

Abstract
RATIONALE
Traditionally, free oligosaccharide internal standards are used to account for variability in glycan relative quantification experiments by mass spectrometry. However, a more suitable internal standard would be a glycoprotein, which could also control for enzymatic cleavage efficiency, allowing for more accurate quantitative experiments.
METHODS
Hydrophobic, hydrazide N-linked glycan reagents (both native and stable-isotope labeled) are used to derivatize and differentially label N-linked glycan samples for relative quantification, and the samples are analyzed by a reversed phase liquid chromatography chip system coupled online to a Q-Exactive mass spectrometer. The inclusion of two internal standards, maltoheptaose (previously used) and HRP (novel) are studied to demonstrate the effectiveness of using a glycoprotein as an internal standard in glycan relative quantification experiments.
RESULTS
HRP is a glycoprotein containing a xylosylated N-linked glycan, which is unique from mammalian N-linked glycans. Thus, the internal standard xylosylated glycan was able to be detected without interference to the sample. Additionally, it was shown that differences in cleavage efficiency can be detected by monitoring the HRP glycan. In a sample where cleavage efficiency variation is minimal, the HRP glycan performs as well as maltoheptaose.
CONCLUSIONS
Because the HRP glycan performs as well as maltoheptaose but is also capable of correcting and accounting for cleavage variability, it is a more versatile internal standard and will be used in all subsequent biological studies. Because of the possible lot to lot variation of an enzyme, differences in biological matrix, and variable enzyme activity over time, it is a necessity to account for glycan cleavage variability in glycan relative quantification experiments.
INTRODUCTION
The roles of glycosylation in biological systems are becoming increasingly evident,[1–3] and numerous strategies have recently been developed using mass spectrometry to relatively quantify the amount of glycans in different samples.[4–13] In this type of experiment, two (or more) samples are differentially labeled, processed in parallel, and mixed prior to analysis. The relative amounts of each glycan in the two samples can then be quantified in one liquid chromatography mass spectrometry (LC-MS) analysis. The disadvantage to these types of techniques is the variability introduced into the quantification by processing the samples in parallel. The analysis of N-linked glycans from a complex biological sample, such as human plasma, involves numerous sample preparation steps that each introduces variability into quantitative analysis. Though efforts are taken to keep conditions constant between samples, slight variances in the digestion efficiency, derivatization efficiency, and solid phase extraction (SPE) recovery can skew the relative amounts of glycans detected. Previous studies have utilized free sugars (simple, branched, and/or complex) to normalize or correct for sample preparation variability.[4, 14–19] However, these spiked-in sugars are unable to account for the N-linked glycan cleavage variability between samples.
The total variability (σ2Total) of these measurements is comprised of both analytical (σ2Analytical) and biological (σ2Biological) variability (Equation 1).
| (1) |
Because the main objective is to measure biological change, it is necessary to minimize and account for the analytical variability such that true and significant biological change can be detected. However, when numerous samples must be processed in parallel, the samples are often grouped into ‘batches’. This allows experiments containing hundreds of samples to be divided into smaller groups in order to facilitate sample preparation. However, ‘batch’ processing has a cost due to all samples not being processed in parallel, and there are two main issues with multiple ‘batch’ processing in the same biological data set: 1) samples are often processed slightly different between samples and (to a larger degree) batches (e.g., cleavage temperature and time can be variable), and 2) often the same lot of enzyme is not able to be used for all batches introducing activity variability. These issues pose inherent problems when trying to compare glycan abundances across not only samples but also across entire batches of data. Thus, an internal standard that is capable of monitoring and correcting for sample preparation variability (including N-linked glycan cleavage variability) becomes a necessity in large-scale biological studies of glycosylation.
It is shown in Equation 2 that many steps in the sample preparation and/or the human error contribute to the total variability of the measurement.
 |
(2) |
The dashed box and the solid box represent the variability that can be accounted for by spiking in a free oligosaccharide and a glycoprotein standard, respectively, at the onset of sample preparation. Thus, if there is significant variability in the enzymatic cleavage efficiency, the currently used free oligosaccharide internal standard would not be able to correct for this, a problem that will have an effect on the relative quantification of batch processed samples. It is necessary to monitor this cleavage variability when analyzing numerous biological samples because often the biological matrix of each sample is different and undefined. The matrix could have protease inhibitors or other physical properties that affect the enzymatic release of N-linked glycans. Additionally, when processing large numbers of samples, lot to lot variation, enzyme activity over time, and incubation conditions can all vary. This variability can significantly contribute to quantification results and must be controlled for in order to elucidate real and significant biological change.
