were clustered among families. It was reported that most methylation changes that occur in the first generation of resynthesized lines in Brassica can be fixed in later generations [20]. These findings show that methylation changes during development might be under genetic control, although how genetic mechanisms regulate the alteration of methylation remains to be elucidated. However, it is worth to mention that the interspecific hybrids between B. napus and B. rapa, especially in seedlings, showed more similarities to the pattern for parental B. napus than for parental B. rapa in this study, indicating that genomic asymmetry possible involves in alteration of methylation.
Association of DNA Methylation with Heterosis
Extensive alterations in methylation have been documented in resynthesized lines in B. napus [20], [24], [41], Arabidopsis [22], and Spartina [23], as well as introgression lines [21]. It was proposed that alterations in cytosine methylation in interspecific hybrids are induced by hybridization [21]. If this is the case, a burst of methylation changes should be detected in F1 hybrids relative to parental lines. However, we did not detect such a burst in both interspecific and intraspecific F1 hybrids of B. napus and B. rapa, in comparison with parental lines. Even the rate of methylation changes in buds in reciprocal crosses between B. napus and B. rapa was lower than that in the parents. Udall et al. [42] have observed that de novo chromosomal rearrangements occur frequently in the vicinity of pre-existing translocations in resynthesized B. napus. This suggests that only a few rearrangements, which are produced during meiosis in S0 plants, give rise to an increasing number of genomic changes in the following generations [43]. A possible explanation is that pairing between nonhomologous chromosomes at first meiosis in an interspecific hybrid, causes genome restructuring in later generations, and that many of the meiosis-driven genetic changes are transmitted to the progeny [43], with accompanying epigenetic changes [44]. Therefore, alterations in DNA methylation in the progeny of interspecific hybrids are possibly triggered by nonhomologous gene–gene interactions, rather than by hybridization.
In this study, we found that interspecific and intraspecific hybrids of Brassica exhibited differential methylation status, including hypo- and hypermethylation, relative to their parental lines at 5′-CCGG methylation sites in seedlings and buds. Differential methylation status relative to the parents has also been observed at 5′-CCGG methylation sites in hybrids [21], [26].
There are three different types of DNA methylation in plant, CG, CHG, and CHH (where H is any nucleotide but G) [45]. It should be remembered in the mind that we only monitored one of DNA methylation type, the DNA methylation at 5′-CCGG sites. We could not detect any obvious correlation between heterosis and genome-wide DNA methylation at 5′-CCGG sites, because the association of patterns of cytosine methylation with heterosis varied among traits and between tissues in this study. However, Shen et al. [18] found that the genome-wide remodeling of CHH sites directed by RNA- directed DNA methylation (RdDM) pathway may play a role in heterosis. In order to answer whether heterosis associated with the different types of DNA methylation, more future studies are required.
Supporting Information
Alterations in cytosine methylation at 5′-CCGG sites as identified by 252 methylation-sensitive amplified polymorphisms among 53 accessions of Brassica in seedlings and buds. The right and left vertical coordinates represent the proportion and number of methylation alterations, respectively.
(PDF)
Pattern and extent of alterations at differentially methylated 5′-CCGG sites from seedlings to buds among parental lines and hybrids of Brassica.
(DOC)
Correlation of hybrid performance (upper triangle) and number of overlapping loci associated with heterosis that were detected in two tissues (lower triangle) for seven traits.
(DOC)
Mid-parent heterosis of agronomic traits among hybrids in Brassica.
(DOC)
Number of cytosine methylation loci in association with heterosis in seedlings and buds, which was selected from 252 loci of MSAP by using single marker analysis.
(DOC)
Acknowledgments
We thank Prof. Li Chen for kind support in the collection of materials.