A glycoprotein containing N-linked glycans would be an ideal candidate for having the potential to account for all aspects of sample preparation variability, including variation in the cleavage efficiency between samples. However, the glycoprotein must have several favorable properties to be able to be used as an internal standard including commercial availability, cost effectiveness, the ability for the glycans to be cleaved by PNGase F, and most importantly, the glycoprotein must contain N-linked glycans that are not already found in the biological system being studied. Horseradish peroxidase (HRP) is a plant glycoprotein that is frequently used as a colorimetric indicator for antigen-antibody analyses. Previous studies have reported that HRP contains N-linked glycans with a xylosylated and often α1,3-fucosylated core.[20–23] This makes it a good candidate for a glycoprotein internal standard due to the inability of most mammals to produce xylosylated glycans.
Herein, we report on the utility of spiking in HRP at the onset of sample preparation. This allows one to monitor the PNGase F cleavage efficiency across samples, whereas maltoheptaose, a simple sugar internal standard, cannot. This leads to skewed quantification results when there is variability introduced in the cleavage reaction due to differences in enzymatic activity, sample matrix, or degradation. A time course digestion experiment of 6 identical pooled human plasma aliquots is used to simulate variability in digestion efficiency across samples. It is shown that comparing the ratios of the two internal standards do not produce the same correction factor when there is a discrepancy in the efficiency of the cleavage reaction (i.e. at short incubation times). Thus, HRP is a successful internal standard for monitoring and controlling for not only the variation in the parallel sample preparation but also any variability in the enzymatic cleavage process.
EXPERIMENTAL
N-linked Glycan Extraction from Pooled Plasma and Hydrazone Derivatization
N-linked glycans cleaved from 50 μL aliquots of pooled human plasma (Sigma Aldrich, St. Louis, MO) were used to simulate variability in cleavage efficiency by allowing different samples to be incubated for various times ranging from 0 to 24 hr. N-linked glycans were cleaved with PNGase F (New England Biolabs, Ipswitch, MA), an ethanol protein precipitation was performed, and the N-linked glycans were separated from the plasma matrix and remaining proteins as previously reported.[24] Both maltoheptaose (1 μg) and HRP (200 μg) were purchased from Sigma Aldrich (St. Louis, MO) and spiked into the plasma aliquots at the onset of sample preparation. Upon completion of the solid phase extraction of the N-linked glycans, derivatization was performed in parallel as previously reported.[4, 24–26] Samples were derivatized with either the ‘light’ (natural) or ‘heavy’ (stable-isotope labeled) P2GPN hydrazide reagent.[4, 25]
nano-Flow Reversed Phase Chromatography Coupled Online to QExactive MS
The derivatized glycan samples were reconstituted in 200μL of a 5% ACN solution. The sample was vortexed for 1 min and centrifuged for 5 min at 14,000 rpm. The supernatant was extracted and samples were mixed together in equal volumes for relative quantification per the experimental design (vide infra). An EASY-nLC II autosampler and liquid chromatography (LC) system (Thermo Fisher Scientific, San Jose, CA) was used to load 10 μL of the sample at 650 nL/min onto a cHiPLC column system (Eksigent, Dublin, CA) set up in the vented column configuration.[27] Immediately after loading, the sample was washed with an additional 2 μL of mobile phase A. A ChromXP C18-CL 75 μm × 15 cm analytical column (Eksigent, Dublin, CA) was used for both the trap and the analytical column, allowing for an effective column length of 30 cm. The EASY-nLC II was used to perform gradient elution, where mobile phase A consisted of a 2% acetonitrile (ACN) and 0.2% formic acid solution, and mobile phase B consisted of a 98% ACN and 0.2% formic acid solution. Glycans were separated at a constant flow of 300 nL/min. The initial solvent condition was 2% mobile phase B and held for 1 min. The mobile phase B composition was then increased to 22% over 1 min and further increased to 35% over 35 min. The column was washed with 90% mobile phase B and then re-equilibrated in the initial conditions for 15 min. The LC eluent was detected online by a QExactive mass spectrometer equipped with a nanospray source (Thermo Fisher Scientific, San Jose, CA). Electrospray ionization was achieved by applying 2.25 kV at the union adjoining the emitter tip and the outlet capillary of the LC. The capillary was heated to 225 °C, and the instrument was calibrated per the manufacturer specifications. Data dependent acquisition was performed, such that up to 5 MS/MS spectra were taken per precursor scan, where the most abundant ions were chosen for fragmentation. After a precursor ion has been chosen for fragmentation, it is put on an exclude list for 25 s. The precursor ions were detected in the orbitrap mass analyzer at 70,000 resolving power at m/z = 200 with a 1 × 106 automatic gain control (AGC) and a maximum injection time of 250 ms. The ions chosen for fragmentation were subjected to higher energy collision dissociation (HCD) at a normalized collision energy (NCE) of 20. The product-ion spectra were collected in the orbitrap as well and detected at 17,500 resolving power at m/z = 200, with a 2 × 104 AGC and a maximum injection time of 120 ms. The unique glycan compositions were identified manually using accurate mass and analyzed using Xcalibur software (v.2.2).
RESULTS and DISCUSSION
Two 200 μg samples of HRP were prepared, derivatized with the natural and SIL hydrazide tags, combined in equal volumes, and analyzed as a control using the same glycan extraction procedure as that in plasma to determine the complexity HRP would add when spiked into a plasma sample. Only two N-linked glycans were detected in the spectra, Hex3HexNAc2Xyl1 and Hex3HexNAc2Fuc1Xyl1, and both of these glycans contain xylose (as reported in the literature)[20–23], which is not found in human biosynthetic pathways. However, the Hex3HexNAc2Fuc1Xyl1 has a low signal to noise ratio, which could introduce technical variation; thus, the more abundant Hex3HexNAc2Xyl1 (Figure 1b) will be referred to as the “HRP” internal standard and used to determine the effectiveness of accounting for the total sample preparation variability using a xylosylated N-linked glycoprotein. Figure 1a shows a generic plant protein with xylosylated N-linked glycan cores. The lack of xylose found in mammalian biosynthetic pathways allows plant N-linked glycans to be unique from the N-linked glycans of interest. An additional property of many plant glycoproteins is the presence of α1–3 fucosylated core, and glycans containing this motif are not able to be cleaved by PNGase F. This is also the reason only two N-linked glycans were detected from HRP. These attributes, along with the commercial availability, make HRP a great candidate for a glycoprotein internal standard.
Figure 1.
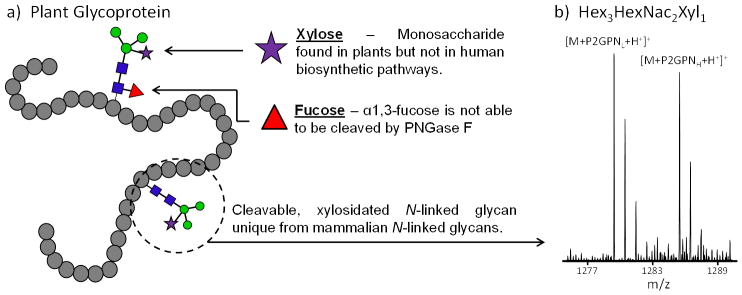
a) Generic plant glycoprotein showing core α1,3-fucose and core xylose containing glycans; b) HRP internal standard, Hex3HexNAc2Xyl1, detected after treating the two control samples with PNGase F, differential derivatization, and combining the samples in equal volumes.
A time course glycan deglycosylation experiment was used to simulate different enzymatic cleavage efficiencies in aliquots of the same pooled human plasma sample. Six pooled human plasma samples were denatured and subjected to PNGase F digestion. One sample was removed at each of the following time points: 0, 4, 8, and 12 hr. Two samples were incubated for a full 24 hr (24a and 24b). When removed from the incubator, each sample was immediately quenched with ethanol and placed in the −80°C freezer. After the final 24 hr samples were removed from the incubator, all samples were allowed to incubate at −80°C for 1 additional hr, centrifuged, and dried. All aliquots were processed identically aside from the amount of time they were incubated for deglycosylation and the amount of time spent in the −80°C ethanol incubation. Each sample will be referred to by the PNGase F deglycosylation incubation time for the sample. The 0, 8, and 24a samples were derivatized with the ‘light’ P2GPN reagent, and the 4, 12, and 24b samples were derivatized with the ‘heavy’ P2GPN reagent. This allowed for comparisons between each adjacent (in time) sample and a comparison between each sample and one of the 24 hr time points.