Funding Statement
This work was supported financially by grants from the Fundamental Research Funds for the Central Universities, National Natural Science Foundation of China (31171585), CSTC 201180001, and 111 Project 13 (B12006). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Melchinger AE (1999) Genetic diversity and heterosis. In: Coors CG, Pandey S (eds) Genetic and exploitation of heterosis in crops. American Society of Agronomy:Madison 99–118. [Google Scholar]
- 2. Hua J, Xing Y, Wu W, Xu C, Sun X, et al. (2003) Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci U S A 100: 2574–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stuber CW, Lincoln SE, Wolff DW, Helentjaris T, Lander ES (1992) Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular markers. Genetics 132: 823–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xiao J, Li J, Yuan L, Tanksley SD (1995) Dominance is the major genetic basis of heterosis in rice as revealed by QTL analysis using molecular markers. Genetics 140: 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu SB, Li JX, Xu CG, Tan YF, Gao YJ, et al. (1997) Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci U S A 94: 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Auger DL, Gray AD, Ream TS, Kato A, Coe EH, et al. (2005) Nonadditive gene expression in diploid and triploid hybrids of maize. Genetics 169: 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo M, Rupe MA, Zinselmeier C, Habben J, Bowen BA, et al. (2004) Allelic variation of gene expression in maize hybrids. Plant Cell 16: 1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song RT, Messing J (2003) Gene expression of a gene family in maize based on noncollinear haplotypes. Proc Natl Acad Sci U S A 100: 9055–9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stupar RM, Springer NM (2006) Cis-transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F-1 hybrid. Genetics 173: 2199–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo M, Rupe MA, Yang XF, Crasta O, et al. (2006) Genome-wide transcript analysis of maize hybrids: allelic additive gene expression and yield heterosis. Theor Appl Genet 113: 831–845. [DOI] [PubMed] [Google Scholar]
- 11. Swanson-Wagner RA, Jia Y, DeCook R, Borsuk LA, Nettleton D, et al. (2006) All possible modes of gene action are observed in a global comparison of gene expression in a maize F-1 hybrid and its inbred parents. Proc Natl Acad Sci U S A 103: 6805–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Birchler JA, Auger DL, Riddle NC (2003) In search of the molecular basis of heterosis. Plant Cell 15: 2236–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Siegfried Z, Simon I (2010) DNA methylation and gene expression. Wiley Interdiscip Rev Syst Biol Med 2: 362–371. [DOI] [PubMed] [Google Scholar]
- 14. Adams S, Vinkenoog R, Spielman M, Dickinson HG, Scott RJ (2000) Parent-of-origin effects on seed development in Arabidopsis thaliana require DNA methylation. Development 127: 2493–2502. [DOI] [PubMed] [Google Scholar]
- 15. Jacobsen SE, Sakai H, Finnegan EJ, Cao X, Meyerowitz EM (2000) Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr Biol 10: 179–186. [DOI] [PubMed] [Google Scholar]
- 16. Xiao WY, Custard KD, Brown RC, Lemmon BE, Harada JJ, et al. (2006) DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 18: 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chinnusamy V, Zhu JK (2009) Epigenetic regulation of stress responses in plants. Curr Opin Plant Biol 12: 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shen H, He H, Li J, Chen W, Wang X, et al. (2012) Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 24: 3875–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chodavarapu RK, Feng S, Ding B, Simon SA, Lopez D, et al. (2012) Transcriptome and methylome interactions in rice hybrids. PNAS 109: 12040–12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaeta RT, Pires JC, Iniguez-Luy F, Leon E, Osborn TC (2007) Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 19: 3403–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin HJ, Hu W, Wei Z, Wan LL, Li G, et al. (2008) Alterations in cytosine methylation and species-specific transcription induced by interspecific hybridization between Oryza sativa and O-officinalis. Theor Appl Genet 117: 1271–1279. [DOI] [PubMed] [Google Scholar]
- 22. Madlung A, Masuelli RW, Watson B, Reynolds SH, Davison J, et al. (2002) Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiology 129: 733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salmon A, Ainouche ML, Wendel JF (2005) Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Molecular Ecology 14: 1163–1175. [DOI] [PubMed] [Google Scholar]
- 24. Xu YH, Zhong L, Wu XM, Fang XP, Wang JB (2009) Rapid alterations of gene expression and cytosine methylation in newly synthesized Brassica napus allopolyploids. Planta 229: 471–483. [DOI] [PubMed] [Google Scholar]
- 25. Banaei Moghaddam AM, Fuchs J, Czauderna T, Houben A, Mette MF (2010) Intraspecific hybrids of Arabidopsis thaliana revealed no gross alterations in endopolyploidy, DNA methylation, histone modifications and transcript levels. Theor Appl Genet 120: 215–226. [DOI] [PubMed] [Google Scholar]
- 26. Xiong LZ, Xu CG, Saghai Maroof MA, Zhang Q (1999) Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol Gen Genet 261: 439–446. [DOI] [PubMed] [Google Scholar]
- 27. Dahal D, Mooney BP, Newton KJ (2012) Specific changes in total and mitochondrial proteomes are associated with higher levels of heterosis in maize hybrids. Plant J. 2012 72: 70–83. [DOI] [PubMed] [Google Scholar]
- 28. Qian W, Liu R, Meng J (2003) Genetic effects on biomass yield in interspecific hybrids between Brassica napus and B. rapa . Euphytica 134: 9–15. [Google Scholar]
- 29. Li MT, Chen X, Meng JL (2006) Potential of intersubgenomic heterosis in rapeseed production with a partial new-typed Brassica napus containing subgenome Ar from B. rapa and Cc from B. carinata . Crop Science 46: 234–242. [Google Scholar]
- 30. Qian W, Chen X, Fu D, Zou J, Meng J (2005) Intersubgenomic heterosis in seed yield potential observed in a new type of Brassica napus introgressed with partial Brassica rapa genome. Theor Appl Genet 110: 1187–1194. [DOI] [PubMed] [Google Scholar]
- 31. Poethig RS (2003) Phase change and the regulation of developmental timing in plants. Science 301: 334–336. [DOI] [PubMed] [Google Scholar]
- 32.Rohlf FJ (1997) NTSYSpc: numerical taxonomy and multivariate analysis system, version 2.02. [Google Scholar]
- 33. Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A 76: 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform Online 1: 47–50. [PMC free article] [PubMed] [Google Scholar]
- 35.Institute S. SAS Users Guide: Statistic. SAS Institute, Cary, NC 1996. [Google Scholar]
- 36. Finnegan EJ, Genger RK, Peacock WJ, Dennis ES (1998) DNA Methylation in Plants. Annu Rev Plant Physiol Plant Mol Biol 49: 223–247. [DOI] [PubMed] [Google Scholar]
- 37. Xiong LZ, Yang GP, Xu CG (1998) Relationships of differential gene expression in leaves with heterosis and heterozygosity in a rice diallel cross. Molecular Breeding 4: 129–136. [Google Scholar]
- 38. Ruiz-Garcia L, Cervera MT, Martinez-Zapater JM (2005) DNA methylation increases throughout Arabidopsis development. Planta 222: 301–306. [DOI] [PubMed] [Google Scholar]
- 39. Zhang MS, Yan HY, Zhao N, Lin XY, Pang JS, et al. (2007) Endosperm-specific hypomethylation, and meiotic inheritance and variation of DNA methylation level and pattern in sorghum (Sorghum bicolor L.) inter-strain hybrids. Theor Appl Genet 115: 195–207. [DOI] [PubMed] [Google Scholar]
- 40. Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, et al. (2008) Intra-individual change over time in DNA methylation with familial clustering. Jama 299: 2877–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lukens LN, Pires JC, Leon E, Vogelzang R, Oslach L, et al. (2006) Patterns of sequence loss and cytosine methylation within a population of newly resynthesized Brassica napus allopolyploids. Plant Physiology 140: 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Udall JA, Quijada PA, Osborn TC (2005) Detection of chromosomal rearrangements derived from homologous recombination in four mapping populations of Brassica napus L. Genetics. 169: 967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Szadkowski E, Eber F, Huteau V, Lode M, Huneau C, et al. (2010) The first meiosis of resynthesized Brassica napus, a genome blender. New Phytol 186: 102–112. [DOI] [PubMed] [Google Scholar]
- 44. Henikoff S, Matzke MA (1997) Exploring and explaining epigenetic effects. Trends Genet 13: 293–295. [DOI] [PubMed] [Google Scholar]
- 45. Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11: 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alterations in cytosine methylation at 5′-CCGG sites as identified by 252 methylation-sensitive amplified polymorphisms among 53 accessions of Brassica in seedlings and buds. The right and left vertical coordinates represent the proportion and number of methylation alterations, respectively.
(PDF)
Pattern and extent of alterations at differentially methylated 5′-CCGG sites from seedlings to buds among parental lines and hybrids of Brassica.
(DOC)
Correlation of hybrid performance (upper triangle) and number of overlapping loci associated with heterosis that were detected in two tissues (lower triangle) for seven traits.
(DOC)
Mid-parent heterosis of agronomic traits among hybrids in Brassica.
(DOC)
Number of cytosine methylation loci in association with heterosis in seedlings and buds, which was selected from 252 loci of MSAP by using single marker analysis.
(DOC)