Figure 2a shows the utility of using a glycoprotein as an internal standard by plotting the HRP glycan (corrected by the maltoheptaose internal standard) at different PNGase F incubation times. It is shown at the early time points that the maltoheptaose is incapable of taking into account poor efficiency in the PNGase F cleavage reaction. This is experimental evidence that the free oligosaccharide can only account for the variability in the dashed box in Equation 2. However, because the PNGase reaction efficiency can be monitored over time using the HRP glycan, all of the variability in the solid box in Equation 2 can be accounted for, making the HRP glycoprotein a more effective internal standard.
Figure 2.
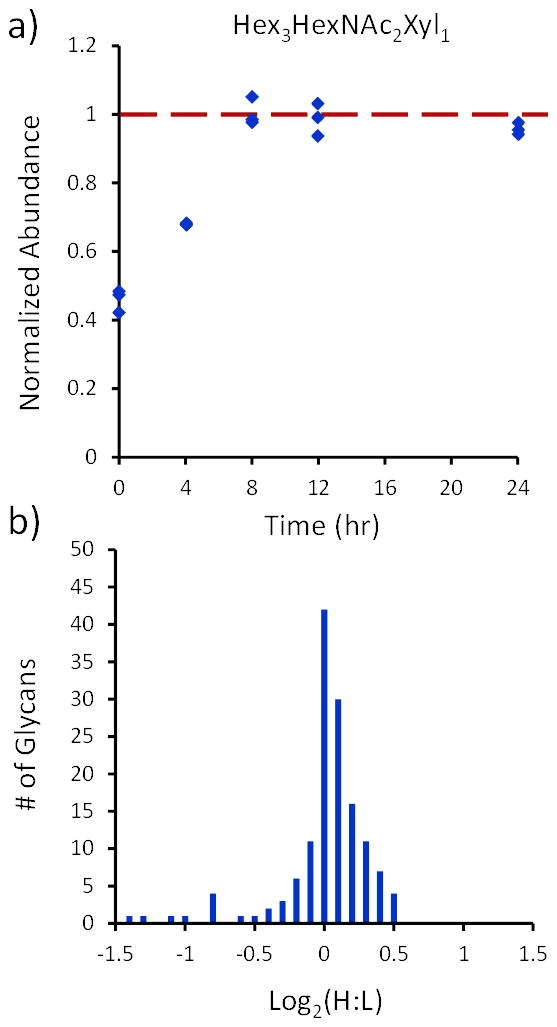
a) The HRP glycan relative abundances, corrected by maltoheptaose and normalized to the 24-hr time point, at each of the 5 PNGase F incubation times. The abundances are normalized to the 24-hr time point, where 100% cleavage efficiency is assumed. b) Histogram plotting the log2(Heavy:Light) for the 24a-24b sample and corrected by HRP.
Though it is shown that the HRP is effective in accounting for enzymatic cleavage variability, in order for it to be a productive internal standard, it must also account for the additional sample preparation variability shown in Equation 2. Thus, the two aliquots of plasma that were incubated for 24 hours were used to determine the effectiveness of the HRP glycan as an internal standard for correcting for σ2Total in glycan relative quantification experiments. The 24a-24b comparison sample is assumed to have no biological variability (σ2Biological = 0). Thus, any systematic variability in the measurement should be able to be corrected using an internal standard. Figure 2b shows the relative quantification histogram of the 50 most abundant N-linked glycans in the 24a-24b comparison corrected by HRP. The mean ± 95% confidence interval for the heavy:light glycan ratio is 1.001 ± 0.037, which statistically encompasses the expected 1:1 ratio. Histograms for other time points have not been shown due to the large variation in the cleavage efficiencies of different glycans at short digestion times. Neither internal standard is capable of reducing the random variability due to the variable cleavage rates of each glycan composition. This is why glycan cleavage is performed for >18 hr. Additionally, matrix effects on the cleavage efficiency would be systematic to all glycans, thus correctable using HRP.
Figure 2b also shows the analytical variability of this relative quantification strategy of N-linked glycans in a complex biological mixture. Because two aliquots of the same plasma sample were used for the 24a-24b time points, zero biological variability can be assumed. Thus, it is shown by the distribution in Figure 2b that a ratio with more than ± 40% deviation from unity can be deemed as a biologically significant change. By introducing HRP as an internal standard, the systematic variability of analytical sample preparation can be minimized, and if there are negligible differences in the cleavage reaction efficiency, HRP as an internal standard performs as well as spiking in a standard oligosaccharide. However, in situations where there is significant variation in the cleavage efficiency, the HRP internal standard out-performs the free oligosaccharide (Figure 2a) and is more suited to correct for analytical systematic variation than a free oligosaccharide. Importantly, for a large number of samples that are carried out in batches, the approach described herein is critical to make quantitative comparisons.
CONCLUSION
The use of a glycoprotein as an internal standard has the potential to correct for both the cleavage and sample preparation variability between two samples, and HRP has been shown to be an effective glycoprotein for this task. HRP will be used in future studies as the primary internal standard for correcting sample preparation and PNGase F cleavage efficiency for N-linked glycan large scale relative quantification studies. Additionally, maltoheptaose will also be used as a secondary internal standard. This will allow one to measure the systematic variability due to parallel sample preparation alone (maltoheptaose ratio), and by comparing the HRP light and heavy abundances to those of the maltoheptaose, the variability solely due to the enzymatic cleavage reaction can be determined. By monitoring this, we can systematically account for any enzyme lot to lot variability, variation in the enzyme activity over time, variation in incubation conditions between batches, and the variability of each undefined biological matrix (e.g. plasma) which can inhibit glycan cleavage. Though it is often assumed that the glycan cleavage efficiency between samples has little variation due to the long incubation times, it would be analytically improper to not incorporate a suitable control for this assumption when studying biological change over a large number of samples. Thus, HRP provides a widely available, cost-effective tool that is easily incorporated into a majority of N-linked glycan relative quantification workflows.
Acknowledgments
The authors would like to gratefully acknowledge the financial support received from the NIH (Grant #R33 CA147988-02), the W. M. Keck Foundation, and North Carolina State University.
References
- 1.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. BBA-Gen Subjects. 1999;1473:4. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 2.Begley TP. Vol. 2. John Wiley & Sons, Inc; Hoboken, New Jersey: 2009. p. 785. [Google Scholar]
- 3.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2009. [Google Scholar]
- 4.Walker SH, Budhathoki-Uprety J, Novak BM, Muddiman DC. Stable-isotope labeled hydrophobic hydrazide reagents for the relative quantification of N-linked glycans by electrospray ionization mass spectrometry. Anal Chem. 2011;83:6738. doi: 10.1021/ac201376q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez-Manilla G, Warren NL, Abney T, Atwood J, Azadi P, York WS, Pierce M, Orlando R. Tools for glycomics: relative quantitation of glycans by isotopic permethylation using (CH3I)-C-13. Glycobiology. 2007:677. doi: 10.1093/glycob/cwm033. [DOI] [PubMed] [Google Scholar]
- 6.Atwood JA, Cheng L, Alvarez-Manilla G, Warren NL, York WS, Orlando R. Quantitation by isobaric labeling: Applications to glycomics. J Proteome Res. 2008:367. doi: 10.1021/pr070476i. [DOI] [PubMed] [Google Scholar]
- 7.Kang P, Mechref Y, Kyselova Z, Goetz JA, Novotny MV. Comparative glycomic mapping through quantitative permethylation and stable-isotope labeling. Anal Chem. 2007:6064. doi: 10.1021/ac062098r. [DOI] [PubMed] [Google Scholar]
- 8.Bowman MJ, Zaia J. Comparative Glycomics Using a Tetraplex Stable-Isotope Coded Tag. Anal Chem. 2010;82:3023. doi: 10.1021/ac100108w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowman MJ, Zaia J. Tags for the stable isotopic labeling of carbohydrates and quantitative analysis by mass spectrometry. Anal Chem. 2007;79:5777. doi: 10.1021/ac070581b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia BY, Feasley CL, Sachdev GP, Smith DF, Cummings RD. Glycan reductive isotope labeling for quantitative glycomics. Anal Biochem. 2009;387:162. doi: 10.1016/j.ab.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlando R, Lim JM, Atwood JA, Angel PM, Fang M, Aoki K, Alvarez-Manilla G, Moremen KW, York WS, Tiemeyer M, Pierce M, Dalton S, Wells L. IDAWG: Metabolic Incorporation of Stable Isotope Labels for Quantitative Glycomics of Cultured Cells. J Proteome Res. 2009:3816. doi: 10.1021/pr8010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahne H, Neubert P, Kuhn K, Etienne C, Bomgarden R, Rogers JC, Kuster B. Carbonyl-Reactive Tandem Mass Tags for the Proteome-Wide Quantification of N-Linked Glycans. Anal Chem. 2012;84:3716. doi: 10.1021/ac300197c. [DOI] [PubMed] [Google Scholar]
- 13.Zhang P, Zhang Y, Xue XD, Wang CJ, Wang ZF, Huang LJ. Relative quantitation of glycans using stable isotopic labels 1-(d0/d5) phenyl-3-methyl-5-pyrazolone by mass spectrometry. Anal Biochem. 2011;418:1. doi: 10.1016/j.ab.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 14.Dixon RB, Bereman MS, Petitte JN, Hawkridge AM, Muddiman DC. One-year plasma N-linked glycome intra-individual and inter-individual variability in the chicken model of spontaneous ovarian adenocarcinoma. Int J Mass Spectrom. 2011;305:79. doi: 10.1016/j.ijms.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bereman MS, Muddiman DC. The effects of abundant plasma protein depletion on global glycan profiling using NanoLC FT-ICR mass spectrometry. Anal Bioanal Chem. 2010;396:1473. doi: 10.1007/s00216-009-3368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bereman MS, Young DD, Deiters A, Muddiman DC. Development of a Robust and High Throughput Method for Profiling N-Linked Glycans Derived from Plasma Glycoproteins by NanoLC-FTICR Mass Spectrometry. J Proteome Res. 2009;8:3764. doi: 10.1021/pr9002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Leoz MLA, Young LJT, An HJ, Kronewitter SR, Kim JH, Miyamoto S, Borowsky AD, Chew HK, Lebrilla CB. High-Mannose Glycans are Elevated during Breast Cancer Progression. Mol Cell Proteomics. 2011;10:1. doi: 10.1074/mcp.M110.002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snovida SI, Perreault H. A 2,5-dihydroxybenzoic acid/N,N-dimethylaniline matrix for the analysis of oligosaccharides by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 2007;21:3711. doi: 10.1002/rcm.3265. [DOI] [PubMed] [Google Scholar]
- 19.Kaneshiro K, Watanabe M, Terasawa K, Uchimura H, Fukuyama Y, Iwamoto S, Sato TA, Shimizu K, Tsujimoto G, Tanaka K. Rapid Quantitative Profiling of N-Glycan by the Glycan-Labeling Method Using 3-Aminoquinoline/alpha-Cyano-4-hydroxycinnamic Acid. Anal Chem. 2012;84:7146. doi: 10.1021/ac301484f. [DOI] [PubMed] [Google Scholar]
- 20.Kamerling JP. Xylose-Containing Carbohydrate Chains Derived from N-Glycoproteins. Pure & Appl Chem. 1991;63:465. [Google Scholar]
- 21.Wuhrer M, Hokke CH, Deelder AM. Glycopeptide analysis by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry reveals novel features of horseradish peroxidase glycosylation. Rapid Commun Mass Spectrom. 2004;18:1741. doi: 10.1002/rcm.1546. [DOI] [PubMed] [Google Scholar]
- 22.Lee BS, Krisnanchettiar S, Lateef SS, Lateef NS, Gupta S. Oligosaccharide analyses of glycopeptides of horseradish peroxidase by thermal-assisted partial acid hydrolysis and mass spectrometry. Carbohydrate Res. 2005;340:1859. doi: 10.1016/j.carres.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 23.Matsuo K. Small-scale, high-throughput method for plant N-glycan preparation for matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis. Anal Biochem. 2011;413:200. doi: 10.1016/j.ab.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 24.Walker SH, Carlisle BC, Muddiman DC. Systematic Comparison of Reverse Phase and Hydrophilic Interaction Liquid Chromatography Platforms for the Analysis of N-Linked Glycans. Anal Chem. 2012;84:8198. doi: 10.1021/ac3012494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker SH, Lilley LM, Enamorado MF, Comins DL, Muddiman DC. Hydrophobic Derivatization of N-linked Glycans for Increased Ion Abundance in Electrospray Ionization Mass Spectrometry. J Am Soc Mass Spectrom. 2011;22:1309. doi: 10.1007/s13361-011-0140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker SH, Papas BN, Comins DL, Muddiman DC. Interplay of Permanent Charge and Hydrophobicity in the Electrospray Ionization of Glycans. Anal Chem. 2010;82:6636. doi: 10.1021/ac101227a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrews GL, Shuford CM, Burnett JC, Hawkridge AM, Muddiman DC. Coupling of a vented column with splitless nanoRPLC-ESI-MS for the improved separation and detection of brain natriuretic peptide-32 and its proteolytic peptides. J Chromatogr B. 2009;877:948. doi: 10.1016/j.jchromb.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]